Adult Stem Cell-Derived Extracellular Vesicles in Cancer Treatment: Opportunities and Challenges

Abstract
:1. Introduction
2. Stem Cell-Derived EVs
3. Unmodified (Native) Adult SCs-EVs Effect on Cancer
3.1. EVs from BM-MSCs
3.2. EVs from UC-MSCs
3.3. EVs from AD-MSCs
3.4. EVs From Other Stem/Progenitor Cells
4. Effects of Modified SC-EVs on Cancer
4.1. Drug Loading
4.2. Protein Loading
4.3. RNA Loading
4.4. SC-EV Modifications to Improve Tissue Targeting
4.5. Increase of SC-EV Yield
5. Adult SCs versus EVs: Advantages and Disadvantages in Cancer Treatment
6. Perspectives and Future Challenges of Adult SC-EV in Cancer Treatment
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Morrison, S.J.; Spradling, A.C. Stem Cells and Niches: Mechanisms That Promote Stem Cell Maintenance throughout Life. Cell 2008, 132, 598–611. [Google Scholar] [CrossRef] [Green Version]
- Clevers, H.; Watt, F.M. Defining Adult Stem Cells by Function, not by Phenotype. Annu. Rev. Biochem. 2018, 87, 1015–1027. [Google Scholar] [CrossRef] [PubMed]
- Méndez-Ferrer, S.; Michurina, T.V.; Ferraro, F.; Mazloom, A.R.; MacArthur, B.D.; Lira, S.A.; Scadden, D.T.; Ma’Ayan, A.; Enikolopov, G.N.; Frenette, P.S. Mesenchymal and haematopoietic stem cells form a unique bone marrow niche. Nature 2010, 466, 829–834. [Google Scholar] [CrossRef] [PubMed]
- Sacchetti, B.; Funari, A.; Michienzi, S.; Di Cesare, S.; Piersanti, S.; Saggio, I.; Tagliafico, E.; Ferrari, S.; Robey, P.; Riminucci, M.; et al. Self-Renewing Osteoprogenitors in Bone Marrow Sinusoids Can Organize a Hematopoietic Microenvironment. Cell 2007, 131, 324–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dominici, M.; Le Blanc, K.; Mueller, I.; Slaper-Cortenbach, I.; Marini, F.; Krause, D.; Deans, R.; Keating, A.; Prockop, D.; Horwitz, E. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy 2006, 8, 315–317. [Google Scholar] [CrossRef]
- Viswanathan, S.; Shi, Y.; Galipeau, J.; Krampera, M.; Leblanc, K.; Martin, I.; Nolta, J.; Phinney, D.; Sensebe, L. Mesenchymal stem versus stromal cells: International Society for Cell & Gene Therapy (ISCT®) Mesenchymal Stromal Cell committee position statement on nomenclature. Cytotherapy 2019, 21, 1019–1024. [Google Scholar] [CrossRef]
- Caplan, A.I. Mesenchymal Stem Cells: Time to Change the Name! Stem Cells Transl. Med. 2017, 6, 1445–1451. [Google Scholar] [CrossRef] [Green Version]
- Reinisch, A.; Etchart, N.; Thomas, D.; Hofmann, N.A.; Fruehwirth, M.; Sinha, S.; Chan, C.K.; Senarath-Yapa, K.; Seo, E.-Y.; Wearda, T.; et al. Epigenetic and in vivo comparison of diverse MSC sources reveals an endochondral signature for human hematopoietic niche formation. Blood 2015, 125, 249–260. [Google Scholar] [CrossRef]
- Sacchetti, B.; Funari, A.; Remoli, C.; Giannicola, G.; Kogler, G.; Liedtke, S.; Cossu, G.; Serafini, M.; Sampaolesi, M.; Tagliafico, E.; et al. No Identical “Mesenchymal Stem Cells” at Different Times and Sites: Human Committed Progenitors of Distinct Origin and Differentiation Potential Are Incorporated as Adventitial Cells in Microvessels. Stem Cell Rep. 2016, 6, 897–913. [Google Scholar] [CrossRef] [Green Version]
