Csf1 Deficiency Dysregulates Glial Responses to Demyelination and Disturbs CNS White Matter Remyelination


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Surgical Procedures
2.3. Tissue Processing
2.4. Immunofluorescence
2.5. Oli-Red-O Staining
2.6. Semi-Thin Resin Sections, Rank Analysis, and Electron Microscopy Analysis
2.7. In Vitro Cell Culture and Treatment
2.8. Hematopoietic Precursor Isolation
2.9. Global Gene Expression Analysis
2.10. Statistical Analysis of Microarray Data
2.11. Quantification and Statistical Analysis
3. Results
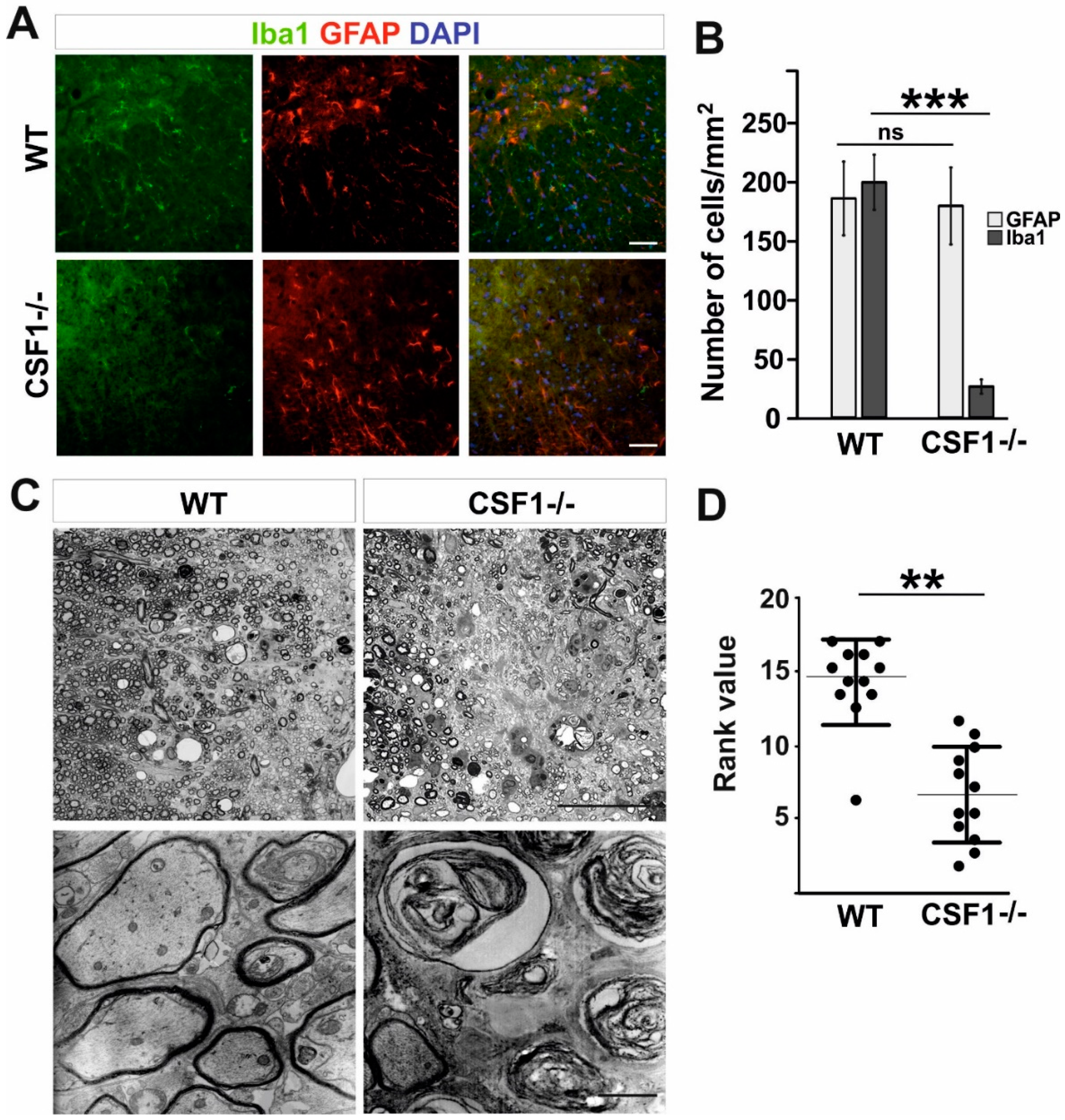
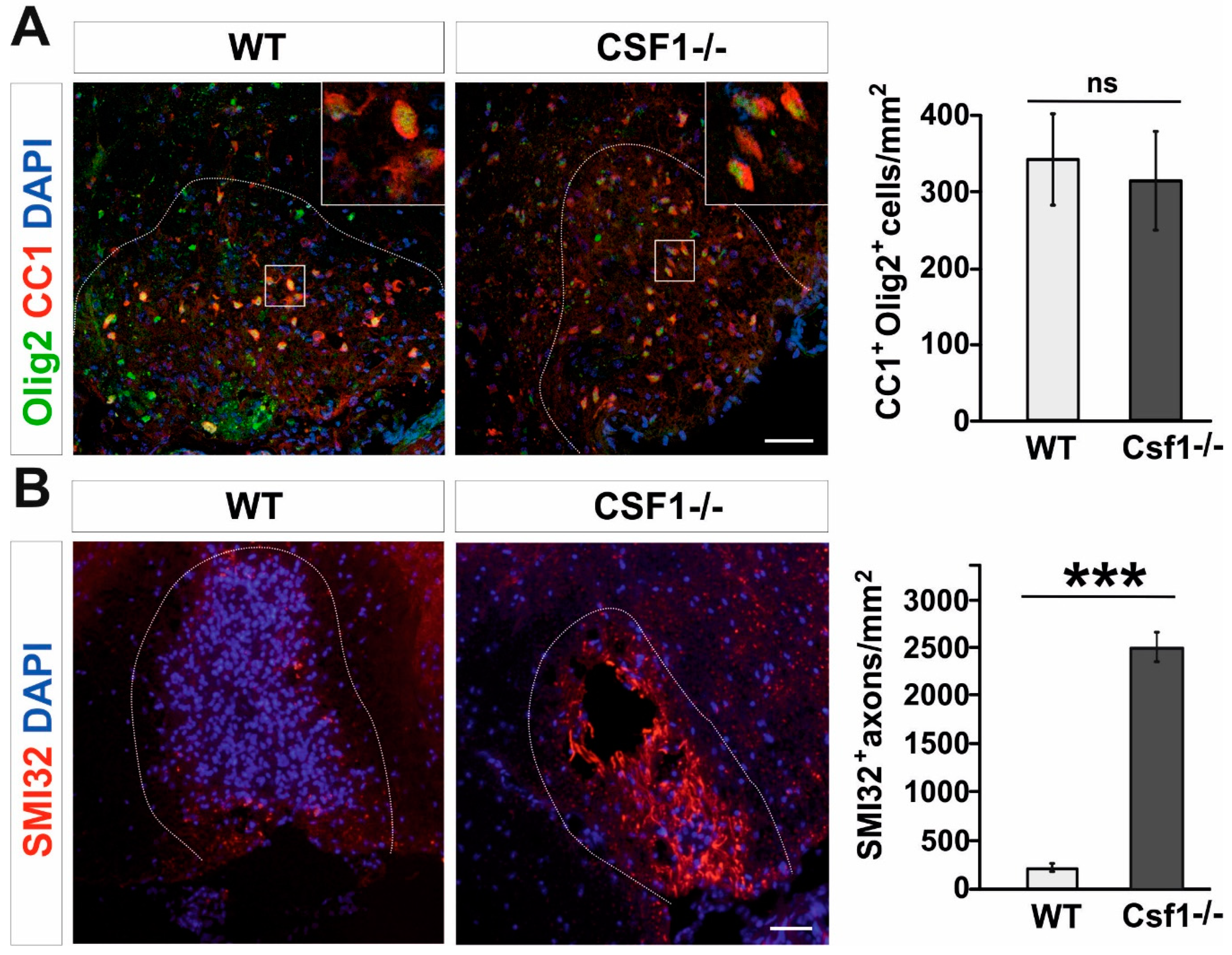
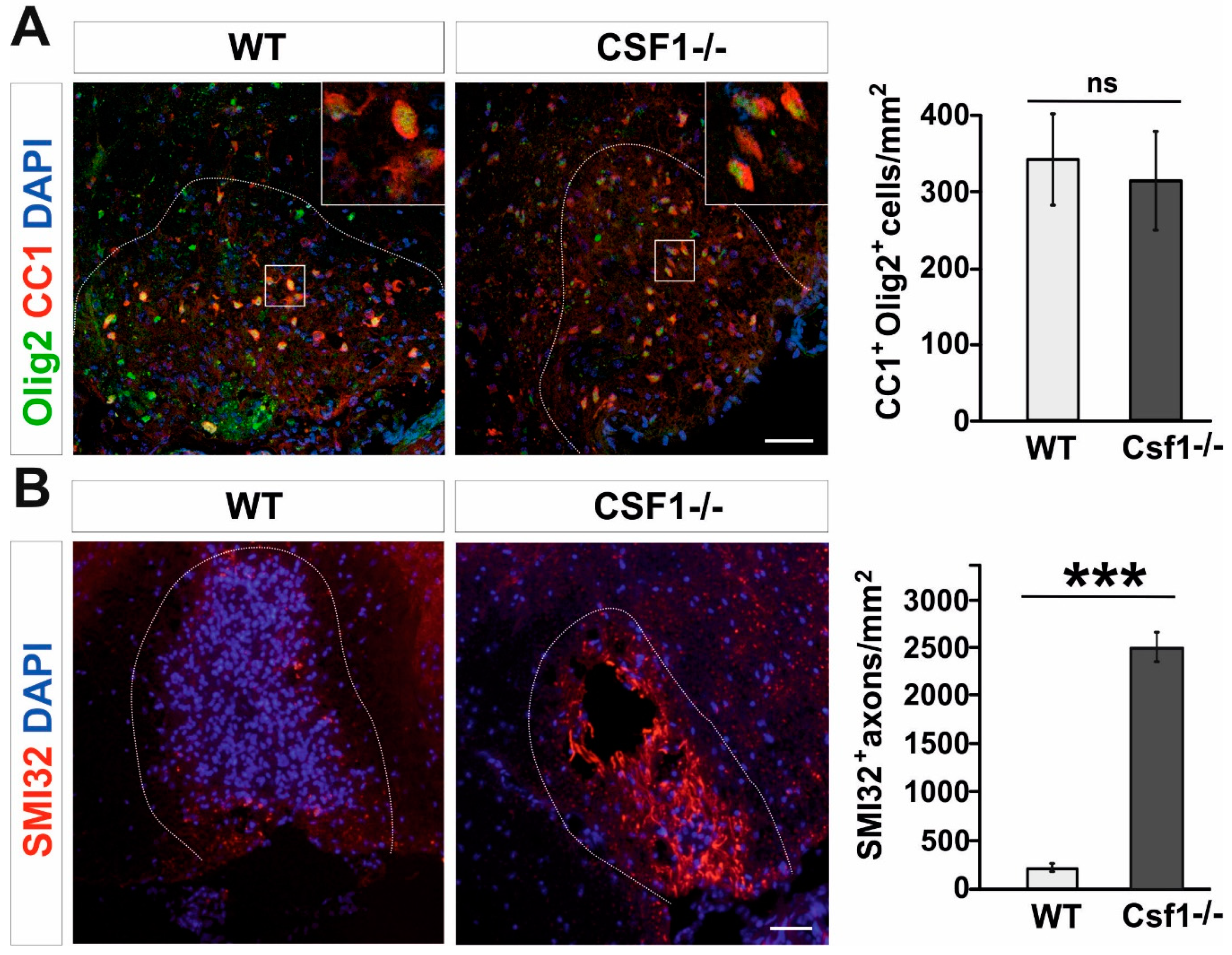
3.1. Csf1 Deficiency Disturbs Remyelination despite Unaltered Recruitment and Differentiation of OPCs
3.2. Csf1 and Csf2 Differentially Alter the Proliferation and Maturation of Monocyte-Lineage Progenitor Cells In Vitro
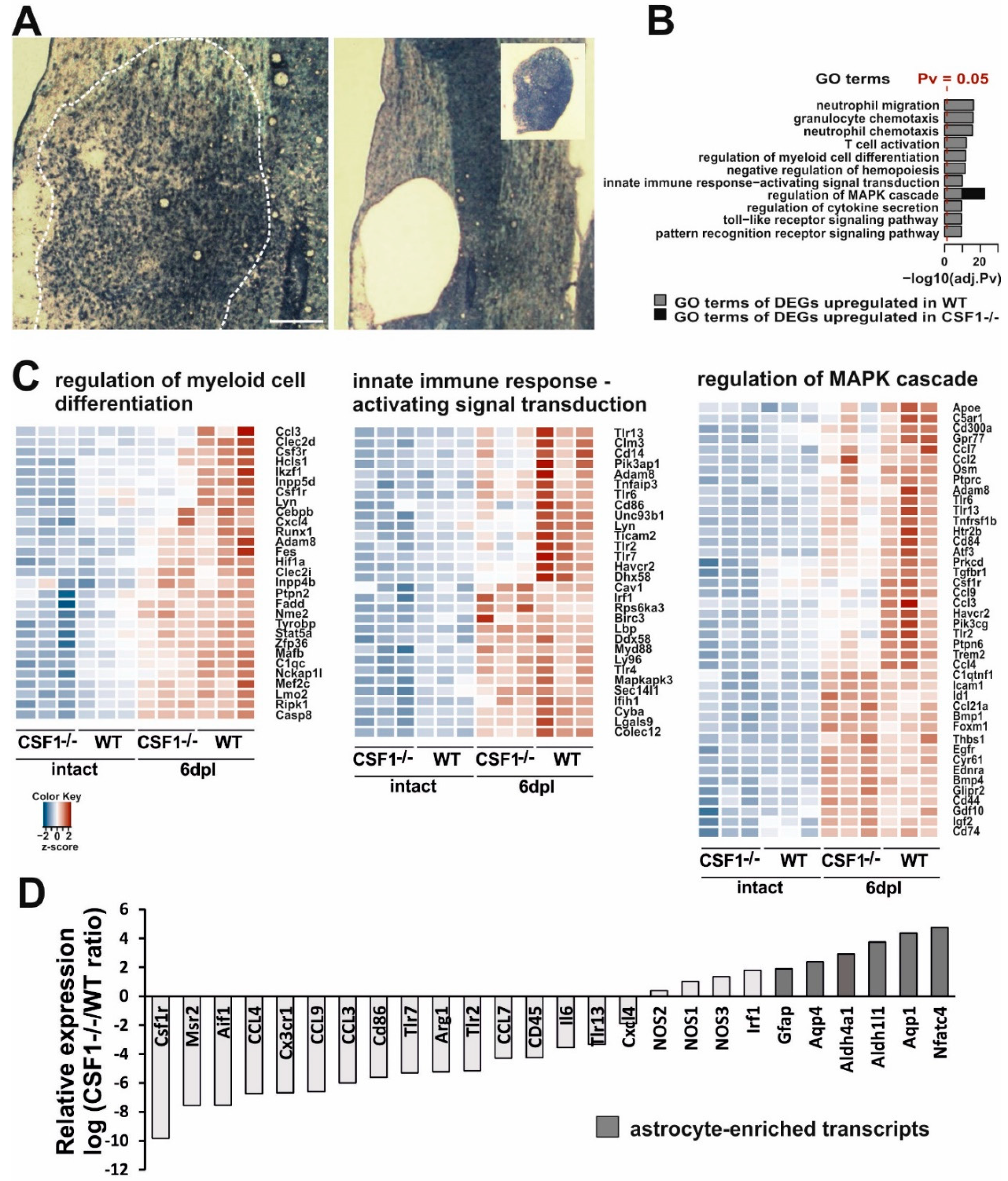
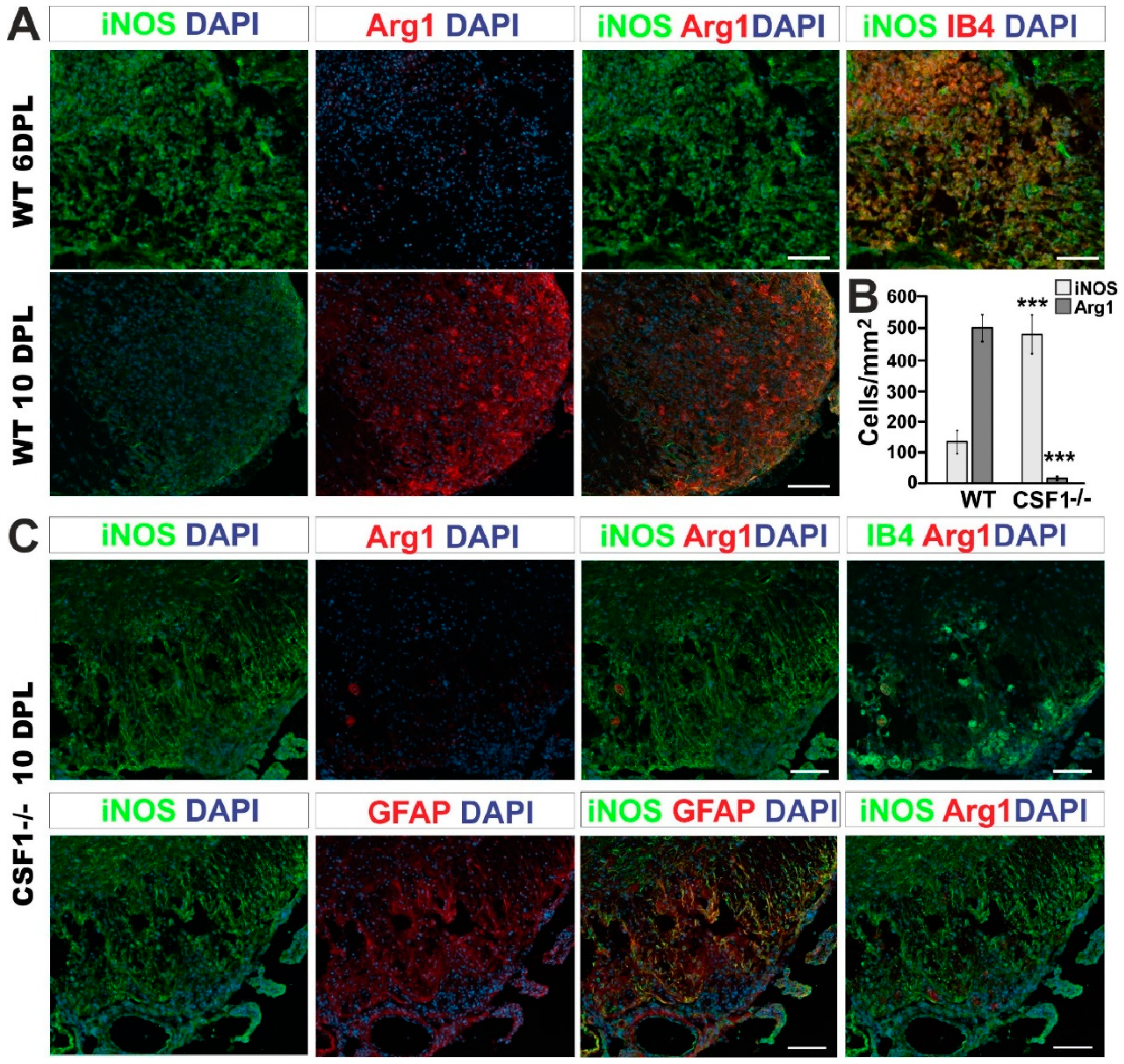
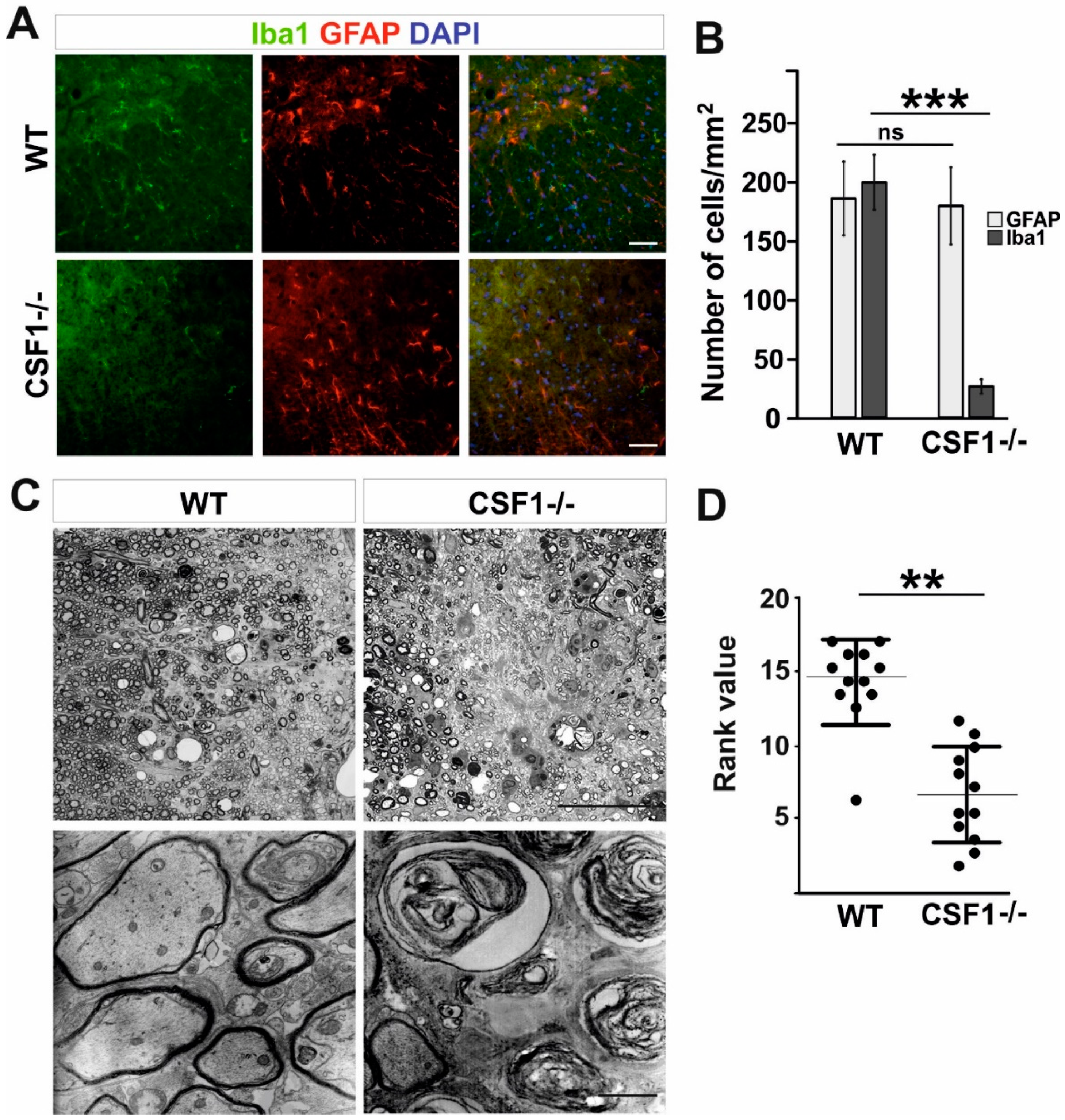
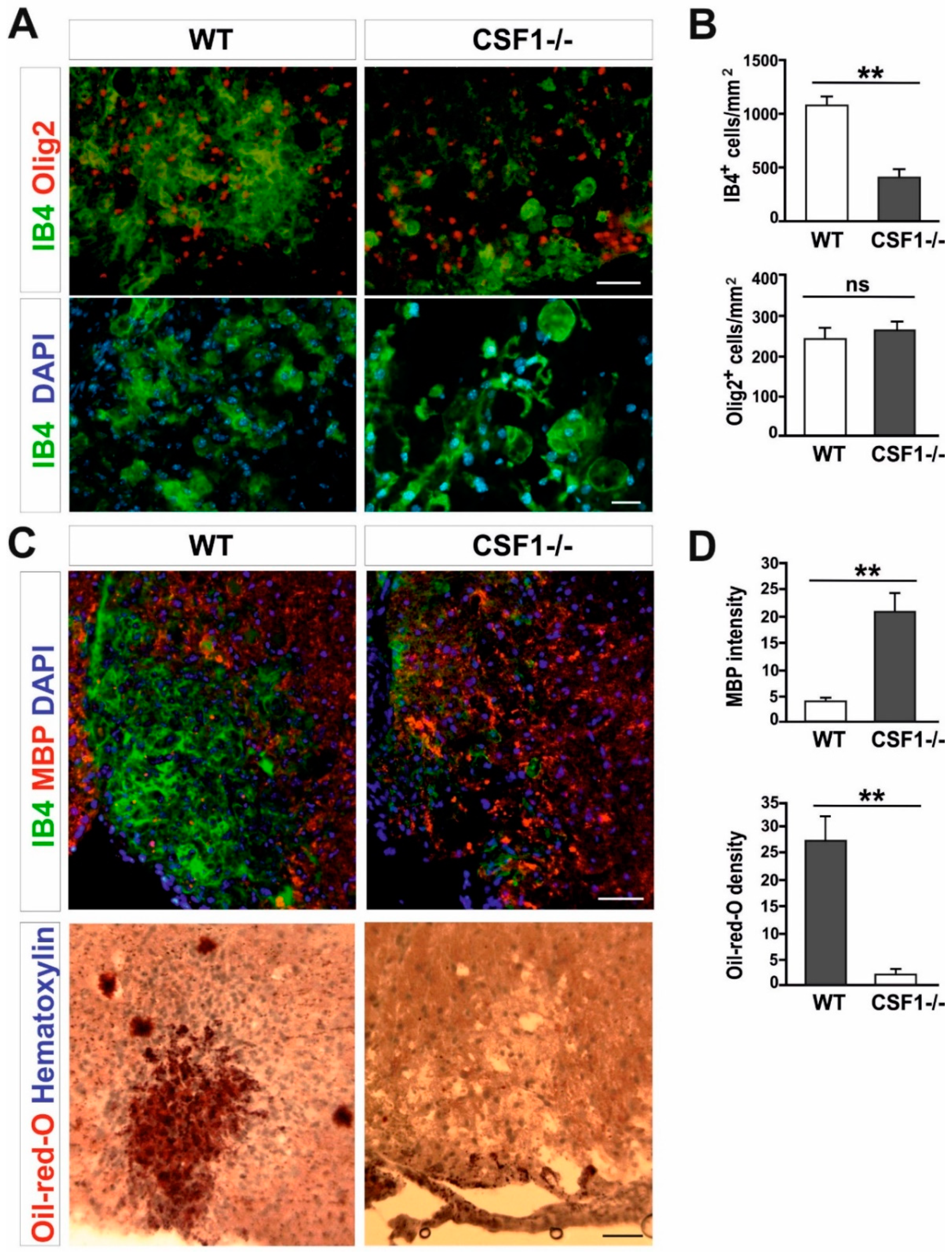
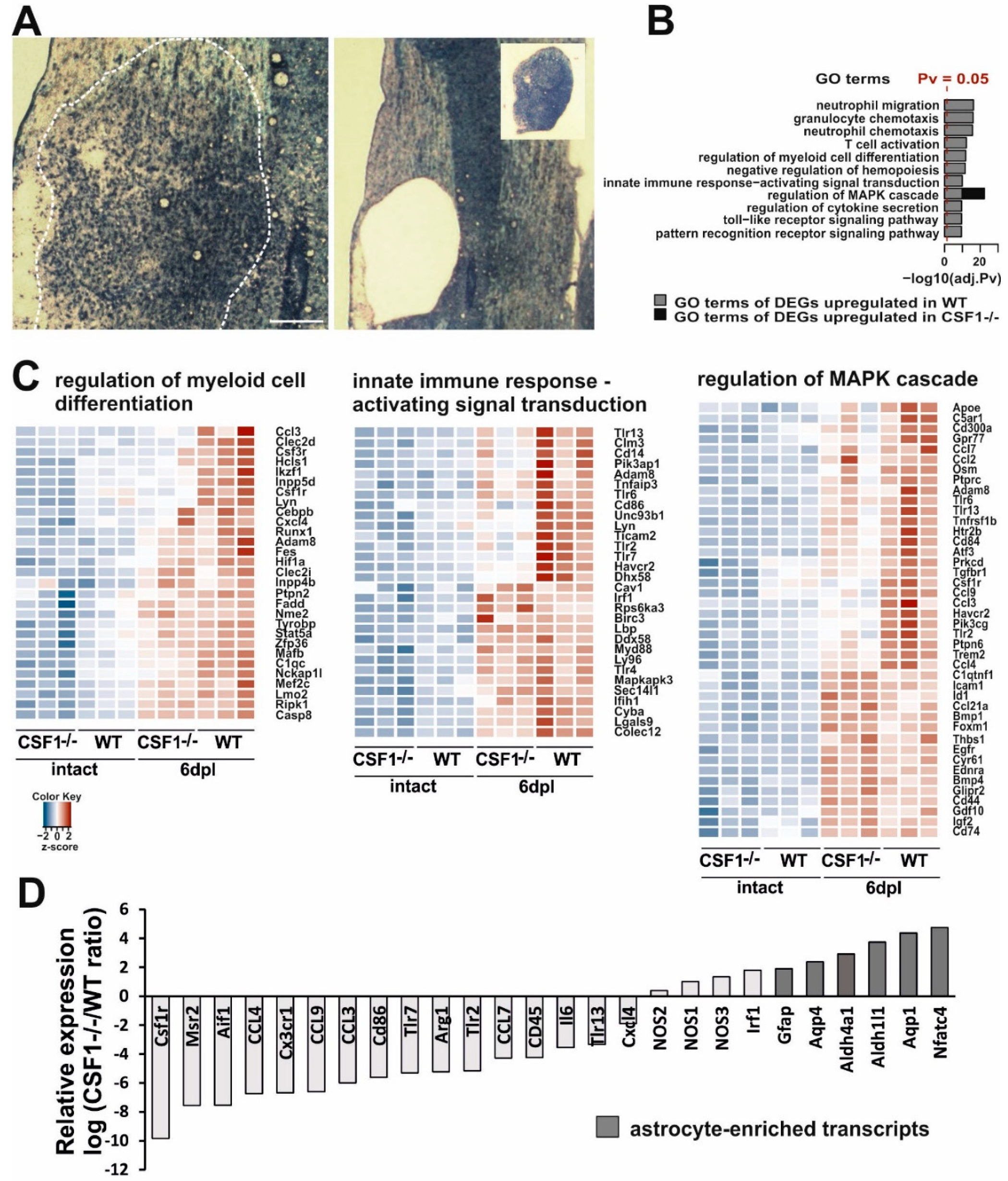
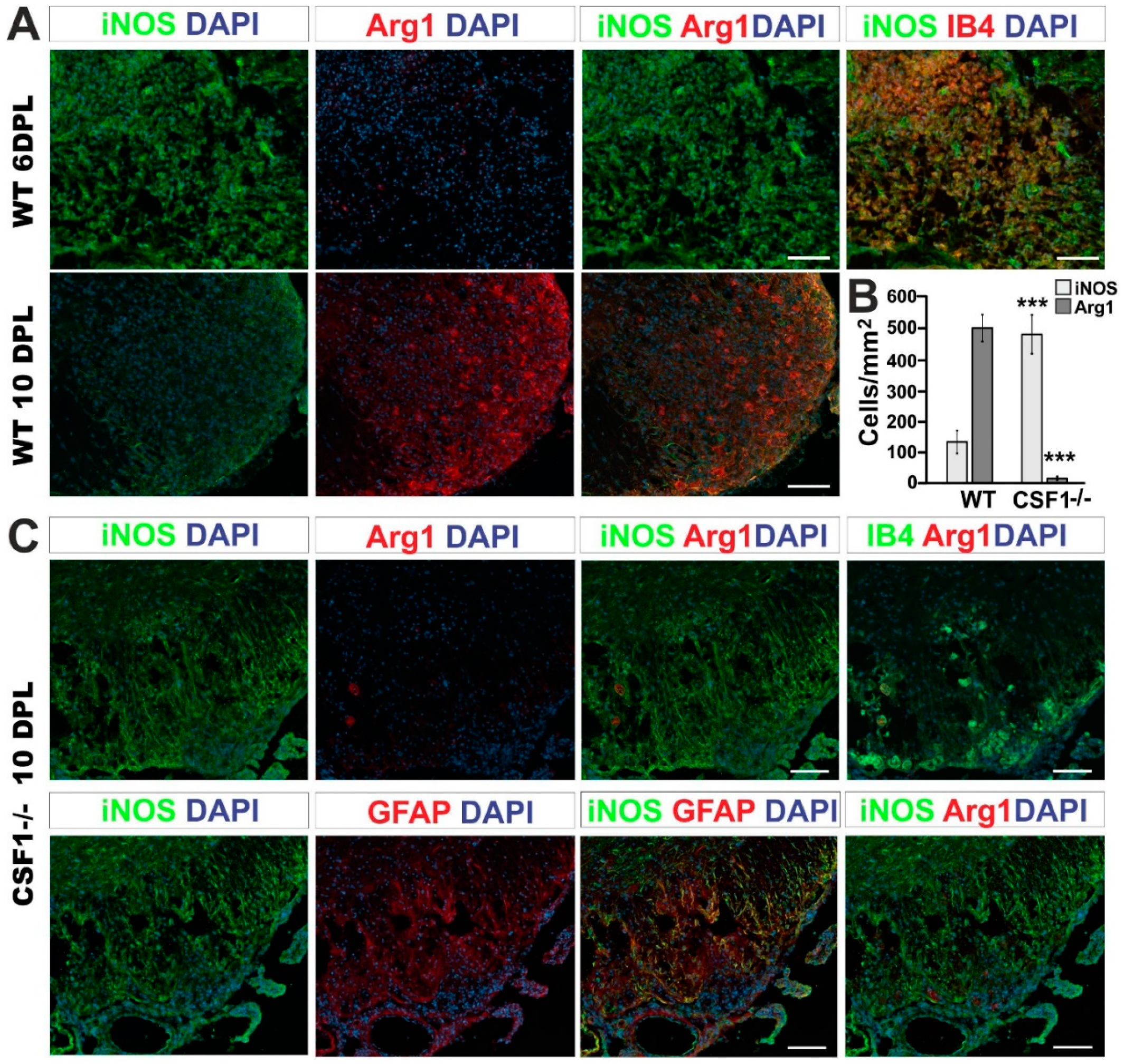
3.3. Reduction of Microglia/Macrophages Alters Pan-Glia Response Dynamic after Demyelination
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Raine, C.S.; Wu, E. Multiple sclerosis: Remyelination in acute lesions. J. Neuropathol. Exp. Neurol. 1993, 52, 199–204. [Google Scholar] [CrossRef] [PubMed]
- Huitinga, I.; van Rooijen, N.; de Groot, C.J.; Uitdehaag, B.M.; Dijkstra, C.D. Suppression of experimental allergic encephalomyelitis in Lewis rats after elimination of macrophages. J. Exp. Med. 1990, 172, 1025–1033. [Google Scholar] [CrossRef] [PubMed]
- Bitsch, A.; Kuhlmann, T.; Da Costa, C.; Bunkowski, S.; Polak, T.; Bruck, W. Tumour necrosis factor alpha mRNA expression in early multiple sclerosis lesions: Correlation with demyelinating activity and oligodendrocyte pathology. Glia 2000, 29, 366–375. [Google Scholar] [CrossRef]
- Becher, B.; Blain, M.; Antel, J.P. CD40 engagement stimulates IL-12 p70 production by human microglial cells: Basis for Th1 polarization in the CNS. J. Neuroimmunol. 2000, 102, 44–50. [Google Scholar] [CrossRef]
- Hill, K.E.; Zollinger, L.V.; Watt, H.E.; Carlson, N.G.; Rose, J.W. Inducible nitric oxide synthase in chronic active multiple sclerosis plaques: Distribution, cellular expression and association with myelin damage. J. Neuroimmunol. 2004, 151, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Lassmann, H.; Bruck, W.; Lucchinetti, C. Heterogeneity of multiple sclerosis pathogenesis: Implications for diagnosis and therapy. Trends Mol. Med. 2001, 7, 115–121. [Google Scholar] [CrossRef]
- Rose, J.W.; Hill, K.E.; Watt, H.E.; Carlson, N.G. Inflammatory cell expression of cyclooxygenase-2 in the multiple sclerosis lesion. J. Neuroimmunol. 2004, 149, 40–49. [Google Scholar] [CrossRef]
- Cannella, B.; Raine, C.S. Multiple sclerosis: Cytokine receptors on oligodendrocytes predict innate regulation. Ann Neurol. 2004, 55, 46–57. [Google Scholar] [CrossRef]
- Matute, C.; Pérez-Cerdá, F. Multiple sclerosis: Novel perspectives on newly forming lesions. Trends Neurosci. 2005, 28, 173–175. [Google Scholar] [CrossRef]
