Adipose Stromal Cell Expansion and Exhaustion: Mechanisms and Consequences

Abstract
:1. Introduction
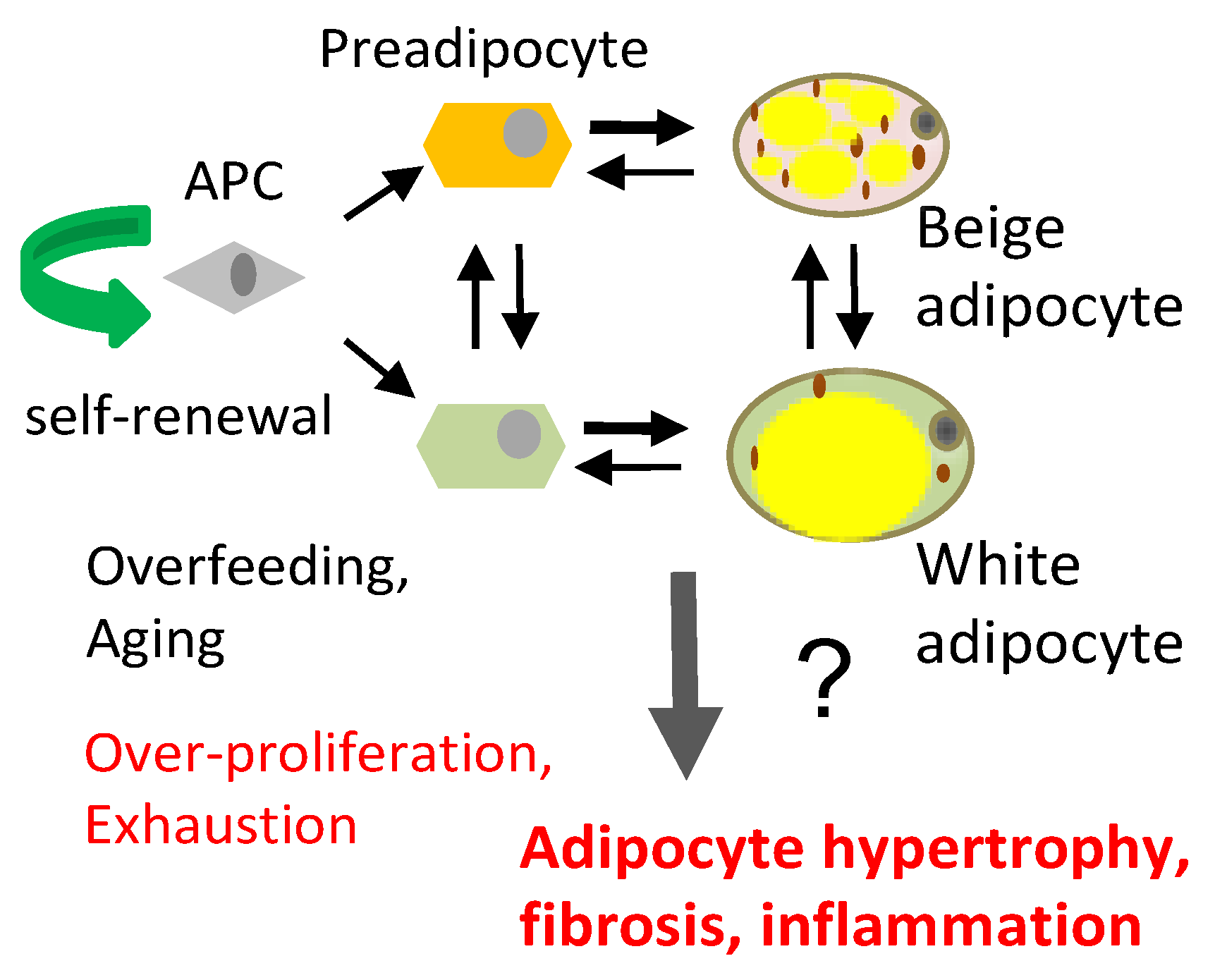
2. Origins and Functions of Adipocyte Progenitors within Fat Tissue
3. Regulation of Adipocyte Progenitor Cell Proliferation
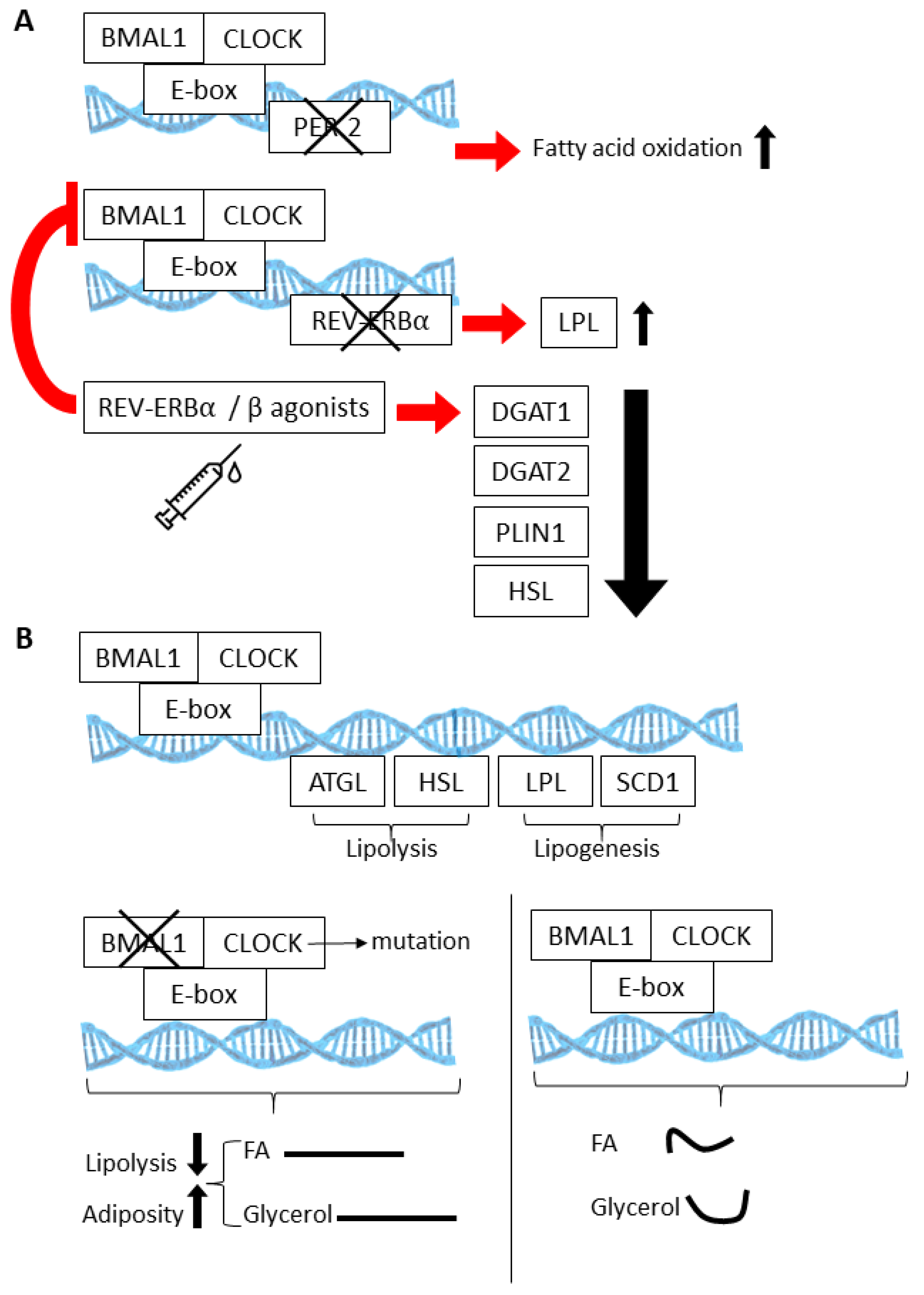
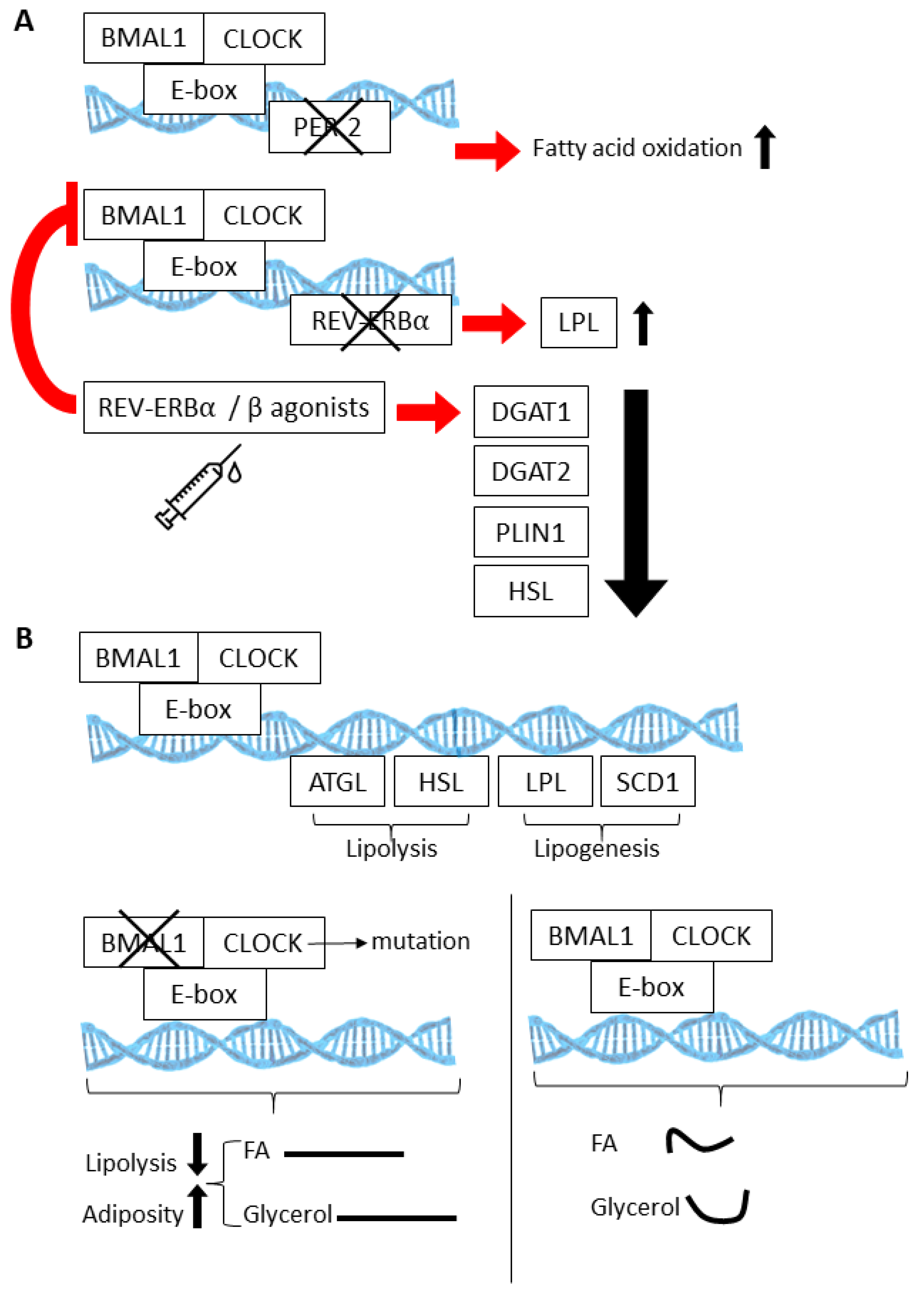
4. Circadian Regulatory Mechanisms within Adipose Tissue
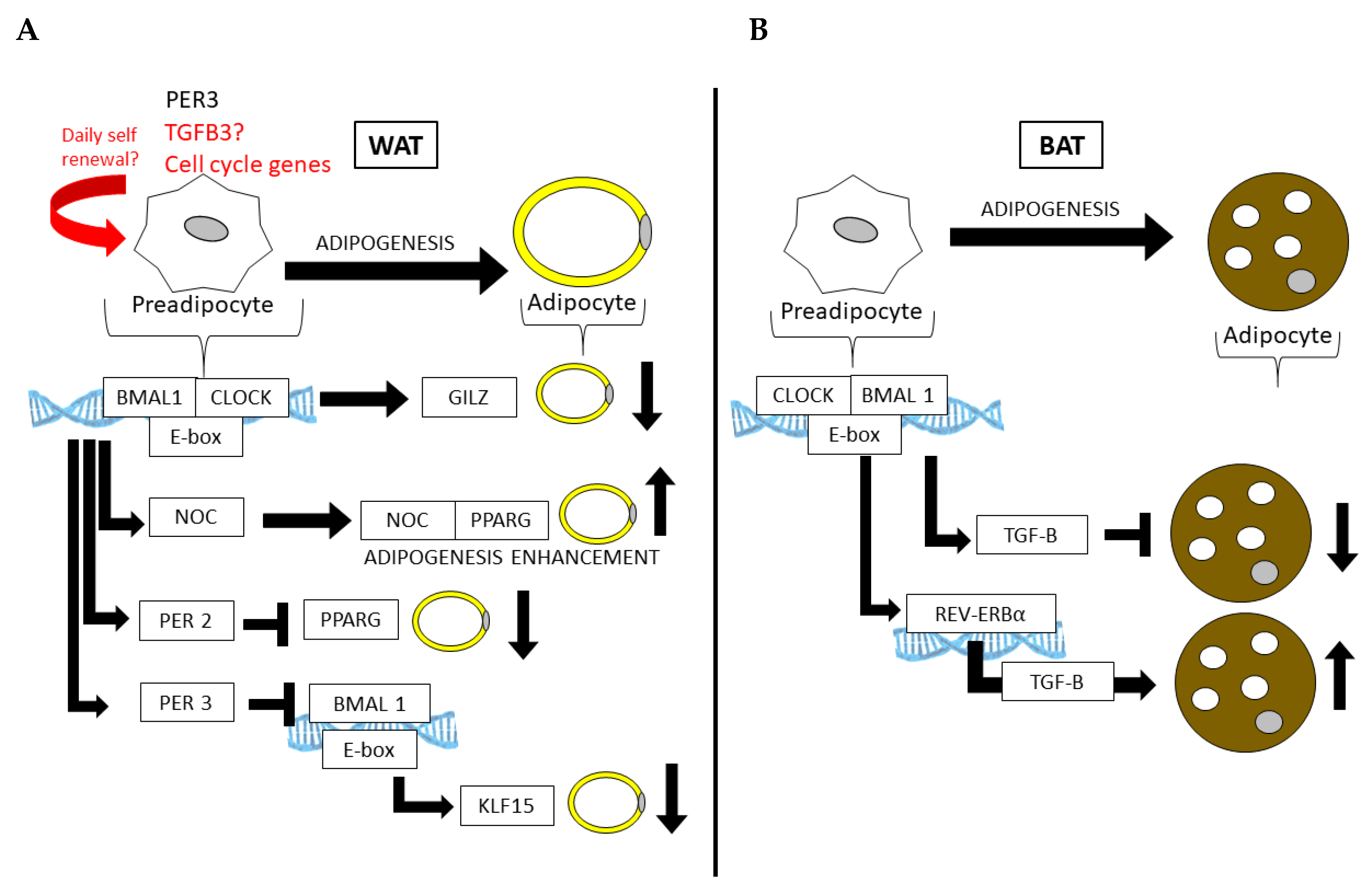
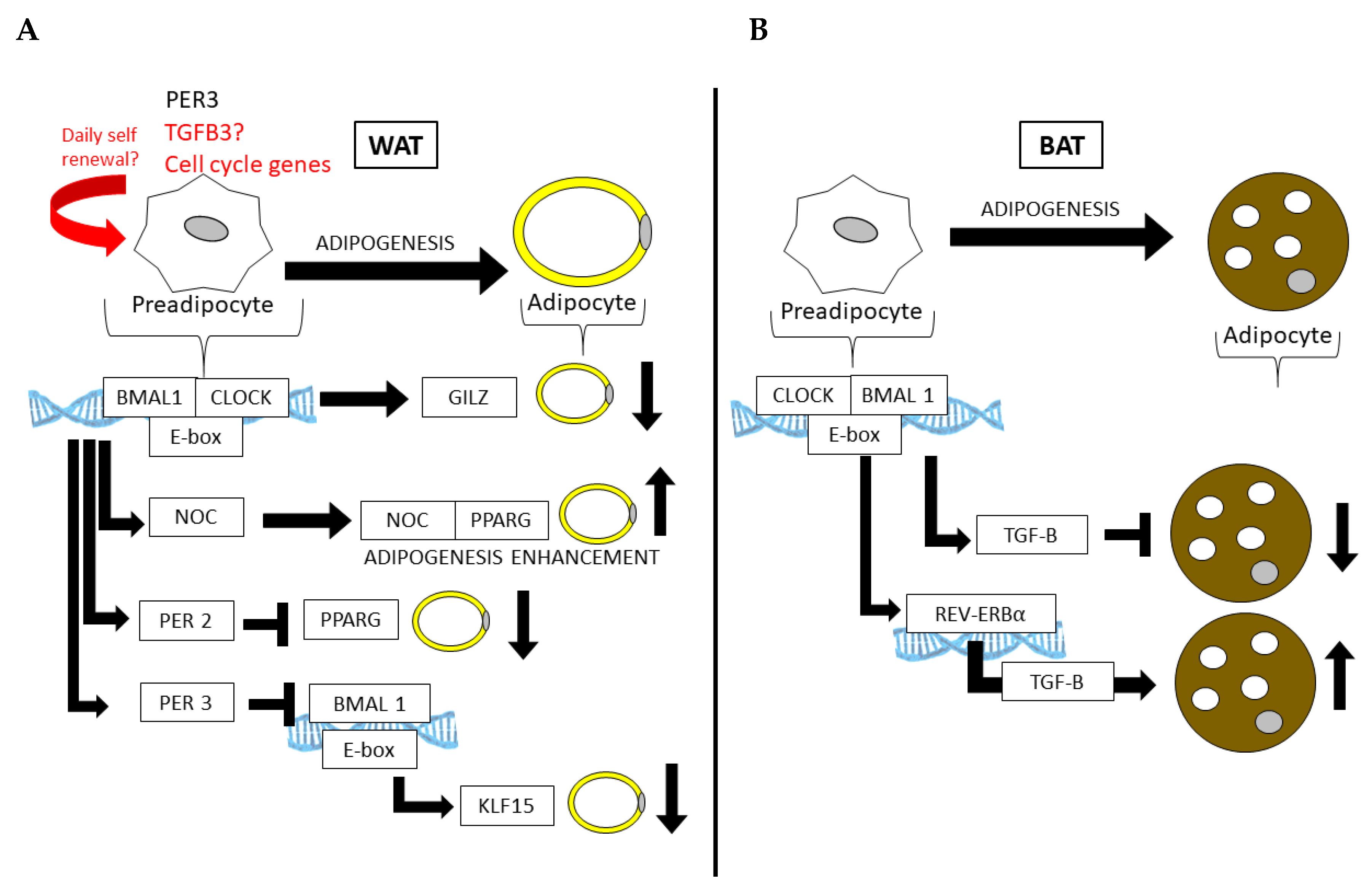
5. The Circadian Clock in Adipocyte Progenitors and Adipogenesis
6. Potential Circadian Regulation of APC Proliferation and Exhaustion?
7. Implications of Adipose Stroma Overactivation
8. Adipose Cell Engagement in Cancer
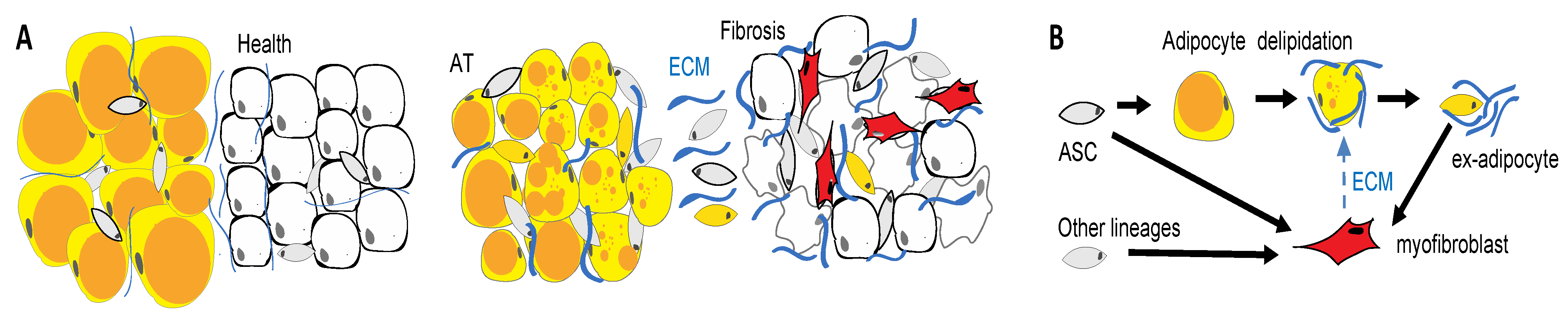
9. Adipose Cell Engagement in Fibrosis
10. Implications of ASC Exhaustion for Adipose Tissue Dysfunction
11. Adipose Cell Targeting
12. Discussion
Funding
Conflicts of Interest
References
- Sun, K.; Kusminski, C.M.; Scherer, P.E. Adipose tissue remodeling and obesity. J. Clin. Investig. 2011, 121, 2094–2101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosen, E.D.; Spiegelman, B.M. What we talk about when we talk about fat. Cell 2014, 156, 20–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Traktuev, D.; Merfeld-Clauss, S.; Li, J.; Kolonin, M.; Arap, W.; Pasqualini, R.; Johnstone, B.H.; March, K.L. A Population of multipotent CD34-positive adipose stromal cells share pericyte and mesenchymal surface markers, reside in a periendothelial location, and stabilize endothelial networks. Circ. Res. 2008, 102, 77–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chau, Y.Y.; Bandiera, R.; Serrels, A.; Martinez-Estrada, O.M.; Qing, W.; Lee, M.; Slight, J.; Thornburn, A.; Berry, R.; McHaffie, S.; et al. Visceral and subcutaneous fat have different origins and evidence supports a mesothelial source. Nat. Cell. Biol. 2014, 16, 367–375. [Google Scholar] [CrossRef] [Green Version]
- Kajimura, S.; Spiegelman, B.M.; Seale, P. Brown and Beige Fat: Physiological Roles beyond Heat Generation. Cell Metab. 2015, 22, 546–559. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.Y.; Attane, C.; Milhas, D.; Dirat, B.; Dauvillier, S.; Guerard, A.; Gilhodes, J.; Lazar, I.; Alet, N.; Laurent, V.; et al. Mammary adipocytes stimulate breast cancer invasion through metabolic remodeling of tumor cells. JCI Insight 2017, 2, e87489. [Google Scholar] [CrossRef] [Green Version]
- Laurent, V.; Guerard, A.; Mazerolles, C.; Le Gonidec, S.; Toulet, A.; Nieto, L.; Zaidi, F.; Majed, B.; Garandeau, D.; Socrier, Y.; et al. Periprostatic adipocytes act as a driving force for prostate cancer progression in obesity. Nat. Commun. 2016, 7, 10230. [Google Scholar] [CrossRef]
- Enerback, S. The origins of brown adipose tissue. N. Engl. J. Med. 2009, 360, 2021–2023. [Google Scholar] [CrossRef]
- Cinti, S. Transdifferentiation properties of adipocytes in the Adipose Organ. Am. J. Physiol. Endocrinol. Metab. 2009, 297, 977–986. [Google Scholar] [CrossRef]
- Zhang, F.; Hao, G.; Shao, M.; Nham, K.; An, Y.; Wang, Q.; Zhu, Y.; Kusminski, C.M.; Hassan, G.; Gupta, R.K.; et al. An Adipose Tissue Atlas: An Image-Guided Identification of Human-like BAT and Beige Depots in Rodents. Cell Metab. 2018, 27, 252–262. [Google Scholar] [CrossRef]
- Wang, Q.A.; Tao, C.; Gupta, R.K.; Scherer, P.E. Tracking adipogenesis during white adipose tissue development, expansion and regeneration. Nat. Med. 2013, 19, 1338–1344. [Google Scholar] [CrossRef] [PubMed]
- Cinti, S. Between brown and white: Novel aspects of adipocyte differentiation. Ann. Med. 2011, 43, 104–115. [Google Scholar] [CrossRef]
- Bagchi, D.P.; Forss, I.; Mandrup, S.; MacDougald, O.A. SnapShot: Niche Determines Adipocyte Character I. Cell Metab. 2018, 27, 264. [Google Scholar] [CrossRef] [PubMed]
- Hankir, M.K.; Klingenspor, M. Brown adipocyte glucose metabolism: A heated subject. EMBO Rep. 2018, 19. [Google Scholar] [CrossRef] [PubMed]
- Cannon, B.; Nedergaard, J. Brown adipose tissue: Function and physiological significance. Physiol. Rev. 2004, 84, 277–359. [Google Scholar] [CrossRef]
- Tseng, Y.H.; Cypess, A.M.; Kahn, C.R. Cellular bioenergetics as a target for obesity therapy. Nat. Rev. Drug Discov. 2010, 9, 465–482. [Google Scholar] [CrossRef] [Green Version]
- Porter, C.; Herndon, D.N.; Chondronikola, M.; Daquinag, A.C.; Kolonin, M.G.; Sidossis, L.S. Human and Mouse Brown Adipose Tissue Mitochondria Have Comparable UCP1 Function. Cell Metab. 2016, 24, 246–255. [Google Scholar] [CrossRef] [Green Version]
- Cypess, A.M.; Haft, C.R.; Laughlin, M.R.; Hu, H.H. Brown Fat in Humans: Consensus Points and Experimental Guidelines. Cell Metab. 2014, 20, 408–415. [Google Scholar] [CrossRef] [Green Version]
- Gray, S.L.; Vidal-Puig, A.J. Adipose tissue expandability in the maintenance of metabolic homeostasis. Nutr. Rev. 2007, 65, S7–S12. [Google Scholar] [CrossRef]
- Orci, L.; Cook, W.S.; Ravazzola, M.; Wang, M.Y.; Park, B.H.; Montesano, R.; Unger, R.H. Rapid transformation of white adipocytes into fat-oxidizing machines. Proc. Natl. Acad. Sci. USA 2004, 101, 2058–2063. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.M.; Lun, M.; Wang, M.; Senyo, S.E.; Guillermier, C.; Patwari, P.; Steinhauser, M.L. Loss of white adipose hyperplastic potential is associated with enhanced susceptibility to insulin resistance. Cell Metab. 2014, 20, 1049–1058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hammarstedt, A.; Gogg, S.; Hedjazifar, S.; Nerstedt, A.; Smith, U. Impaired Adipogenesis and Dysfunctional Adipose Tissue in Human Hypertrophic Obesity. Physiol. Rev. 2018, 98, 1911–1941. [Google Scholar] [CrossRef] [PubMed]
- Daquinag, A.C.; Zhang, Y.; Kolonin, M.G. Vascular targeting of adipose tissue as an anti-obesity approach. Trends Pharmacol. Sci. 2011, 32, 300–307. [Google Scholar] [CrossRef]
- Mariman, E.C.; Wang, P. Adipocyte extracellular matrix composition, dynamics and role in obesity. Cell Mol. Life Sci. 2010, 67, 1277–1292. [Google Scholar] [CrossRef] [Green Version]
- Eto, H.; Suga, H.; Inoue, K.; Aoi, N.; Kato, H.; Araki, J.; Doi, K.; Higashino, T.; Yoshimura, K. Adipose injury-associated factors mitigate hypoxia in ischemic tissues through activation of adipose-derived stem/progenitor/stromal cells and induction of angiogenesis. Am. J. Pathol. 2011, 178, 2322–2332. [Google Scholar] [CrossRef] [Green Version]
- Frayn, K.N.; Karpe, F.; Fielding, B.A.; Macdonald, I.A.; Coppack, S.W. Integrative physiology of human adipose tissue. Int. J. Obes. Relat. Metab. Disord. 2003, 27, 875–888. [Google Scholar] [CrossRef] [Green Version]