- Corselli, M.; Chen, W.C.; Sun, B.; Yap, S.; Rubin, J.P.; Péault, B. The Tunica Adventitia of Human Arteries and Veins As a Source of Mesenchymal Stem Cells. Stem Cells Dev. 2011, 21, 1299–1308. [Google Scholar] [CrossRef] [Green Version]
- Crisan, M.; Yap, S.; Casteilla, L.; Chen, C.-W.; Corselli, M.; Park, T.S.; Andriolo, G.; Sun, B.; Zheng, B.; Zhang, L.; et al. A Perivascular Origin for Mesenchymal Stem Cells in Multiple Human Organs. Cell Stem Cell 2008, 3, 301–313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campagnolo, P.; Katare, R.; Beltrami, A.P. Realities and misconceptions on the pericytes role in tissue repair. Regen. Med. 2018, 13, 119–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guimarães-Camboa, N.; Cattaneo, P.; Sun, Y.; Moore-Morris, T.; Gu, Y.; Dalton, N.D.; Rockenstein, E.; Masliah, E.; Peterson, K.L.; Stallcup, W.B.; et al. Pericytes of Multiple Organs Do Not Behave as Mesenchymal Stem Cells In Vivo. Cell Stem Cell 2017, 20, 345–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caplan, A. Adult Mesenchymal Stem Cells: When, Where, and How. Stem Cells Int. 2015, 2015, 1–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cornelissen, A.S.; Maijenburg, M.W.; Nolte, M.A.; Voermans, C. Organ-specific migration of mesenchymal stromal cells: Who, when, where and why? Immunol. Lett. 2015, 168, 159–169. [Google Scholar] [CrossRef]
- Galipeau, J.; Sensebé, L. Mesenchymal Stromal Cells: Clinical Challenges and Therapeutic Opportunities. Cell Stem Cell 2018, 22, 824–833. [Google Scholar] [CrossRef] [Green Version]
- Timaner, M.; Tsai, K.K.; Shaked, Y. The multifaceted role of mesenchymal stem cells in cancer. Semin. Cancer Boil. 2020, 60, 225–237. [Google Scholar] [CrossRef]
- Quante, M.; Tu, S.P.; Tomita, H.; Gonda, T.; Wang, S.S.; Takashi, S.; Baik, G.H.; Shibata, W.; DiPrete, B.; Betz, K.S.; et al. Bone Marrow-Derived Myofibroblasts Contribute to the Mesenchymal Stem Cell Niche and Promote Tumor Growth. Cancer Cell 2011, 19, 257–272. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.T.; Ji, D.; Wu, S.; Jiang, J.; Zhao, H.; Lin, C.; Cai, X. Human mesenchymal stem cells promote tumor growth via MAPK pathway and metastasis by epithelial mesenchymal transition and integrin alpha5 in hepatocellular carcinoma. Cell Death Dis. 2019, 2019, 425. [Google Scholar] [CrossRef] [Green Version]
- Xue, Z.; Wu, X.; Liu, Y.; Chen, X.; Wang, X.; Wu, K.; Nie, Y.; Fan, D. Mesenchymal Stem Cells Promote Epithelial to Mesenchymal Transition and Metastasis in Gastric Cancer Though Paracrine Cues and Close Physical Contact. J. Cell. Biochem. 2015, 116, 618–627. [Google Scholar] [CrossRef]
- Direkze, N.C. Bone Marrow Contribution to Tumor-Associated Myofibroblasts and Fibroblasts. Cancer Res. 2004, 64, 8492–8495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papaccio, F.; Paino, F.; Regad, T.; Papaccio, G.; Desiderio, V.; Tirino, V. Concise Review: Cancer Cells, Cancer Stem Cells, and Mesenchymal Stem Cells: Influence in Cancer Development. Stem Cells Transl. Med. 2017, 6, 2115–2125. [Google Scholar] [CrossRef] [PubMed]