- Sospedra, M.; Martin, R. Immunology of multiple sclerosis. Annu. Rev. Immunol. 2005, 23, 683–747. [Google Scholar] [CrossRef] [Green Version]
- Prineas, J.; McDonald, W.; Franklin, R.J.M. Demyelinating diseases. In Greenfield’s Neuropathology; Graham, D.I., Lantos, P.L., Eds.; CRC Press: London, UK, 2002; pp. 471–550. [Google Scholar]
- Raine, C.S. The Norton Lecture: A review of the oligodendrocyte in the multiple sclerosis lesion. J. Neuroimmunol. 1997, 77, 135–152. [Google Scholar] [CrossRef]
- Wolswijk, G. Oligodendrocyte precursor cells in the demyelinated multiple sclerosis spinal cord. Brain 2002, 125, 338–349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graça, D.L.; Blakemore, W.F. Delayed remyelination in rat spinal cord following ethidium bromide injection. Neuropathol. Appl. Neurobiol. 1986, 12, 593–605. [Google Scholar] [CrossRef] [PubMed]
- Hinks, G.L.; Franklin, R.J.M. Distinctive patterns of PDGF-A, FGF-2, IGF-I, and TGF-beta1 gene expression during remyelination of experimentally-induced spinal cord demyelination. Mol. Cell. Neurosci. 1999, 14, 153–168. [Google Scholar] [CrossRef] [PubMed]
- Ludwin, S.K. Chronic demyelination inhibits remyelination in the central nervous system. An analysis of contributing factors. Lab. Investig. 1980, 43, 382–387. [Google Scholar]
- Morell, P.; Barrett, C.V.; Mason, J.L.; Toews, A.D.; Hostettler, J.D.; Knapp, G.W.; Matsushima, G.K. Gene expression in brain during cuprizone-induced demyelination and remyelination. Mol. Cell. Neurosci. 1998, 12, 220–227. [Google Scholar] [CrossRef]
- Foote, A.K.; Blakemore, W.F. Inflammation stimulates remyelination in areas of chronic demyelination. Brain 2005, 128, 528–539. [Google Scholar] [CrossRef] [Green Version]
- Kuhlmann, T.; Ludwin, S.; Prat, A.; Antel, J.; Bruck, W.; Lassmann, H. An updated histological classification system for multiple sclerosis lesions. Acta Neuropathol. 2017, 133, 13–24. [Google Scholar] [CrossRef]
- Arnett, H.A.; Mason, J.; Marino, M.; Suzuki, K.; Matsushima, G.K.; Ting, J.P. TNF alpha promotes proliferation of oligodendrocyte progenitors and remyelination. Nat. Neurosci. 2001, 4, 1116–1122. [Google Scholar] [CrossRef]
- Mason, J.L.; Suzuki, K.; Chaplin, D.D.; Matsushima, G.K. Interleukin-1beta promotes repair of the CNS. J. Neurosci. 2001, 21, 7046–7052. [Google Scholar] [CrossRef]
- Li, W.W.; Setzu, A.; Zhao, C.; Franklin, R.J. Minocycline-mediated inhibition of microglia activation impairs oligodendrocyte progenitor cell responses and remyelination in a non-immune model of demyelination. J. Neuroimmunol. 2005, 158, 58–66. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, N.; Pham, L.D.; Hayakawa, K.; Matsuzaki, T.; Seo, J.H.; Magnain, C. Age-related decline in oligodendrogenesis retards white matter repair in mice. Stroke 2013, 44, 2573–2578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, C.; Li, W.W.; Franklin, R.J. Differences in the early inflammatory responses to toxin-induced demyelination are associated with the age-related decline in CNS remyelination. Neurobiol. Aging 2006, 27, 1298–1307. [Google Scholar] [CrossRef] [PubMed]