- Blogowski, W.; Ratajczak, M.Z.; Zyzniewska-Banaszak, E.; Dolegowska, B.; Starzynska, T. Adipose tissue as a potential source of hematopoietic stem/progenitor cells. Obesity 2012, 20, 923–931. [Google Scholar] [CrossRef]
- Zhang, Y.; Bellows, C.F.; Kolonin, M.G. Adipose tissue-derived progenitor cells and cancer. World J. Stem Cells 2010, 2, 103–113. [Google Scholar] [CrossRef]
- Spalding, K.L.; Arner, E.; Westermark, P.O.; Bernard, S.; Buchholz, B.A.; Bergmann, O.; Blomqvist, L.; Hoffstedt, J.; Naslund, E.; Britton, T.; et al. Dynamics of fat cell turnover in humans. Nature 2008, 453, 783–787. [Google Scholar] [CrossRef]
- White, U.; Ravussin, E. Dynamics of adipose tissue turnover in human metabolic health and disease. Diabetologia 2019, 62, 17–23. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Y.; Jo, A.Y.; Graff, J.M. SnapShot: Adipocyte life cycle. Cell 2012, 150, 234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neese, R.A.; Misell, L.M.; Turner, S.; Chu, A.; Kim, J.; Cesar, D.; Hoh, R.; Antelo, F.; Strawford, A.; McCune, J.M.; et al. Measurement in vivo of proliferation rates of slow turnover cells by 2H2O labeling of the deoxyribose moiety of DNA. Proc. Natl. Acad. Sci. USA 2002, 99, 15345–15350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rigamonti, A.; Brennand, K.; Lau, F.; Cowan, C.A. Rapid cellular turnover in adipose tissue. PLoS ONE 2011, 6, e17637. [Google Scholar] [CrossRef] [Green Version]
- Ghaben, A.L.; Scherer, P.E. Adipogenesis and metabolic health. Nat. Rev. Mol. Cell Biol. 2019, 20, 242–258. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.H.; Petkova, A.P.; Konkar, A.A.; Granneman, J.G. Cellular origins of cold-induced brown adipocytes in adult mice. FASEB J. 2015, 29, 286–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berry, D.C.; Jiang, Y.; Graff, J.M. Emerging Roles of Adipose Progenitor Cells in Tissue Development, Homeostasis, Expansion and Thermogenesis. Trends Endocrinol. Metab. 2016, 27, 574–585. [Google Scholar] [CrossRef] [PubMed]
- Rosenwald, M.; Perdikari, A.; Rulicke, T.; Wolfrum, C. Bi-directional interconversion of brite and white adipocytes. Nat. Cell Biol. 2013, 15, 659–667. [Google Scholar] [CrossRef]
- Paulo, E.; Wu, D.; Hecker, P.; Zhang, Y.; Wang, B. Adipocyte HDAC4 activation leads to beige adipocyte expansion and reduced adiposity. J. Endocrinol. 2018, 239, 153–165. [Google Scholar] [CrossRef] [Green Version]
- Kras, K.M.; Hausman, D.B.; Hausman, G.J.; Martin, R.J. Adipocyte development is dependent upon stem cell recruitment and proliferation of preadipocytes. Obes. Res. 1999, 7, 491–497. [Google Scholar] [CrossRef]
- Vishvanath, L.; MacPherson, K.A.; Hepler, C.; Wang, Q.A.; Shao, M.; Spurgin, S.B.; Wang, M.Y.; Kusminski, C.M.; Morley, T.S.; Gupta, R.K. Pdgfrbeta Mural Preadipocytes Contribute to Adipocyte Hyperplasia Induced by High-Fat-Diet Feeding and Prolonged Cold Exposure in Adult Mice. Cell Metab. 2016, 23, 350–359. [Google Scholar] [CrossRef] [Green Version]
- Tang, W.; Zeve, D.; Suh, J.M.; Bosnakovski, D.; Kyba, M.; Hammer, R.E.; Tallquist, M.D.; Graff, J.M. White fat progenitor cells reside in the adipose vasculature. Science 2008, 322, 583–586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berry, R.; Jeffery, E.; Rodeheffer, M.S. Weighing in on adipocyte precursors. Cell Metab. 2014, 19, 8–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodeheffer, M.S.; Birsoy, K.; Friedman, J.M. Identification of white adipocyte progenitor cells in vivo. Cell 2008, 135, 240–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, C.; Medley, S.C.; Hu, T.; Hinsdale, M.E.; Lupu, F.; Virmani, R.; Olson, L.E. PDGFRbeta signalling regulates local inflammation and synergizes with hypercholesterolaemia to promote atherosclerosis. Nat. Commun. 2015, 6, 7770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwayama, T.; Steele, C.; Yao, L.; Dozmorov, M.G.; Karamichos, D.; Wren, J.D.; Olson, L.E. PDGFRalpha signaling drives adipose tissue fibrosis by targeting progenitor cell plasticity. Genes Dev. 2015, 29, 1106–1119. [Google Scholar] [CrossRef] [Green Version]
- Sun, K.; Gao, Z.; Kolonin, M.G. Transient inflammatory signaling promotes beige adipogenesis. Sci. Signal. 2018, 11. [Google Scholar] [CrossRef]
- Hepler, C.; Shan, B.; Zhang, Q.; Henry, G.H.; Shao, M.; Vishvanath, L.; Ghaben, A.L.; Mobley, A.B.; Strand, D.; Hon, G.C.; et al. Identification of functionally distinct fibro-inflammatory and adipogenic stromal subpopulations in visceral adipose tissue of adult mice. Elife 2018, 7. [Google Scholar] [CrossRef]
- Berry, R.; Rodeheffer, M.S. Characterization of the adipocyte cellular lineage in vivo. Nat. Cell Biol. 2013, 15, 302–308. [Google Scholar] [CrossRef]
- Raajendiran, A.; Ooi, G.; Bayliss, J.; O’Brien, P.E.; Schittenhelm, R.B.; Clark, A.K.; Taylor, R.A.; Rodeheffer, M.S.; Burton, P.R.; Watt, M.J. Identification of Metabolically Distinct Adipocyte Progenitor Cells in Human Adipose Tissues. Cell Rep. 2019, 27, 1528–1540. [Google Scholar] [CrossRef] [Green Version]
- Gimble, J.M.; Bunnell, B.A.; Chiu, E.S.; Guilak, F. Concise review: Adipose-derived stromal vascular fraction cells and stem cells: Let’s not get lost in translation. Stem Cells 2011, 29, 749–754. [Google Scholar] [CrossRef]
- Cawthorn, W.P.; Scheller, E.L.; MacDougald, O.A. Adipose tissue stem cells: The great WAT hope. Trends Endocrinol. Metab. 2012, 23, 270–277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daquinag, A.C.; Salameh, A.; Zhang, Y.; Tong, Q.; Kolonin, M.G. Depletion of white adipocyte progenitors induces beige adipocyte differentiation and suppresses obesity development. Cell Death Diff. 2015, 22, 351–363. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.H.; Petkova, A.P.; Mottillo, E.P.; Granneman, J.G. In vivo identification of bipotential adipocyte progenitors recruited by beta3-adrenoceptor activation and high-fat feeding. Cell Metab. 2012, 15, 480–491. [Google Scholar] [CrossRef] [Green Version]
- Sun, C.; Berry, W.L.; Olson, L.E. PDGFRalpha controls the balance of stromal and adipogenic cells during adipose tissue organogenesis. Development 2017, 144, 83–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seki, T.; Hosaka, K.; Lim, S.; Fischer, C.; Honek, J.; Yang, Y.; Andersson, P.; Nakamura, M.; Naslund, E.; Yla-Herttuala, S.; et al. Endothelial PDGF-CC regulates angiogenesis-dependent thermogenesis in beige fat. Nat. Commun. 2016, 7, 12152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, Z.; Daquinag, A.C.; Su, F.; Snyder, B.; Kolonin, M.G. PDGFRalpha/PDGFRbeta signaling balance modulates progenitor cell differentiation into white and beige adipocytes. Development 2018, 145. [Google Scholar] [CrossRef] [Green Version]
- Hoch, R.V.; Soriano, P. Roles of PDGF in animal development. Development 2003, 130, 4769–4784. [Google Scholar] [CrossRef] [Green Version]