- Poggi, A.; Giuliani, M. Mesenchymal Stromal Cells Can Regulate the Immune Response in the Tumor Microenvironment. Vaccines 2016, 4, 41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roodhart, J.M.; Daenen, L.G.; Stigter, E.C.; Prins, H.-J.; Gerrits, J.; Houthuijzen, J.M.; Gerritsen, M.G.; Schipper, H.S.; Backer, M.J.; Van Amersfoort, M.; et al. Mesenchymal Stem Cells Induce Resistance to Chemotherapy through the Release of Platinum-Induced Fatty Acids. Cancer Cell 2011, 20, 370–383. [Google Scholar] [CrossRef] [Green Version]
- Ridge, S.M.; Sullivan, F.; Glynn, S. Mesenchymal stem cells: Key players in cancer progression. Mol. Cancer 2017, 16, 31. [Google Scholar] [CrossRef] [Green Version]
- Galipeau, J. The Bottom LineMesenchymal Stromal Cells for Graft-versus-Host Disease: A Trilogy. Boil. Blood Marrow Transplant. 2020. [Google Scholar] [CrossRef]
- Carvello, M.M.; Lightner, A.L.; Yamamoto, T.; Kotze, L.M.D.S.; Spinelli, A. Mesenchymal Stem Cells for Perianal Crohn’s Disease. Cells 2019, 8, 764. [Google Scholar] [CrossRef] [Green Version]
- Hoogduijn, M.J.; Lombardo, E. Mesenchymal Stromal Cells Anno 2019: Dawn of the Therapeutic Era? Concise Review. Stem Cells Transl. Med. 2019, 8, 1126–1134. [Google Scholar] [CrossRef] [Green Version]
- Pittenger, M.F.; Discher, D.E.; Péault, B.; Phinney, D.; Hare, J.M.; Caplan, A.I. Mesenchymal stem cell perspective: Cell biology to clinical progress. npj Regen. Med. 2019, 4, 22. [Google Scholar] [CrossRef] [Green Version]
- Phinney, D.; Pittenger, M.F. Concise Review: MSC-Derived Exosomes for Cell-Free Therapy. STEM CELLS 2017, 35, 851–858. [Google Scholar] [CrossRef] [Green Version]
- Yáñez-Mó, M.; Siljander, P.R.M.; Andreu, Z.; Zavec, A.B.; Borràs, F.E.; Buzas, E.I.; Buzas, K.; Casal, E.; Cappello, F.; Carvalho, J.; et al. Biological properties of extracellular vesicles and their physiological functions. J. Extracell. Vesicles 2015, 4, 27066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chargaff, E.; West, R. The biological significance of the thromboplastic protein of blood. J. Boil. Chem. 1946, 166, 189–197. [Google Scholar]
- Van Niel, G.; D’Angelo, G.; Raposo, G. Shedding light on the cell biology of extracellular vesicles. Nat. Rev. Mol. Cell Boil. 2018, 19, 213–228. [Google Scholar] [CrossRef] [PubMed]
- Colombo, M.; Raposo, G.; Théry, C. Biogenesis, Secretion, and Intercellular Interactions of Exosomes and Other Extracellular Vesicles. Annu. Rev. Cell Dev. Boil. 2014, 30, 255–289. [Google Scholar] [CrossRef]
- Del Fattore, A.; Luciano, R.; Saracino, R.; Battafarano, G.; Rizzo, C.; Pascucci, L.; Alessandri, G.; Pessina, A.; Perrotta, A.; Fierabracci, A.; et al. Differential effects of extracellular vesicles secreted by mesenchymal stem cells from different sources on glioblastoma cells. Expert Opin. Boil. Ther. 2014, 15, 495–504. [Google Scholar] [CrossRef]
- Lee, J.-K.; Park, S.-R.; Jung, B.-K.; Jeon, Y.-K.; Lee, Y.-S.; Kim, M.-K.; Kim, Y.-G.; Jang, J.-Y.; Kim, C.-W. Exosomes Derived from Mesenchymal Stem Cells Suppress Angiogenesis by Down-Regulating VEGF Expression in Breast Cancer Cells. PLoS ONE 2013, 8, e84256. [Google Scholar] [CrossRef] [Green Version]