- Ruckh, J.M.; Zhao, J.W.; Shadrach, J.L.; van Wijngaarden, P.; Rao, T.N.; Wagers, A.J.; Franklin, R.J. Rejuvenation of regeneration in the aging central nervous system. Cell Stem Cell 2012, 10, 96–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kotter, M.R.; Setzu, A.; Sim, F.J.; Van Rooijen, N.; Franklin, R.J. Macrophage depletion impairs oligodendrocyte remyelination following lysolecithin-induced demyelination. Glia 2001, 35, 204–212. [Google Scholar] [CrossRef]
- Kotter, M.R.; Li, W.W.; Zhao, C.; Franklin, R.J. Myelin Impairs CNS Remyelination by Inhibiting Oligodendrocyte Precursor Cell Differentiation. J. Neurosci. 2006, 26, 328–332. [Google Scholar] [CrossRef]
- Olah, M.; Amor, S.; Brouwer, N.; Vinet, J.; Eggen, B.; Biber, K.; Boddeke, H.W. Identification of a microglia phenotype supportive of remyelination. Glia 2012, 60, 306–321. [Google Scholar] [CrossRef]
- Sica, A.; Mantovani, A. Macrophage plasticity and polarization: In vivo veritas. J. Clin. Investig. 2012, 122, 787–795. [Google Scholar] [CrossRef]
- Martinez, F.O.; Gordon, S. The M1 and M2 paradigm of macrophage activation: Time for reassessment. F1000Prime Rep. 2014, 6, 6–13. [Google Scholar] [CrossRef] [Green Version]
- Miron, V.E.; Boyd, A.; Zhao, J.W.; Yuen, T.J.; Ruckh, J.M.; Shadrach, J.L.; van Wijngaarden, P.; Wagers, A.J.; Williams, A.; Franklin, R.J.M.; et al. M2 microglia and macrophages drive oligodendrocyte differentiation during CNS remyelination. Nat. Neurosci. 2013, 16, 1211–1218. [Google Scholar] [CrossRef] [Green Version]
- Dombrowski, Y.; O’Hagan, T.; Dittmer, M.; Penalva, R.; Mayoral, S.R.; Bankhead, P.; Fleville, S.; Eleftheriadis, G.; Zhao, C.; Naughton, M.; et al. Regulatory T cells promote myelin regeneration in the central nervous system. Nat. Neurosci. 2017, 20, 674–680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chitu, V.; Gokhan, Ş.; Nandi, S.; Mehler, M.F.; Stanley, E.R. Emerging roles for CSF-1 receptor and its ligands in the nervous system. Trends Neurosci. 2016, 39, 378–393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiktor-Jedrzejczak, W.; Bartocci, A.; Ferrante, A.W.; Ahmed-Ansari, A.; Sell, K.W.; Pollard, J.W.; Stanley, E.R. Total absence of colony-stimulating factor 1 in the macrophage-deficient osteopetrotic (op/op) mouse. Proc. Natl. Acad. Sci. USA 1990, 87, 4828–4832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshida, H.; Hayashi, S.; Kunisada, T.; Ogawa, M.; Nishikawa, S.; Okamura, H.; Sudo, T.; Shultz, L.D.; Nishikawa, S. The murine mutation osteopetrosis is in the coding region of the macrophage colony stimulating factor gene. Nature 1990, 345, 442–444. [Google Scholar] [CrossRef]
- Raivich, G.; Moreno-Flores, T.; Moller, J.; Kreutzberg, G. Inhibition of post-traumatic microglial proliferation in a genetic model of macrophage colony-stimulating factor deficiency in the mouse. Eur. J. Neurosci. 1994, 6, 1615–1618. [Google Scholar] [CrossRef]
- Berezovskaya, O.; Maysinger, D.; Federoff, S. The hematopoietic cytokine, colony-stimulating factor 1, is also a growth factor in the CNS: Congenital absence of Csf-1 in mice results in abnormal microglial response and increased neuron vulnerability to injury. Int. J. Dev. Neurosci. 1995, 13, 285–299. [Google Scholar] [CrossRef]
- Rogove, A.D.; Lu, W.; Tsirka, S.E. Microglial activation and recruitment, but not proliferation, suffice to mediate neurodegeneration. Cell Death Differ. 2002, 9, 801–806. [Google Scholar] [CrossRef]