- Kloting, N.; Bluher, M. Adipocyte dysfunction, inflammation and metabolic syndrome. Rev. Endocr. Metab. Disord. 2014, 15, 277–287. [Google Scholar] [CrossRef]
- Bartness, T.J.; Shrestha, Y.B.; Vaughan, C.H.; Schwartz, G.J.; Song, C.K. Sensory and sympathetic nervous system control of white adipose tissue lipolysis. Mol. Cell Endocrinol. 2010, 318, 34–43. [Google Scholar] [CrossRef] [Green Version]
- Jeffery, E.; Church, C.D.; Holtrup, B.; Colman, L.; Rodeheffer, M.S. Rapid depot-specific activation of adipocyte precursor cells at the onset of obesity. Nature Cell Biol. 2015, 17, 376–385. [Google Scholar] [CrossRef]
- Merrick, D.; Sakers, A.; Irgebay, Z.; Okada, C.; Calvert, C.; Morley, M.P.; Percec, I.; Seale, P. Identification of a mesenchymal progenitor cell hierarchy in adipose tissue. Science 2019, 364. [Google Scholar] [CrossRef] [PubMed]
- Hoffstedt, J.; Andersson, D.P.; Eriksson Hogling, D.; Theorell, J.; Naslund, E.; Thorell, A.; Ehrlund, A.; Ryden, M.; Arner, P. Long-term Protective Changes in Adipose Tissue After Gastric Bypass. Diabetes Care 2017, 40, 77–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petrus, P.; Mejhert, N.; Corrales, P.; Lecoutre, S.; Li, Q.; Maldonado, E.; Kulyte, A.; Lopez, Y.; Campbell, M.; Acosta, J.R.; et al. Transforming Growth Factor-beta3 Regulates Adipocyte Number in Subcutaneous White Adipose Tissue. Cell Rep. 2018, 25, 551–560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmierer, B.; Hill, C.S. TGFbeta-SMAD signal transduction: Molecular specificity and functional flexibility. Nat. Rev. Mol. Cell Biol. 2007, 8, 970–982. [Google Scholar] [CrossRef]
- Eckel-Mahan, K.; Sassone-Corsi, P. Metabolism and the circadian clock converge. Physiol. Rev. 2013, 93, 107–135. [Google Scholar] [CrossRef]
- Huang, W.; Ramsey, K.M.; Marcheva, B.; Bass, J. Circadian rhythms, sleep, and metabolism. J. Clin. Investig. 2011, 121, 2133–2141. [Google Scholar] [CrossRef]
- Ding, G.; Gong, Y.; Eckel-Mahan, K.L.; Sun, Z. Central Circadian Clock Regulates Energy Metabolism. Adv. Exp. Med. Biol. 2018, 1090, 79–103. [Google Scholar] [CrossRef]
- Longo, V.D.; Panda, S. Fasting, Circadian Rhythms, and Time-Restricted Feeding in Healthy Lifespan. Cell Metab. 2016, 23, 1048–1059. [Google Scholar] [CrossRef] [Green Version]
- Ribas-Latre, A.; Eckel-Mahan, K. Interdependence of nutrient metabolism and the circadian clock system: Importance for metabolic health. Mol. Metab. 2016, 5, 133–152. [Google Scholar] [CrossRef]
- Asher, G.; Sassone-Corsi, P. Time for food: The intimate interplay between nutrition, metabolism, and the circadian clock. Cell 2015, 161, 84–92. [Google Scholar] [CrossRef] [Green Version]
- Roenneberg, T.; Allebrandt, K.V.; Merrow, M.; Vetter, C. Social jetlag and obesity. Curr. Biol. 2012, 22, 939–943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roenneberg, T.; Merrow, M. The Circadian Clock and Human Health. Curr. Biol. 2016, 26, R432–R443. [Google Scholar] [CrossRef] [PubMed]
- Froy, O.; Garaulet, M. The Circadian Clock in White and Brown Adipose Tissue: Mechanistic, Endocrine, and Clinical Aspects. Endocr. Rev. 2018, 39, 261–273. [Google Scholar] [CrossRef]
- Patel, V.R.; Ceglia, N.; Zeller, M.; Eckel-Mahan, K.; Sassone-Corsi, P.; Baldi, P. The Pervasiveness and Plasticity of Circadian Oscillations: The Coupled Circadian-Oscillators Framework. Bioinformatics 2015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christou, S.; Wehrens, S.M.T.; Isherwood, C.; Moller-Levet, C.S.; Wu, H.; Revell, V.L.; Bucca, G.; Skene, D.J.; Laing, E.E.; Archer, S.N.; et al. Circadian regulation in human white adipose tissue revealed by transcriptome and metabolic network analysis. Sci. Rep. 2019, 9, 2641. [Google Scholar] [CrossRef]
- Kohsaka, A.; Laposky, A.D.; Ramsey, K.M.; Estrada, C.; Joshu, C.; Kobayashi, Y.; Turek, F.W.; Bass, J. High-fat diet disrupts behavioral and molecular circadian rhythms in mice. Cell Metab. 2007, 6, 414–421. [Google Scholar] [CrossRef] [Green Version]
- Dyar, K.A.; Lutter, D.; Artati, A.; Ceglia, N.J.; Liu, Y.; Armenta, D.; Jastroch, M.; Schneider, S.; de Mateo, S.; Cervantes, M.; et al. Atlas of Circadian Metabolism Reveals System-wide Coordination and Communication between Clocks. Cell 2018, 174, 1571–1585. [Google Scholar] [CrossRef] [Green Version]
- Kennaway, D.J.; Varcoe, T.J.; Voultsios, A.; Boden, M.J. Global loss of bmal1 expression alters adipose tissue hormones, gene expression and glucose metabolism. PLoS ONE 2013, 8, e65255. [Google Scholar] [CrossRef] [Green Version]
- Paschos, G.K.; Ibrahim, S.; Song, W.L.; Kunieda, T.; Grant, G.; Reyes, T.M.; Bradfield, C.A.; Vaughan, C.H.; Eiden, M.; Masoodi, M.; et al. Obesity in mice with adipocyte-specific deletion of clock component Arntl. Nat. Med. 2012, 18, 1768–1777. [Google Scholar] [CrossRef] [Green Version]
- Zvonic, S.; Ptitsyn, A.A.; Conrad, S.A.; Scott, L.K.; Floyd, Z.E.; Kilroy, G.; Wu, X.; Goh, B.C.; Mynatt, R.L.; Gimble, J.M. Characterization of peripheral circadian clocks in adipose tissues. Diabetes 2006, 55, 962–970. [Google Scholar] [CrossRef] [Green Version]
- Zhang, R.; Lahens, N.F.; Ballance, H.I.; Hughes, M.E.; Hogenesch, J.B. A circadian gene expression atlas in mammals: Implications for biology and medicine. Proc. Natl. Acad. Sci. USA 2014, 111, 16219–16224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Otway, D.T.; Mantele, S.; Bretschneider, S.; Wright, J.; Trayhurn, P.; Skene, D.J.; Robertson, M.D.; Johnston, J.D. Rhythmic diurnal gene expression in human adipose tissue from individuals who are lean, overweight, and type 2 diabetic. Diabetes 2011, 60, 1577–1581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerhart-Hines, Z.; Feng, D.; Emmett, M.J.; Everett, L.J.; Loro, E.; Briggs, E.R.; Bugge, A.; Hou, C.; Ferrara, C.; Seale, P.; et al. The nuclear receptor Rev-erbalpha controls circadian thermogenic plasticity. Nature 2013, 503, 410–413. [Google Scholar] [CrossRef] [PubMed]
- Mure, L.S.; Le, H.D.; Benegiamo, G.; Chang, M.W.; Rios, L.; Jillani, N.; Ngotho, M.; Kariuki, T.; Dkhissi-Benyahya, O.; Cooper, H.M.; et al. Diurnal transcriptome atlas of a primate across major neural and peripheral tissues. Science 2018, 359. [Google Scholar] [CrossRef] [Green Version]
- Delezie, J.; Dumont, S.; Dardente, H.; Oudart, H.; Grechez-Cassiau, A.; Klosen, P.; Teboul, M.; Delaunay, F.; Pevet, P.; Challet, E. The nuclear receptor REV-ERBalpha is required for the daily balance of carbohydrate and lipid metabolism. FASEB J. 2012, 26, 3321–3335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grimaldi, B.; Bellet, M.M.; Katada, S.; Astarita, G.; Hirayama, J.; Amin, R.H.; Granneman, J.G.; Piomelli, D.; Leff, T.; Sassone-Corsi, P. PER2 controls lipid metabolism by direct regulation of PPARgamma. Cell Metab. 2010, 12, 509–520. [Google Scholar] [CrossRef] [Green Version]