- Vallabhaneni, K.C.; Penfornis, P.; Dhule, S.; Guillonneau, F.; Adams, K.V.; Mo, Y.Y.; Xu, R.; Liu, Y.; Watabe, K.; Vemuri, M.C.; et al. Extracellular vesicles from bone marrow mesenchymal stem/stromal cells transport tumor regulatory microRNA, proteins, and metabolites. Oncotarget 2014, 6, 4953–4967. [Google Scholar] [CrossRef] [Green Version]
- Roccaro, A.; Sacco, A.; Maiso, P.; Azab, A.K.; Tai, Y.-T.; Reagan, M.R.; Azab, F.; Flores, L.M.; Campigotto, F.; Weller, E.; et al. BM mesenchymal stromal cell-derived exosomes facilitate multiple myeloma progression. J. Clin. Investig. 2013, 123, 1542–1555. [Google Scholar] [CrossRef]
- Théry, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G.K.; et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J. Extracell. Vesicles 2018, 7, 1535750. [Google Scholar] [CrossRef] [Green Version]
- Yang, T.; Martin, P.; Fogarty, B.; Brown, A.; Schurman, K.; Phipps, R.; Yin, Y.P.; Lochman, P.; Bai, S. Exosome delivered anti-cancer drugs across the blood-brain barrier for brain cancer therapy in Danio rerio. Pharm. Res. 2015, 32, 2003–2014. [Google Scholar] [CrossRef]
- Théry, C.; Amigorena, S.; Raposo, G.; Clayton, A. Isolation and Characterization of Exosomes from Cell Culture Supernatants and Biological Fluids. Curr. Protoc. Cell Boil. 2006, 30, 3–22. [Google Scholar] [CrossRef] [PubMed]
- Heijnen, H.F.; E Schiel, A.; Fijnheer, R.; Geuze, H.J.; Sixma, J.J. Activated platelets release two types of membrane vesicles: Microvesicles by surface shedding and exosomes derived from exocytosis of multivesicular bodies and alpha-granules. Blood 1999, 94, 3791–3799. [Google Scholar] [CrossRef] [PubMed]
- Al-Nedawi, K.; Meehan, B.; Micallef, J.; Lhotak, V.; May, L.; Guha, A.; Rak, J. Intercellular transfer of the oncogenic receptor EGFRvIII by microvesicles derived from tumour cells. Nat. Cell Biol. 2008, 10, 619–624. [Google Scholar] [CrossRef] [PubMed]
- Hristov, M.; Erl, W.; Linder, S.; Weber, P.C. Apoptotic bodies from endothelial cells enhance the number and initiate the differentiation of human endothelial progenitor cells in vitro. Blood 2004, 104, 2761–2766. [Google Scholar] [CrossRef]
- Greening, D.; Simpson, R. Understanding extracellular vesicle diversity – current status. Expert Rev. Proteom. 2018, 15, 887–910. [Google Scholar] [CrossRef]
- Latifkar, A.; Hur, Y.H.; Sanchez, J.C.; Cerione, R.A.; Antonyak, M.A. New insights into extracellular vesicle biogenesis and function. J. Cell Sci. 2019, 132, jcs222406. [Google Scholar] [CrossRef] [Green Version]
- Mulcahy, L.; Pink, R.C.; Carter, D.R.F. Routes and mechanisms of extracellular vesicle uptake. J. Extracell. Vesicles 2014, 3, 1093. [Google Scholar] [CrossRef] [Green Version]
- Hessvik, N.P.; Llorente, A. Current knowledge on exosome biogenesis and release. Cell. Mol. Life Sci. 2017, 75, 193–208. [Google Scholar] [CrossRef] [Green Version]
- Bruno, S.; Grange, C.; Deregibus, M.C.; Calogero, R.A.; Saviozzi, S.; Collino, F.; Morando, L.; Busca, A.; Falda, M.; Bussolati, B.; et al. Mesenchymal stem cell-derived microvesicles protect against acute tubular injury. J. Am. Soc. Nephrol. 2009, 20, 1053–1067. [Google Scholar] [CrossRef] [Green Version]