- Sielska, M.; Przanowsk, I.P.; Wylot, B.; Gabrusiewicz, K.; Maleszewska, M.; Kijewska, M.; Zawadzka, M.; Kucharska, J.; Vinnakota, K.; Kettenmann, H.; et al. Distinct roles of Csf family cytokines in macrophage infiltration and activation in glioma progression and injury response. J. Pathol. 2013, 230, 310–321. [Google Scholar] [CrossRef]
- Zawadzka, M.; Rivers, L.E.; Fancy, S.P.; Zhao, C.; Tripathi, R.; Jamen, F.; Young, K.; Goncharevich, A.; Pohl, H.; Rizzi, M.; et al. CNS-resident glial progenitor/stem cells produce schwann cells as well as oligodendrocytes during repair of CNS demyelination. Cell Stem Cell 2010, 6, 578–590. [Google Scholar] [CrossRef] [Green Version]
- McMurran, C.E.; Zhao, C.; Franklin, R.J.M. Toxin-based models to investigate demyelination and remyelination. Methods Mol. Biol. 2019, 1936, 377–396. [Google Scholar]
- Zawadzka, M.; Kaminska, B. Immunosuppressant FK506 affects multiple signaling pathways and modulates gene expression in astrocytes. Mol. Cell. Neurosci. 2003, 22, 202–209. [Google Scholar] [CrossRef]
- Randall, T.D.; Weissman, I.L. Phenotypic and functional changes induced at the clonal level in hematopoietic stem cells after 5-fluorouracil treatment. Blood 1997, 89, 3596–3606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Jeffery, N.D.; Blakemore, W.F. Remyelination of mouse spinal cord axons demyelinated by local injection of lysolecithin. J. Neurocytol. 1995, 24, 775–781. [Google Scholar] [CrossRef]
- Sanchez, I.; Hassinger, L.; Sihag, R.; Cleveland, D.; Mohan, P.; Nixon, R. Local control of neurofilament accumulation during radial growth of myelinating axons in vivo. Selective role of site-specific phosphorylation. J. Cell Biol. 2000, 151, 1013–1024. [Google Scholar] [CrossRef] [PubMed]
- Petzold, A.; Gveric, D.; Groves, M.; Schmierer, K.; Grant, D.; Chapman, M.; Keir, G.; Cuzner, L.; Thompson, E.J. Phosphorylation and compactness of neurofilaments in multiple sclerosis: Indicators of axonal pathology. Exp. Neurol. 2008, 213, 326–335. [Google Scholar] [CrossRef] [Green Version]
- Dziedzic, T.; Metz, I.; Dallenga, T.; König, F.B.; Müller, S.; Stadelmann, C.; Brück, W. Wallerian degeneration: A major component of early axonal pathology in multiple sclerosis. Brain Pathol. 2010, 20, 976–985. [Google Scholar] [CrossRef]
- Sternberger, L.A.; Sternberger, N.H. Monoclonal antibodies distinguish phosphorylated and nonphosphorylated forms of neurofilaments in situ. Proc. Natl. Acad. Sci. USA 1983, 80, 6126–6130. [Google Scholar] [CrossRef] [Green Version]
- Su, J.H.; Cummings, B.J.; Cotman, C.W. Plaque biogenesis in brain aging and Alzheimer’s disease. I. Progressive changes in phosphorylation states of paired helical filaments and neurofilaments. Brain Res. 1996, 739, 79–87. [Google Scholar] [CrossRef]
- Nihei, K.; McKee, A.C.; Kowall, N.W. Patterns of neuronal degeneration in the motor cortex of amyotrophic lateral sclerosis patients. Acta Neuropathol. 1993, 86, 55–64. [Google Scholar] [CrossRef]
- Wegner, C.; Esiri, M.M.; Chance, S.A.; Palace, J.; Matthews, P.M. Neocortical neuronal, synaptic, and glial loss in multiple sclerosis. Neurology 2006, 67, 960–967. [Google Scholar] [CrossRef] [PubMed]