- Solt, L.A.; Wang, Y.; Banerjee, S.; Hughes, T.; Kojetin, D.J.; Lundasen, T.; Shin, Y.; Liu, J.; Cameron, M.D.; Noel, R.; et al. Regulation of circadian behaviour and metabolism by synthetic REV-ERB agonists. Nature 2012, 485, 62–68. [Google Scholar] [CrossRef]
- Shostak, A.; Meyer-Kovac, J.; Oster, H. Circadian regulation of lipid mobilization in white adipose tissues. Diabetes 2013, 62, 2195–2203. [Google Scholar] [CrossRef] [Green Version]
- van den Berg, R.; Kooijman, S.; Noordam, R.; Ramkisoensing, A.; Abreu-Vieira, G.; Tambyrajah, L.L.; Dijk, W.; Ruppert, P.; Mol, I.M.; Kramar, B.; et al. A Diurnal Rhythm in Brown Adipose Tissue Causes Rapid Clearance and Combustion of Plasma Lipids at Wakening. Cell Rep. 2018, 22, 3521–3533. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Gan, L.; Luo, D.; Sun, C. Melatonin promotes circadian rhythm-induced proliferation through Clock/histone deacetylase 3/c-Myc interaction in mouse adipose tissue. J. Pineal Res. 2017, 62. [Google Scholar] [CrossRef]
- Kiehn, J.T.; Tsang, A.H.; Heyde, I.; Leinweber, B.; Kolbe, I.; Leliavski, A.; Oster, H. Circadian Rhythms in Adipose Tissue Physiology. Compr. Physiol. 2017, 7, 383–427. [Google Scholar] [CrossRef] [PubMed]
- Shimba, S.; Ishii, N.; Ohta, Y.; Ohno, T.; Watabe, Y.; Hayashi, M.; Wada, T.; Aoyagi, T.; Tezuka, M. Brain and muscle Arnt-like protein-1 (BMAL1), a component of the molecular clock, regulates adipogenesis. Proc. Natl. Acad. Sci. USA 2005, 102, 12071–12076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turek, F.W.; Joshu, C.; Kohsaka, A.; Lin, E.; Ivanova, G.; McDearmon, E.; Laposky, A.; Losee-Olson, S.; Easton, A.; Jensen, D.R.; et al. Obesity and metabolic syndrome in circadian Clock mutant mice. Science 2005, 308, 1043–1045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, B.; Chatterjee, S.; Li, L.; Kim, J.M.; Lee, J.; Yechoor, V.K.; Minze, L.J.; Hsueh, W.; Ma, K. The clock gene, brain and muscle Arnt-like 1, regulates adipogenesis via Wnt signaling pathway. FASEB J. 2012, 26, 3453–3463. [Google Scholar] [CrossRef]
- Nam, D.; Yechoor, V.K.; Ma, K. Molecular clock integration of brown adipose tissue formation and function. Adipocyte 2016, 5, 243–250. [Google Scholar] [CrossRef] [Green Version]
- Kawai, M.; Green, C.B.; Lecka-Czernik, B.; Douris, N.; Gilbert, M.R.; Kojima, S.; Ackert-Bicknell, C.; Garg, N.; Horowitz, M.C.; Adamo, M.L.; et al. A circadian-regulated gene, Nocturnin, promotes adipogenesis by stimulating PPAR-gamma nuclear translocation. Proc. Natl. Acad. Sci. USA 2010, 107, 10508–10513. [Google Scholar] [CrossRef] [Green Version]
- Onder, Y.; Laothamatas, I.; Berto, S.; Sewart, K.; Kilaru, G.; Bordieanu, B.; Stubblefield, J.J.; Konopka, G.; Mishra, P.; Green, C.B. The Circadian Protein Nocturnin Regulates Metabolic Adaptation in Brown Adipose Tissue. iScience 2019, 19, 83–92. [Google Scholar] [CrossRef] [Green Version]
- Aggarwal, A.; Costa, M.J.; Rivero-Gutierrez, B.; Ji, L.; Morgan, S.L.; Feldman, B.J. The Circadian Clock Regulates Adipogenesis by a Per3 Crosstalk Pathway to Klf15. Cell Rep. 2017, 21, 2367–2375. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Z.; Xu, L.; Cai, T.; Yuan, G.; Sun, N.; Lu, C.; Qian, R. Clock represses preadipocytes adipogenesis via GILZ. J. Cell Physiol. 2018, 233, 6028–6040. [Google Scholar] [CrossRef]
- Nam, D.; Guo, B.; Chatterjee, S.; Chen, M.H.; Nelson, D.; Yechoor, V.K.; Ma, K. The adipocyte clock controls brown adipogenesis through the TGF-beta and BMP signaling pathways. J. Cell Sci. 2015, 128, 1835–1847. [Google Scholar] [CrossRef] [Green Version]
- Chang, L.; Xiong, W.; Zhao, X.; Fan, Y.; Guo, Y.; Garcia-Barrio, M.; Zhang, J.; Jiang, Z.; Lin, J.D.; Chen, Y.E. Bmal1 in Perivascular Adipose Tissue Regulates Resting-Phase Blood Pressure Through Transcriptional Regulation of Angiotensinogen. Circulation 2018, 138, 67–79. [Google Scholar] [CrossRef] [PubMed]
- Jager, J.; Wang, F.; Fang, B.; Lim, H.W.; Peed, L.C.; Steger, D.J.; Won, K.J.; Kharitonenkov, A.; Adams, A.C.; Lazar, M.A. The Nuclear Receptor Rev-erbalpha Regulates Adipose Tissue-specific FGF21 Signaling. J. Biol. Chem. 2016, 291, 10867–10875. [Google Scholar] [CrossRef] [Green Version]
- Adlanmerini, M.; Carpenter, B.J.; Remsberg, J.R.; Aubert, Y.; Peed, L.C.; Richter, H.J.; Lazar, M.A. Circadian lipid synthesis in brown fat maintains murine body temperature during chronic cold. Proc. Natl. Acad. Sci. USA 2019, 116, 18691–18699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Green, C.B.; Douris, N.; Kojima, S.; Strayer, C.A.; Fogerty, J.; Lourim, D.; Keller, S.R.; Besharse, J.C. Loss of Nocturnin, a circadian deadenylase, confers resistance to hepatic steatosis and diet-induced obesity. Proc. Natl. Acad. Sci. USA 2007, 104, 9888–9893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoo, S.H.; Yamazaki, S.; Lowrey, P.L.; Shimomura, K.; Ko, C.H.; Buhr, E.D.; Siepka, S.M.; Hong, H.K.; Oh, W.J.; Yoo, O.J.; et al. PERIOD2::LUCIFERASE real-time reporting of circadian dynamics reveals persistent circadian oscillations in mouse peripheral tissues. Proc. Natl. Acad. Sci. USA 2004, 101, 5339–5346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costa, M.J.; So, A.Y.; Kaasik, K.; Krueger, K.C.; Pillsbury, M.L.; Fu, Y.H.; Ptacek, L.J.; Yamamoto, K.R.; Feldman, B.J. Circadian rhythm gene period 3 is an inhibitor of the adipocyte cell fate. J. Biol. Chem. 2011, 286, 9063–9070. [Google Scholar] [CrossRef] [Green Version]
- Knarr, M.; Nagaraj, A.B.; Kwiatkowski, L.J.; DiFeo, A. miR-181a modulates circadian rhythm in immortalized bone marrow and adipose derived stromal cells and promotes differentiation through the regulation of PER3. Sci. Rep. 2019, 9, 307. [Google Scholar] [CrossRef]
- Friedrichs, M.; Kolbe, I.; Seemann, J.; Tsang, A.H.; Cherradi, L.; Klein, J.; Oster, H. Circadian clock rhythms in different adipose tissue model systems. Chronobiol. Int. 2018, 35, 1543–1552. [Google Scholar] [CrossRef]
- Wu, X.; Zvonic, S.; Floyd, Z.E.; Kilroy, G.; Goh, B.C.; Hernandez, T.L.; Eckel, R.H.; Mynatt, R.L.; Gimble, J.M. Induction of circadian gene expression in human subcutaneous adipose-derived stem cells. Obesity 2007, 15, 2560–2570. [Google Scholar] [CrossRef]
- Matsuo, T.; Yamaguchi, S.; Mitsui, S.; Emi, A.; Shimoda, F.; Okamura, H. Control mechanism of the circadian clock for timing of cell division in vivo. Science 2003, 302, 255–259. [Google Scholar] [CrossRef] [Green Version]