- Haines, J.D.; Inglese, M.; Casaccia, P. Axonal damage in multiple sclerosis. Mt Sinai J. Med. 2011, 78, 231–243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matcovitch-Natan, O.; Winter, D.R.; Giladi, A.; Vargas Aguilar, S.; Spinrad, A.; Sarrazin, S.; Ben-Yehuda, H.; David, E.; Zelada González, F.; Perrin, P.; et al. Microglia development follows a stepwise program to regulate brain homeostasis. Science 2016, 353, 8670. [Google Scholar] [CrossRef] [PubMed]
- van der Poel, M.; Ulas, T.; Mizee, M.R.; Hsiao, C.C.; Miedema, S.S.M.; Adelia; Schuurman, K.G.; Helder, B.; Tas, S.W.; Schultze, J.L.; et al. Transcriptional profiling of human microglia reveals grey-white matter heterogeneity and multiple sclerosis-associated changes. Nat. Commun. 2019, 10, 1139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saijo, K.; Glass, C.K. Microglial cell origin and phenotypes in health and disease. Nat. Rev. Immunol. 2011, 11, 775–787. [Google Scholar] [CrossRef]
- Stratoulias, V.; Venero, J.L.; Tremblay, M.È.; Joseph, B. Microglial subtypes: Diversity within the microglial community. EMBO J. 2019, 38, e101997. [Google Scholar] [CrossRef]
- Cahoy, J.D.; Emery, B.; Kaushal, A.; Foo, L.C.; Zamanian, J.L.; Christopherson, K.S.; Xing, Y.; Lubischer, J.L.; Krieg, P.A.; Krupenko, S.A.; et al. A transcriptome database for astrocytes, neurons, and oligodendrocytes: A new resource for understanding brain development and function. J. Neurosci. 2008, 28, 264–278. [Google Scholar] [CrossRef] [Green Version]
- Ulanska-Poutanen, J.; Mieczkowski, J.; Zhao, C.; Konarzewska, K.; Kaza, B.; Pohl, H.B.; Bugajski, L.; Kaminska, B.; Franklin, R.J.; Zawadzka, M. Injury-induced perivascular niche supports alternative differentiation of adult rodent CNS progenitor cells. Elife 2018, 7, e30325. [Google Scholar] [CrossRef]
- Stanley, E.R.; Chitu, V. Csf-1 receptor signaling in myeloid cells. Cold Spring Harb. Perspect. Biol. 2014, 6, a021857. [Google Scholar] [CrossRef] [Green Version]
- Priller, J.; Prinz, M. Targeting microglia in brain disorders. Science 2019, 365, 32–33. [Google Scholar] [CrossRef]
- Dai, X.; Ryan, G.R.; Hapel, A.J.; Dominguez, M.G.; Russell, R.G.; Kapp, S.; Sylvestre, V.; Stanley, E.R. Targeted disruption of the mouse CSF-1 receptor gene results in osteopetrosis, mononuclear phagocyte deficiency, increased primitive progenitor cell frequencies and reproductive defects. Blood 2002, 99, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Ginhoux, F.; Greter, M.; Leboeuf, M.; Nandi, S.; See, P.; Gokhan, S.; Mehler, M.F.; Conway, S.J.; Ng, L.G.; Stanley, E.R.; et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 2010, 330, 841–845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erblich, B.; Zhu, L.; Etgen, A.M.; Dobrenis, K.; Pollard, J.W. Absence of colony stimulation factor-1 receptor results in loss of microglia, disrupted brain development and olfactory deficits. PLoS ONE 2011, 6, e26317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elmore, M.R.; Najafi, A.R.; Koike, M.A.; Dagher, N.N.; Spangenberg, E.E.; Rice, R.A.; Kitazawa, M.; Matusow, B.; Nguyen, H.; West, B.L.; et al. Colony-stimulating factor 1 receptor signaling is necessary for microglia viability, unmasking a microglia progenitor cell in the adult brain. Neuron 2014, 82, 380–397. [Google Scholar] [CrossRef] [Green Version]
- Spangenberg, E.E.; Lee, R.J.; Najafi, A.R.; Rice, R.A.; Elmore, M.R.; Blurton-Jones, M.; West, B.L.; Green, K.N. Eliminating microglia in Alzheimer’s mice prevents neuronal loss without modulating amyloid-β pathology. Brain 2016, 139, 1265–1281. [Google Scholar] [CrossRef] [Green Version]
- Prinz, M.; Erny, D.; Hagemeyer, N. Ontogeny and homeostasis of CNS myeloid cells. Nat. Immunol. 2017, 18, 385–392. [Google Scholar] [CrossRef]
- Wegiel, J.; Wiśniewski, H.M.; Dziewiatkowski, J.; Tarnawski, M.; Kozielski, R.; Trenkner, E.; Wiktor-Jedrzejczak, W. Reduced number and altered morphology of microglial cells in colony stimulating factor-1-deficient osteopetrotic op/op mice. Brain Res. 1998, 804, 135–139. [Google Scholar] [CrossRef]
- Kondo, Y.; Duncan, I.D. Selective reduction in microglia density and function in the white matter of Colony-stimulating factor-1–deficient mice. J. Neurosci. Res. 2009, 87, 2686–2695. [Google Scholar] [CrossRef] [Green Version]
- Lin, H.; Lee, E.; Hestir, K.; Leo, C.; Huang, M.; Bosch, E.; Halenbeck, R.; Wu, G.; Zhou, A. Discovery of a cytokine and its receptor by functional screening of the extracellular proteome. Science 2008, 320, 807–811. [Google Scholar] [CrossRef] [Green Version]
- Easley-Neal, C.; Foreman, O.; Sharma, N.; Zarrin, A.A.; Weimer, R.M. CSF1R Ligands IL-34 and CSF1 Are Differentially Required for Microglia Development and Maintenance in White and Gray Matter Brain Regions. Front. Immunol. 2019, 10, 2199. [Google Scholar] [CrossRef]
- Kana, V.; Desland, F.A.; Casanova-Acebes, M.; Ayata, P.; Badimon, A.; Nabel, E.; Yamamuro, K.; Sneeboer, M.; Tan, I.L.; Flanigan, M.E.; et al. Csf-1 controls cerebellar microglia and is required for motor function and social interaction. J. Exp. Med. 2019, 216, 2265–2281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, J.; Harris, R.A.; Zhang, X.M. An updated assessment of microglia depletion: Current concepts and future directions. Mol. Brain 2017, 10, 25. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Xu, Z.; Xiong, S.; Sun, F.; Qin, G.; Hu, G.; Wang, J.; Zhao, L.; Liang, Y.X.; Wu, T.; et al. Repopulated microglia are solely derived from the proliferation of residual microglia after acute depletion. Nat. Neurosci. 2018, 21, 530–540. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Given, K.S.; Dickson, E.L.; Owens, G.P.; Macklin, W.B.; Bennett, J.L. Concentration-dependent effects of Csf1R inhibitors on oligodendrocyte progenitor cells ex vivo and in vivo. Exp. Neurol. 2019, 318, 32–41. [Google Scholar] [CrossRef] [PubMed]
- Rademakers, R.; Baker, M.; Nicholson, A.M.; Rutherford, N.J.; Finch, N.; Soto-Ortolaza, A.; Lash, J.; Wider, C.; Wojtas, A.; DeJesus-Hernandez, M.; et al. Mutations in the colony stimulating factor 1 receptor (CSF1R) gene cause hereditary diffuse leukoencephalopathy with spheroids. Nat. Genet. 2011, 44, 200–205. [Google Scholar] [CrossRef] [PubMed]
- Oosterhof, N.; Chang, I.J.; Karimiani, E.G.; Kuil, L.E.; Jensen, D.M.; Daza, R.; Young, E.; Astle, L.; van der Linde, H.C.; Shivaram, G.M.; et al. Homozygous mutations in CSF1R cause a pediatric-onset leukoencephalopathy and can result in congenital absence of microglia. Am. J. Hum. Genet. 2019, 104, 936–947. [Google Scholar] [CrossRef] [Green Version]