- Fekry, B.; Ribas-Latre, A.; Baumgartner, C.; Deans, J.R.; Kwok, C.; Patel, P.; Fu, L.; Berdeaux, R.; Sun, K.; Kolonin, M.G.; et al. Incompatibility of the circadian protein BMAL1 and HNF4alpha in hepatocellular carcinoma. Nat. Commun. 2018, 9, 4349. [Google Scholar] [CrossRef] [PubMed]
- Gaucher, J.; Montellier, E.; Sassone-Corsi, P. Molecular Cogs: Interplay between Circadian Clock and Cell Cycle. Trends Cell Biol. 2018, 28, 368–379. [Google Scholar] [CrossRef] [PubMed]
- Egli, M.; Leeners, B.; Kruger, T.H. Prolactin secretion patterns: Basic mechanisms and clinical implications for reproduction. Reproduction 2010, 140, 643–654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dickmeis, T. Glucocorticoids and the circadian clock. J. Endocrinol. 2009, 200, 3–22. [Google Scholar] [CrossRef]
- Qian, J.; Morris, C.J.; Caputo, R.; Garaulet, M.; Scheer, F. Ghrelin is impacted by the endogenous circadian system and by circadian misalignment in humans. Int. J. Obes. 2019, 43, 1644–1649. [Google Scholar] [CrossRef]
- Bahrami-Nejad, Z.; Zhao, M.L.; Tholen, S.; Hunerdosse, D.; Tkach, K.E.; van Schie, S.; Chung, M.; Teruel, M.N. A Transcriptional Circuit Filters Oscillating Circadian Hormonal Inputs to Regulate Fat Cell Differentiation. Cell Metab. 2018, 27, 854–868. [Google Scholar] [CrossRef]
- Tchoukalova, Y.D.; Sarr, M.G.; Jensen, M.D. Measuring committed preadipocytes in human adipose tissue from severely obese patients by using adipocyte fatty acid binding protein. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2004, 287, R1132–R1140. [Google Scholar] [CrossRef] [Green Version]
- Feillet, C.; van der Horst, G.T.; Levi, F.; Rand, D.A.; Delaunay, F. Coupling between the Circadian Clock and Cell Cycle Oscillators: Implication for Healthy Cells and Malignant Growth. Front. Neurol. 2015, 6, 96. [Google Scholar] [CrossRef] [Green Version]
- Damiola, F.; Le Minh, N.; Preitner, N.; Kornmann, B.; Fleury-Olela, F.; Schibler, U. Restricted feeding uncouples circadian oscillators in peripheral tissues from the central pacemaker in the suprachiasmatic nucleus. Genes Dev. 2000, 14, 2950–2961. [Google Scholar] [CrossRef] [Green Version]
- Eckel-Mahan, K.L.; Patel, V.R.; de Mateo, S.; Orozco-Solis, R.; Ceglia, N.J.; Sahar, S.; Dilag-Penilla, S.A.; Dyar, K.A.; Baldi, P.; Sassone-Corsi, P. Reprogramming of the circadian clock by nutritional challenge. Cell 2013, 155, 1464–1478. [Google Scholar] [CrossRef] [Green Version]
- Vollmers, C.; Gill, S.; DiTacchio, L.; Pulivarthy, S.R.; Le, H.D.; Panda, S. Time of feeding and the intrinsic circadian clock drive rhythms in hepatic gene expression. Proc. Natl. Acad. Sci. USA 2009, 106, 21453–21458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hatori, M.; Vollmers, C.; Zarrinpar, A.; DiTacchio, L.; Bushong, E.A.; Gill, S.; Leblanc, M.; Chaix, A.; Joens, M.; Fitzpatrick, J.A.; et al. Time-restricted feeding without reducing caloric intake prevents metabolic diseases in mice fed a high-fat diet. Cell Metab. 2012, 15, 848–860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaix, A.; Lin, T.; Le, H.D.; Chang, M.W.; Panda, S. Time-Restricted Feeding Prevents Obesity and Metabolic Syndrome in Mice Lacking a Circadian Clock. Cell Metab. 2019, 29, 303–319. [Google Scholar] [CrossRef] [PubMed]
- Tognini, P.; Murakami, M.; Liu, Y.; Eckel-Mahan, K.L.; Newman, J.C.; Verdin, E.; Baldi, P.; Sassone-Corsi, P. Distinct Circadian Signatures in Liver and Gut Clocks Revealed by Ketogenic Diet. Cell Metab. 2017, 26, 523–538. [Google Scholar] [CrossRef] [PubMed]
- Pendergast, J.S.; Branecky, K.L.; Yang, W.; Ellacott, K.L.; Niswender, K.D.; Yamazaki, S. High-fat diet acutely affects circadian organisation and eating behavior. Eur. J. Neurosci. 2013, 37, 1350–1356. [Google Scholar] [CrossRef] [Green Version]
- Ribas-Latre, A.; Fekry, B.; Kwok, C.; Baumgartner, C.; Shivshankar, S.; Sun, K.; Chen, Z.; Eckel-Mahan, K. Rosiglitazone reverses high fat diet-induced changes in BMAL1 function in muscle, fat, and liver tissue in mice. Int. J. Obes. 2018. [Google Scholar] [CrossRef] [Green Version]
- Klyde, B.J.; Hirsch, J. Increased cellular proliferation in adipose tissue of adult rats fed a high-fat diet. J. Lipid Res. 1979, 20, 705–715. [Google Scholar]
- Zuk, P.A.; Zhu, M.; Mizuno, H.; Huang, J.; Futrell, J.W.; Katz, A.J.; Benhaim, P.; Lorenz, H.P.; Hedrick, M.H. Multilineage cells from human adipose tissue: Implications for cell-based therapies. Tissue Eng. 2001, 7, 211–228. [Google Scholar] [CrossRef] [Green Version]
- Bianco, P.; Robey, P.G.; Simmons, P.J. Mesenchymal stem cells: Revisiting history, concepts, and assays. Cell Stem Cell 2008, 2, 313–319. [Google Scholar] [CrossRef] [Green Version]
- Gimble, J.M.; Bunnell, B.A.; Casteilla, L.; Jung, J.S.; Yoshimura, K. Phases I-III Clinical Trials Using Adult Stem Cells. Stem Cells Int. 2011, 2010, 604–613. [Google Scholar] [CrossRef] [Green Version]
- Bertolini, F.; Petit, J.Y.; Kolonin, M.G. Stem cells from adipose tissue and breast cancer: Hype, risks and hope. Br. J. Cancer 2015, 112, 419–423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez, R.L.; Frazier, T.; Bunnell, B.A.; Mouton, C.A.; March, K.L.; Katz, A.J.; Rubin, J.P.; Llull, R.; Sorensen, J.A.; Gimble, J.M. Arguments for a Different Regulatory Categorization and Framework for Stromal Vascular Fraction. Stem Cells Dev. 2020. [Google Scholar] [CrossRef] [PubMed]
- Wagner, J.; Kean, T.; Young, R.; Dennis, J.E.; Caplan, A.I. Optimizing mesenchymal stem cell-based therapeutics. Curr. Opin. Biotechnol. 2009, 20, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Bellows, C.F.; Zhang, Y.; Simmons, P.J.; Khalsa, A.S.; Kolonin, M.G. Influence of BMI on level of circulating progenitor cells. Obesity 2011, 19, 1722–1726. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Lei, H.; Dong, P.; Fu, X.; Yang, Z.; Yang, Y.; Ma, J.; Liu, X.; Cao, Y.; Xiao, R. Adipose-Derived Mesenchymal Stem Cells from the Elderly Exhibit Decreased Migration and Differentiation Abilities with Senescent Properties. Cell Transplant. 2017, 26, 1505–1519. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, S.; Hughes, D.; Parma, D.L.; Ramirez, A.; Li, R. Association of obesity and circulating adipose stromal cells among breast cancer survivors. Mol. Biol. Rep. 2014, 41, 2907–2916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baker, H.A.; Fabian, C.J.; Hastings, R.C.; Dixon, D.A.; Nydegger, J.L.; Phillips, T.A.; Powers, K.R.; Kimler, B.F. Circulating adipose stromal cells as a response biomarker in phase II energy balance trials of obese breast cancer survivors and high-risk women. Breast Cancer Res. Treat. 2019, 176, 387–394. [Google Scholar] [CrossRef]