- Wiktor-Jedrzejczak, W.; Ratajczak, M.Z.; Ptasznik, A.; Sell, K.W.; Ahmed-Ansari, A.; Ostertag, W. Csf-1 deficiency in the op/op mouse has differential effects on macrophage populations and differentiation stages. Exp. Hematol. 1992, 20, 1004–1010. [Google Scholar]
- Simard, A.R.; Rivest, S. Bone marrow stem cells have the ability to populate the entire central nervous system into fully differentiated parenchymal microglia. FASEB J. 2004, 18, 998–1000. [Google Scholar] [CrossRef] [Green Version]
- Ousman, S.S.; David, S. MIP-1alpha, MCP-1, GM-CSF, and TNF-alpha control the immune cell response that mediates rapid phagocytosis of myelin from the adult mouse spinal cord. J. Neurosci. 2001, 21, 4649–4656. [Google Scholar] [CrossRef]
- Neumann, H.; Kotter, M.R.; Franklin, R.J. Debris clearance by microglia: An essential link between degeneration and regeneration. Brain 2009, 132, 288–295. [Google Scholar] [CrossRef]
- Kotter, M.R.; Zhao, C.; van Rooijen, N.; Franklin, R.J. Macrophage-depletion induced impairment of experimental CNS remyelination is associated with a reduced oligodendrocyte progenitor cell response and altered growth factor expression. Neurobiol. Dis. 2005, 18, 166–175. [Google Scholar] [CrossRef] [PubMed]
- Miller, B.A.; Crum, J.M.; Tovar, C.A.; Ferguson, A.R.; Bresnahan, J.C.; Beattie, M.S. Developmental stage of oligodendrocytes determines their response to activated microglia in vitro. J. Neuroinflamm. 2007, 4, 28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, D.L.; Pirianov, G.; Holland, S.; McGinnity, C.J.; Norman, A.L.; Reali, C.; Diemel, L.T.; Gveric, D.; Yeung, D.; Mehmet, H. Attenuation of proliferation in oligodendrocyte precursor cells by activated microglia. J. Neurosci. Res. 2010, 88, 1632–1644. [Google Scholar] [CrossRef] [PubMed]
- Mosser, D.M.; Edwards, J.P. Exploring the full spectrum of macrophage activation. Nat. Rev. Immunol. 2008, 8, 958–969. [Google Scholar] [CrossRef] [PubMed]
- Miron, V.E.; Franklin, R.J.M. Macrophages and CNS remyelination. J. Neurochem. 2014, 130, 165–171. [Google Scholar] [CrossRef] [PubMed]
- Locatelli, G.; Theodorou, D.; Kendirli, A.; Joana, M.; Jordão, C.; Staszewski, O.; Phulphagar, K.; Cantuti-castelvetri, L.; Dagkalis, A.; Bessis, A.; et al. Mononuclear phagocytes locally specify and adapt their phenotype in a multiple sclerosis model. Nat. Neurosci. 2018, 21, 1196–1208. [Google Scholar] [CrossRef]
- Vogel, D.Y.S.; Vereyken, E.J.F.; Glim, J.E.; Heijnen, P.D.A.M.; Moeton, M.; van der Valk, P.; Amor, S.; Teunissen, C.E.; van Horssen, J.; Dijkstra, C.D. Macrophages in inflammatory multiple sclerosis lesions have an intermediate activation status. J. Neuroinflamm. 2013, 10, 35. [Google Scholar] [CrossRef] [Green Version]
- Giles, D.A.; Washnock-Schmid, J.M.; Duncker, P.C.; Dahlawi, S.; Ponath, G.; Pitt, D.; Segal, B.M. Myeloid cell plasticity in the evolution of Central Nervous System autoimmunity. Ann. Neurol. 2018, 83, 131–141. [Google Scholar] [CrossRef]
- Trapp, B.D.; Peterson, J.; Ransohoff, R.M.; Rudick, R.; Mörk, S.; Bö, L. Axonal transection in the lesions of multiple sclerosis. N. Engl. J. Med. 1998, 338, 278–285. [Google Scholar] [CrossRef]
- Bjartmar, C.; Wujek, J.R.; Trapp, B.D. Axonal loss in the pathology of MS: Consequences for understanding the progressive phase of the disease. J. Neurol Sci. 2003, 206, 165–171. [Google Scholar] [CrossRef]