- Zhang, T.; Tseng, C.; Daquinag, A.C.; Corn, P.G.; Troncoso, P.; Pettaway, C.; Logothetis, C.; Kolonin, M.G. CXCL1 mediates obesity-associated adipose stromal cell trafficking and function in the tumor microenvironment. Nature Comm. 2016, 7, 11674–11690. [Google Scholar] [CrossRef]
- Bellows, C.F.; Zhang, Y.; Chen, J.; Frazier, M.L.; Kolonin, M.G. Circulation of progenitor cells in obese and lean colorectal cancer patients. Cancer Epidemiol. Biomarkers Prev. 2011, 20, 2461–2468. [Google Scholar] [CrossRef] [Green Version]
- Lengyel, E.; Makowski, L.; DiGiovanni, J.; Kolonin, M.G. Cancer as a Matter of Fat: The Crosstalk between Adipose Tissue and Tumors. Trends Cancer 2018, 4, 374–384. [Google Scholar] [CrossRef]
- Lauby-Secretan, B.; Scoccianti, C.; Loomis, D.; Grosse, Y.; Bianchini, F.; Straif, K. Body Fatness and Cancer--Viewpoint of the IARC Working Group. N. Engl. J. Med. 2016, 375, 794–798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Daquinag, A.C.; Amaya-Manzanares, F.; Sirin, O.; Tseng, C.; Kolonin, M.G. Stromal progenitor cells from endogenous adipose tissue contribute to pericytes and adipocytes that populate the tumor microenvironment. Cancer Res. 2012, 72, 5198–5208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sirin, O.; Kolonin, M.G. Treatment of obesity as a potential complementary approach to cancer therapy. Drug Discov. Today 2013, 11, 567–573. [Google Scholar] [CrossRef]
- Hanahan, D.; Coussens, L.M. Accessories to the crime: Functions of cells recruited to the tumor microenvironment. Cancer Cell 2012, 21, 309–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karnoub, A.E.; Dash, A.B.; Vo, A.P.; Sullivan, A.; Brooks, M.W.; Bell, G.W.; Richardson, A.L.; Polyak, K.; Tubo, R.; Weinberg, R.A. Mesenchymal stem cells within tumour stroma promote breast cancer metastasis. Nature 2007, 449, 557–563. [Google Scholar] [CrossRef]
- Bhowmick, N.A.; Neilson, E.G.; Moses, H.L. Stromal fibroblasts in cancer initiation and progression. Nature 2004, 432, 332–337. [Google Scholar] [CrossRef]
- Sahai, E.; Astsaturov, I.; Cukierman, E.; DeNardo, D.; Egeblad, M.; Evans, R.; Fearon, D.; Greten, F.; Hingorani, S.; Hunter, T.; et al. A Framework for Advancing our Understanding of Cancer-Associated Fibroblasts. Nat. Rev. Cancer 2020, in press. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Daquinag, A.; Traktuev, D.O.; Amaya, F.; Simmons, P.J.; March, K.L.; Pasqualini, R.; Arap, W.; Kolonin, M.G. White adipose tissue cells are recruited by experimental tumors and promote cancer progression in mouse models. Cancer Res. 2009, 69, 5259–5266. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Zhou, D.; Strakovsky, R.; Zhang, Y.; Pan, Y.X. Hepatic cellular senescence pathway genes are induced through histone modifications in a diet-induced obese rat model. Am. J. Physiol. Gastrointest. Liver Physiol. 2011, 302, G558–G564. [Google Scholar] [CrossRef]
- Tseng, C.; Kolonin, M.G. Proteolytic Isoforms of SPARC Induce Adipose Stromal Cell Mobilization in Obesity. Stem Cells 2015, 34, 174–190. [Google Scholar] [CrossRef] [Green Version]
- Klopp, A.H.; Zhang, Y.; Solley, T.; Amaya-Manzanares, F.; Marini, F.; Andreeff, M.; Debeb, B.; Woodward, W.; Schmandt, R.; Broaddus, R.; et al. Omental adipose tissue-derived stromal cells promote vascularization and growth of endometrial tumors. Clin. Cancer Res. 2012, 18, 771–782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Kolonin, M.G. Cytokine signaling regulating adipose stromal cell trafficking. Adipocyte 2016, 5, 369–374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arendt, L.M.; McCready, J.; Keller, P.J.; Baker, D.D.; Naber, S.P.; Seewaldt, V.; Kuperwasser, C. Obesity promotes breast cancer by CCL2-mediated macrophage recruitment and angiogenesis. Cancer Res. 2013, 73, 6080–6093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ebmeier, S.; Horsley, V. Origin of fibrosing cells in systemic sclerosis. Curr. Opin. Rheumatol. 2015, 27, 555–562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seo, B.R.; Bhardwaj, P.; Choi, S.; Gonzalez, J.; Andresen Eguiluz, R.C.; Wang, K.; Mohanan, S.; Morris, P.G.; Du, B.; Zhou, X.K.; et al. Obesity-dependent changes in interstitial ECM mechanics promote breast tumorigenesis. Sci. Transl. Med. 2015, 7, 301–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Bonzo, J.A.; Gonzalez, F.J.; Wang, L. Diurnal regulation of the early growth response 1 (Egr-1) protein expression by hepatocyte nuclear factor 4alpha (HNF4alpha) and small heterodimer partner (SHP) cross-talk in liver fibrosis. J. Biol. Chem. 2011, 286, 29635–29643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, D.; Liu, S.; Ma, H.; Liang, X.; Ma, H.; Yan, X.; Yang, B.; Wei, J.; Liu, X. Paracrine factors from adipose-mesenchymal stem cells enhance metastatic capacity through Wnt signaling pathway in a colon cancer cell co-culture model. Cancer Cell Int. 2015, 15, 42–47. [Google Scholar] [CrossRef] [Green Version]
- Saha, A.; Ahn, S.; Blando, J.; Su, F.; Kolonin, M.G.; DiGiovanni, J. Proinflammatory CXCL12-CXCR4/CXCR7 signaling axis drives Myc-induced prostate cancer in obese mice. Cancer Res. 2017, 77, 5158–5168. [Google Scholar] [CrossRef] [Green Version]
- Nowicka, A.; Marini, F.C.; Solley, T.N.; Elizondo, P.B.; Zhang, Y.; Sharp, H.J.; Broaddus, R.; Kolonin, M.; Mok, S.C.; Thompson, M.S.; et al. Human omental-derived adipose stem cells increase ovarian cancer proliferation, migration, and chemoresistance. PLoS ONE 2013, 8, e81859. [Google Scholar] [CrossRef]
- Orecchioni, S.; Gregato, G.; Martin-Padura, I.; Reggiani, F.; Braidotti, P.; Mancuso, P.; Calleri, A.; Quarna, J.; Marighetti, P.; Aldeni, C.; et al. Complementary populations of human adipose CD34+ progenitor cells promote growth, angiogenesis, and metastasis of breast cancer. Cancer Res. 2013, 73, 5880–5891. [Google Scholar] [CrossRef] [Green Version]
- Rowan, B.G.; Gimble, J.M.; Sheng, M.; Anbalagan, M.; Jones, R.K.; Frazier, T.P.; Asher, M.; Lacayo, E.A.; Friedlander, P.L.; Kutner, R.; et al. Human adipose tissue-derived stromal/stem cells promote migration and early metastasis of triple negative breast cancer xenografts. PLoS ONE 2014, 9, e89595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salimian Rizi, B.; Caneba, C.; Nowicka, A.; Nabiyar, A.W.; Liu, X.; Chen, K.; Klopp, A.; Nagrath, D. Nitric oxide mediates metabolic coupling of omentum-derived adipose stroma to ovarian and endometrial cancer cells. Cancer Res. 2015, 75, 456–4571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zimmerlin, L.; Park, T.S.; Zambidis, E.T.; Donnenberg, V.S.; Donnenberg, A.D. Mesenchymal stem cell secretome and regenerative therapy after cancer. Biochimie 2013, 95, 2235–2245. [Google Scholar] [CrossRef]
- Su, F.; Ahn, S.; Saha, A.; DiGiovanni, J.; Kolonin, M.G. Adipose stromal cell targeting suppresses prostate cancer epithelial-mesenchymal transition and chemoresistance. Oncogene 2019, 38, 1979–1988. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Gu, X.; Zhang, N.; Kolonin, M.G.; An, Z.; Sun, K. Divergent functions of endotrophin on different cell populations in adipose tissue. Am. J. Physiol. Endocrinol. Metab. 2016, 311, 952–963. [Google Scholar] [CrossRef]
- Sun, K.; Park, J.; Gupta, O.T.; Holland, W.L.; Auerbach, P.; Zhang, N.; Goncalves Marangoni, R.; Nicoloro, S.M.; Czech, M.P.; Varga, J.; et al. Endotrophin triggers adipose tissue fibrosis and metabolic dysfunction. Nat. Commun. 2014, 5, 3485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, K.; Tordjman, J.; Clement, K.; Scherer, P.E. Fibrosis and adipose tissue dysfunction. Cell Metab. 2013, 18, 470–477. [Google Scholar] [CrossRef] [Green Version]
- Marangoni, R.G.; Korman, B.D.; Wei, J.; Wood, T.A.; Graham, L.V.; Whitfield, M.L.; Scherer, P.E.; Tourtellotte, W.G.; Varga, J. Myofibroblasts in murine cutaneous fibrosis originate from adiponectin-positive intradermal progenitors. Arthritis Rheumatol. 2015, 67, 1062–1073. [Google Scholar] [CrossRef]
- Sachse, A.; Wolf, G. New aspects of the relationship among hypertension, obesity, and the kidneys. Curr. Hypertens. Rep. 2008, 10, 138–142. [Google Scholar] [CrossRef]
- Sarwar, R.; Pierce, N.; Koppe, S. Obesity and nonalcoholic fatty liver disease: Current perspectives. Diabetes Metab. Syndr. Obes. 2018, 11, 533–542. [Google Scholar] [CrossRef] [Green Version]
- Uezumi, A.; Ito, T.; Morikawa, D.; Shimizu, N.; Yoneda, T.; Segawa, M.; Yamaguchi, M.; Ogawa, R.; Matev, M.M.; Miyagoe-Suzuki, Y.; et al. Fibrosis and adipogenesis originate from a common mesenchymal progenitor in skeletal muscle. J. Cell Sci. 2011, 124, 3654–3664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mozzetta, C.; Consalvi, S.; Saccone, V.; Tierney, M.; Diamantini, A.; Mitchell, K.J.; Marazzi, G.; Borsellino, G.; Battistini, L.; Sassoon, D.; et al. Fibroadipogenic progenitors mediate the ability of HDAC inhibitors to promote regeneration in dystrophic muscles of young, but not old Mdx mice. EMBO Mol Med. 2013, 5, 626–639. [Google Scholar] [CrossRef] [PubMed]
- Gilbane, A.J.; Denton, C.P.; Holmes, A.M. Scleroderma pathogenesis: A pivotal role for fibroblasts as effector cells. Arthritis Res. Ther. 2013, 15, 215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwayama, T.; Olson, L.E. Involvement of PDGF in fibrosis and scleroderma: Recent insights from animal models and potential therapeutic opportunities. Curr. Rheumatol. Rep. 2013, 15, 304. [Google Scholar] [CrossRef] [Green Version]
- Sohn, J.; Lu, A.; Tang, Y.; Wang, B.; Huard, J. Activation of non-myogenic mesenchymal stem cells during the disease progression in dystrophic dystrophin/utrophin knockout mice. Hum. Mol. Genet. 2015, 24, 3814–3829. [Google Scholar] [CrossRef] [Green Version]
- Alakhras, M.; Decker, P.A.; Nadrous, H.F.; Collazo-Clavell, M.; Ryu, J.H. Body mass index and mortality in patients with idiopathic pulmonary fibrosis. Chest 2007, 131, 1448–1453. [Google Scholar] [CrossRef]
- Kolonin, M.G.; Simmons, P.J. Combinatorial stem cell mobilization. Nat. Biotechnol. 2009, 27, 252–253. [Google Scholar] [CrossRef]
- Schwalie, P.C.; Dong, H.; Zachara, M.; Russeil, J.; Alpern, D.; Akchiche, N.; Caprara, C.; Sun, W.; Schlaudraff, K.U.; Soldati, G.; et al. A stromal cell population that inhibits adipogenesis in mammalian fat depots. Nature 2018, 559, 103–108. [Google Scholar] [CrossRef]
- Kruglikov, I.L.; Scherer, P.E. Adipocyte-myofibroblast transition as a possible pathophysiological step in androgenetic alopecia. Exp. Dermatol. 2017, 26, 522–523. [Google Scholar] [CrossRef]
- Dani, C.; Pfeifer, A. The complexity of PDGFR signaling: Regulation of adipose progenitor maintenance and adipocyte-myofibroblast transition. Stem Cell Investig. 2017, 4, 28. [Google Scholar] [CrossRef] [Green Version]
- Campisi, J. Aging, cellular senescence, and cancer. Annu. Rev. Physiol. 2013, 75, 685–705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Y.; Tchkonia, T.; Pirtskhalava, T.; Gower, A.C.; Ding, H.; Giorgadze, N.; Palmer, A.K.; Ikeno, Y.; Hubbard, G.B.; Lenburg, M.; et al. The Achilles’ heel of senescent cells: From transcriptome to senolytic drugs. Aging Cell 2015, 14, 644–658. [Google Scholar] [CrossRef] [PubMed]
- Xue, R.; Lynes, M.D.; Dreyfuss, J.M.; Shamsi, F.; Schulz, T.J.; Zhang, H.; Huang, T.L.; Townsend, K.L.; Li, Y.; Takahashi, H.; et al. Clonal analyses and gene profiling identify genetic biomarkers of the thermogenic potential of human brown and white preadipocytes. Nat. Med. 2015, 21, 760–768. [Google Scholar] [CrossRef] [PubMed]
- Palmer, A.K.; Xu, M.; Zhu, Y.; Pirtskhalava, T.; Weivoda, M.M.; Hachfeld, C.M.; Prata, L.G.; van Dijk, T.H.; Verkade, E.; Casaclang-Verzosa, G.; et al. Targeting senescent cells alleviates obesity-induced metabolic dysfunction. Aging Cell 2019, 18, e12950. [Google Scholar] [CrossRef]
- Childs, B.G.; Durik, M.; Baker, D.J.; van Deursen, J.M. Cellular senescence in aging and age-related disease: From mechanisms to therapy. Nat. Med. 2015, 21, 1424–1435. [Google Scholar] [CrossRef] [Green Version]
- Demaria, M.; Ohtani, N.; Youssef, S.A.; Rodier, F.; Toussaint, W.; Mitchell, J.R.; Laberge, R.M.; Vijg, J.; Van Steeg, H.; Dolle, M.E.; et al. An essential role for senescent cells in optimal wound healing through secretion of PDGF-AA. Dev. Cell 2014, 31, 722–733. [Google Scholar] [CrossRef] [Green Version]
- Ewald, J.A.; Desotelle, J.A.; Wilding, G.; Jarrard, D.F. Therapy-induced senescence in cancer. J. Natl. Cancer Inst. 2010, 102, 1536–1546. [Google Scholar] [CrossRef] [Green Version]
- Baker, D.J.; Childs, B.G.; Durik, M.; Wijers, M.E.; Sieben, C.J.; Zhong, J.; Saltness, R.A.; Jeganathan, K.B.; Verzosa, G.C.; Pezeshki, A.; et al. Naturally occurring p16(Ink4a)-positive cells shorten healthy lifespan. Nature 2016, 530, 184–189. [Google Scholar] [CrossRef] [Green Version]
- Minamino, T.; Orimo, M.; Shimizu, I.; Kunieda, T.; Yokoyama, M.; Ito, T.; Nojima, A.; Nabetani, A.; Oike, Y.; Matsubara, H.; et al. A crucial role for adipose tissue p53 in the regulation of insulin resistance. Nat. Med. 2009, 15, 1082–1087. [Google Scholar] [CrossRef]
- Mitterberger, M.C.; Lechner, S.; Mattesich, M.; Zwerschke, W. Adipogenic differentiation is impaired in replicative senescent human subcutaneous adipose-derived stromal/progenitor cells. J. Gerontol. A Biol. Sci. Med. Sci. 2014, 69, 13–24. [Google Scholar] [CrossRef] [Green Version]