- Hirrlinger, J.; Nave, K.A. Adapting brain metabolism to myelination and long-range signal transduction. Glia 2014, 62, 1749–1761. [Google Scholar] [CrossRef] [PubMed]
- Morrison, B.M.; Lee, Y.; Rothstein, J.D. Oligodendroglia: Metabolic supporters of axons. Trends Cell Biol. 2013, 23, 644–651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saab, A.S.; Tzvetanova, I.D.; Nave, K.A. The role of myelin and oligodendrocytes in axonal energy metabolism. Curr. Opin. Neurobiol. 2013, 23, 1065–1072. [Google Scholar] [CrossRef] [PubMed]
- Mei, F.; Lehmann-Horn, K.; Shen, Y.; Rankin, K.A.; Stebbins, K.J.; Lorrain, D.S.; Pekarek, K.A.; Sagan, S.; Xiao, L.; Teuscher, C.; et al. Accelerated remyelination during inflammatory demyelination prevents axonal loss and improves functional recovery. Elife 2016, 5, e18246. [Google Scholar] [CrossRef] [PubMed]
- Mildner, A.; Mack, M.; Schmidt, H.; Bruck, W.; Djukic, M.; Zabel, M.D.; Hille, A.; Priller, J.; Prinz, M. CCR2+Ly-6Chi monocytes are crucial for the effector phase of autoimmunity in the central nervous system. Brain 2009, 132, 2487–2500. [Google Scholar] [CrossRef] [Green Version]
- Heppner, F.L.; Greter, M.; Marino, D.; Falsig, J.; Raivich, G.; Hovelmeyer, N.; Waisman, A.; Rulicke, T.; Prinz, M.; Priller, J.; et al. Experimental autoimmune encephalomyelitis repressed by microglial paralysis. Nat. Med. 2005, 11, 146–152. [Google Scholar] [CrossRef]
- Nissen, J.C.; Thompson, K.K.; West, B.L.; Tsirka, S.E. Csf1R inhibition attenuates experimental autoimmune encephalomyelitis and promotes recovery. Exp. Neurol. 2018, 307, 24–36. [Google Scholar] [CrossRef]
- Gushchina, S.; Pryce, G.; Yip, P.K.; Wu, D.; Pallier, P.; Giovannoni, G.; Baker, D.; Bo, X. Increased expression of colony-stimulating factor-1 in mouse spinal cord with experimental autoimmune encephalomyelitis correlates with microglial activation and neuronal loss. Glia 2018, 66, 2108–2125. [Google Scholar] [CrossRef]
- Tahmasebi, F.; Pasbakhsh, P.; Mortezaee, K.; Madadi, S.; Barati, S.; Kashani, I.R. Effect of the CSF1R inhibitor PLX3397 on remyelination of corpus callosum in a cuprizone-induced demyelination mouse model. J. Cell. Biochem. 2019, 120, 10576–10586. [Google Scholar] [CrossRef]

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wylot, B.; Mieczkowski, J.; Niedziolka, S.; Kaminska, B.; Zawadzka, M. Csf1 Deficiency Dysregulates Glial Responses to Demyelination and Disturbs CNS White Matter Remyelination. Cells 2020, 9, 99. https://doi.org/10.3390/cells9010099
Wylot B, Mieczkowski J, Niedziolka S, Kaminska B, Zawadzka M. Csf1 Deficiency Dysregulates Glial Responses to Demyelination and Disturbs CNS White Matter Remyelination. Cells. 2020; 9(1):99. https://doi.org/10.3390/cells9010099
Chicago/Turabian StyleWylot, Bartosz, Jakub Mieczkowski, Sylwia Niedziolka, Bozena Kaminska, and Malgorzata Zawadzka. 2020. "Csf1 Deficiency Dysregulates Glial Responses to Demyelination and Disturbs CNS White Matter Remyelination" Cells 9, no. 1: 99. https://doi.org/10.3390/cells9010099
APA StyleWylot, B., Mieczkowski, J., Niedziolka, S., Kaminska, B., & Zawadzka, M. (2020). Csf1 Deficiency Dysregulates Glial Responses to Demyelination and Disturbs CNS White Matter Remyelination. Cells, 9(1), 99. https://doi.org/10.3390/cells9010099