- Mitterberger, M.C.; Mattesich, M.; Zwerschke, W. Bariatric surgery and diet-induced long-term caloric restriction protect subcutaneous adipose-derived stromal/progenitor cells and prolong their life span in formerly obese humans. Exp. Gerontol. 2014, 56, 106–113. [Google Scholar] [CrossRef] [PubMed]
- Berry, D.C.; Jiang, Y.; Arpke, R.W.; Close, E.L.; Uchida, A.; Reading, D.; Berglund, E.D.; Kyba, M.; Graff, J.M. Cellular Aging Contributes to Failure of Cold-Induced Beige Adipocyte Formation in Old Mice and Humans. Cell Metab. 2017, 25, 166–181. [Google Scholar] [CrossRef] [PubMed]
- Schafer, M.J.; White, T.A.; Evans, G.; Tonne, J.M.; Verzosa, G.C.; Stout, M.B.; Mazula, D.L.; Palmer, A.K.; Baker, D.J.; Jensen, M.D.; et al. Exercise Prevents Diet-Induced Cellular Senescence in Adipose Tissue. Diabetes 2016, 65, 1606–1615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gorgoulis, V.; Adams, P.D.; Alimonti, A.; Bennett, D.C.; Bischof, O.; Bishop, C.; Campisi, J.; Collado, M.; Evangelou, K.; Ferbeyre, G.; et al. Cellular Senescence: Defining a Path Forward. Cell 2019, 179, 813–827. [Google Scholar] [CrossRef] [PubMed]
- Whittemore, K.; Vera, E.; Martinez-Nevado, E.; Sanpera, C.; Blasco, M.A. Telomere shortening rate predicts species life span. Proc. Natl. Acad. Sci. USA 2019, 116, 15122–15127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blackburn, E.H.; Greider, C.W.; Szostak, J.W. Telomeres and telomerase: The path from maize, Tetrahymena and yeast to human cancer and aging. Nat. Med. 2006, 12, 1133–1138. [Google Scholar] [CrossRef]
- Calado, R.T.; Dumitriu, B. Telomere dynamics in mice and humans. Semin. Hematol. 2013, 50, 165–174. [Google Scholar] [CrossRef] [Green Version]
- Blasco, M.A. Telomere length, stem cells and aging. Nat. Chem. Biol. 2007, 3, 640–649. [Google Scholar] [CrossRef]
- Lakowa, N.; Trieu, N.; Flehmig, G.; Lohmann, T.; Schon, M.R.; Dietrich, A.; Zeplin, P.H.; Langer, S.; Stumvoll, M.; Bluher, M.; et al. Telomere length differences between subcutaneous and visceral adipose tissue in humans. Biochem. Biophys. Res. Commun. 2015, 457, 426–432. [Google Scholar] [CrossRef]
- Joe, A.W.; Yi, L.; Even, Y.; Vogl, A.W.; Rossi, F.M. Depot-specific differences in adipogenic progenitor abundance and proliferative response to high-fat diet. Stem Cells 2009, 27, 2563–2570. [Google Scholar] [CrossRef]
- Kusminski, C.M.; Holland, W.L.; Sun, K.; Park, J.; Spurgin, S.B.; Lin, Y.; Askew, G.R.; Simcox, J.A.; McClain, D.A.; Li, C.; et al. MitoNEET-driven alterations in adipocyte mitochondrial activity reveal a crucial adaptive process that preserves insulin sensitivity in obesity. Nat. Med. 2012, 18, 1539–1549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kipling, D.; Cooke, H.J. Hypervariable ultra-long telomeres in mice. Nature 1990, 347, 400–402. [Google Scholar] [CrossRef]
- Sacco, A.; Mourkioti, F.; Tran, R.; Choi, J.; Llewellyn, M.; Kraft, P.; Shkreli, M.; Delp, S.; Pomerantz, J.H.; Artandi, S.E.; et al. Short telomeres and stem cell exhaustion model Duchenne muscular dystrophy in mdx/mTR mice. Cell 2010, 143, 1059–1071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolonin, M.G.; Saha, P.K.; Chan, L.; Pasqualini, R.; Arap, W. Reversal of obesity by targeted ablation of adipose tissue. Nat. Med. 2004, 10, 625–632. [Google Scholar] [CrossRef]
- Daquinag, A.C.; Zhang, Y.; Amaya-Manzanares, F.; Simmons, P.J.; Kolonin, M.G. An isoform of decorin is a resistin receptor on the surface of adipose progenitor cells. Cell Stem Cell 2011, 9, 74–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daquinag, A.C.; Tseng, C.; Zhang, Y.; Amaya-Manzanares, F.; Florez, F.; Dadbin, A.; Zhang, T.; Kolonin, M.G. Targeted Pro-apoptotic Peptides Depleting Adipose Stromal Cells Inhibit Tumor Growth. Mol. Ther. 2016, 1, 34–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daquinag, A.C.; Dadbin, A.; Snyder, B.; Wang, X.; Sahin, A.; Uneo, N.T.; Kolonin, M.G. Non-glycanated Decorin is a Drug Target on Human Adipose Stromal Cells. Mol. Ther. Oncolytics 2017, 6, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Song, E. Turning foes to friends: Targeting cancer-associated fibroblasts. Nat. Rev. Drug Discov. 2019, 18, 99–115. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | WAT | BAT |
|---|---|---|
| BMAL1 (whole body) | Increased adiposity but impaired adipogenesis Adipocyte hypertrophy | Increase in BAT mass and heightened cold tolerance [95] |
| BMAL1 (adipocyte-specific; adipocyte protein 2 [aP2] driver) | WAT expansion and loss of rhythmicity in polyunsaturated fatty acid release, driving arrhythmic eating [79] | Enhanced cold tolerance [100] |
| BMAL1 (brown adipocyte-specific, perivascular adipose tissue; Ucp1 driver) | Defective angiotensin production in PVAT. Reduced resting blood pressure, resulting in “superdipper” phenotype [101] | |
| ClockΔ19 mutant (whole body) | Increased mass and exaggerated WAT adipocyte hypertrophy on high fat diet [93] Increased adipogenesis in vivo and in cultured adipose-derived stem cells. Upregulation of adipogenic factors due to loss of transcription factor GILZ expression [99] Blunted lipolysis, resulting in loss of rhythmic glycerol and FA release [88] | |
| REV-ERBα (whole body) | More prone to diet-induced increases in fat mass Upregulation of βKlotho and FGF21 signaling in WAT [102] | Blocks neonatal BAT formation due to loss of brown lineage commitment [100] Improves cold tolerance in a zeitgeber-specific manner [83] |
| REV-ERBα/β (BAT-specific; Ucp1 driver) | Enhanced cold tolerance (via loss of suppression at Srebp1) [103] | |
| PER2 (whole body) | Reduced fat mass, increased oxidative capacity in WAT Increase in adipogenesis-related genes (activation of PPARG targets) [86] | |
| PER3 (whole body) | Increased adipogenesis Increase proliferation of APCs in vivo (SAT) [98] | |
| Nocturnin (NOC) (whole body) | Protection from diet induced obesity, reduced visceral fat [104] | Altered long-term metabolic adaptation in to cold exposure [97] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Eckel-Mahan, K.; Ribas Latre, A.; Kolonin, M.G. Adipose Stromal Cell Expansion and Exhaustion: Mechanisms and Consequences. Cells 2020, 9, 863. https://doi.org/10.3390/cells9040863
Eckel-Mahan K, Ribas Latre A, Kolonin MG. Adipose Stromal Cell Expansion and Exhaustion: Mechanisms and Consequences. Cells. 2020; 9(4):863. https://doi.org/10.3390/cells9040863
Chicago/Turabian StyleEckel-Mahan, Kristin, Aleix Ribas Latre, and Mikhail G. Kolonin. 2020. "Adipose Stromal Cell Expansion and Exhaustion: Mechanisms and Consequences" Cells 9, no. 4: 863. https://doi.org/10.3390/cells9040863
APA StyleEckel-Mahan, K., Ribas Latre, A., & Kolonin, M. G. (2020). Adipose Stromal Cell Expansion and Exhaustion: Mechanisms and Consequences. Cells, 9(4), 863. https://doi.org/10.3390/cells9040863