- Bruno, S.; Collino, F.; Deregibus, M.C.; Grange, C.; Tetta, C.; Camussi, G. Microvesicles Derived from Human Bone Marrow Mesenchymal Stem Cells Inhibit Tumor Growth. Stem Cells Dev. 2013, 22, 758–771. [Google Scholar] [CrossRef]
- Deregibus, M.C.; Cantaluppi, V.; Calogero, R.A.; Andreani, G.; Tetta, C.; Biancone, L.; Bruno, S.; Bussolati, B.; Camussi, G. Endothelial progenitor cell–derived microvesicles activate an angiogenic program in endothelial cells by a horizontal transfer of mRNA. Blood 2007, 110, 2440–2448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rong, Y.; Liu, W.; Wang, J.; Fan, J.; Luo, Y.; Li, L.; Kong, F.; Chen, J.; Tang, P.; Cai, W. Neural stem cell-derived small extracellular vesicles attenuate apoptosis and neuroinflammation after traumatic spinal cord injury by activating autophagy. Cell Death Dis. 2019, 10, 340. [Google Scholar] [CrossRef] [PubMed]
- Povero, D.; Pinatel, E.M.; Leszczynska, A.; Goyal, N.P.; Nishio, T.; Kim, J.; Kneiber, D.B.; Horcel, L.D.A.; Eguchi, A.; Ordonez, P.M.; et al. Human induced pluripotent stem cell-derived extracellular vesicles reduce hepatic stellate cell activation and liver fibrosis. JCI Insight 2019, 5, 5. [Google Scholar] [CrossRef] [PubMed]
- Adamiak, M.; Cheng, G.; Bobis-Wozowicz, S.; Zhao, L.; Kedracka-Krok, S.; Samanta, A.; Karnas, E.; Xuan, Y.-T.; Skupien-Rabian, B.; Chen, X.; et al. Induced Pluripotent Stem Cell (iPSC)–Derived Extracellular Vesicles Are Safer and More Effective for Cardiac Repair Than iPSCs. Circ. Res. 2018, 122, 296–309. [Google Scholar] [CrossRef]
- Mc Gough, I.; Vincent, J.-P. Exosomes in developmental signalling. Development 2016, 143, 2482–2493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ratajczak, J.; Miekus, K.; Kucia, M.; Zhang, J.; Reca, R.; Dvorak, P.; Ratajczak, M.Z. Embryonic stem cell-derived microvesicles reprogram hematopoietic progenitors: Evidence for horizontal transfer of mRNA and protein delivery. Leukemia 2006, 20, 847–856. [Google Scholar] [CrossRef] [Green Version]
- Vrijsen, K.R.; Sluijter, J.P.G.; Schuchardt, M.W.L.; Van Balkom, B.W.M.; Noort, W.A.; Chamuleau, S.A.J.; Doevendans, P.A.F.M. Cardiomyocyte progenitor cell-derived exosomes stimulate migration of endothelial cells. J. Cell. Mol. Med. 2010, 14, 1064–1070. [Google Scholar] [CrossRef] [Green Version]
- Sansone, P.; Savini, C.; Kurelac, I.; Chang, Q.; Amato, L.B.; Strillacci, A.; Stepanova, A.; Iommarini, L.; Mastroleo, C.; Daly, L.; et al. Packaging and transfer of mitochondrial DNA via exosomes regulate escape from dormancy in hormonal therapy-resistant breast cancer. Proc. Natl. Acad. Sci. USA 2017, 114, E9066–E9075. [Google Scholar] [CrossRef] [Green Version]
- Kilpinen, L.; Impola, U.; Sankkila, L.; Ritamo, I.; Aatonen, M.; Kilpinen, S.; Tuimala, J.; Valmu, L.; Levijoki, J.; Finckenberg, P.; et al. Extracellular membrane vesicles from umbilical cord blood-derived MSC protect against ischemic acute kidney injury, a feature that is lost after inflammatory conditioning. J. Extracell. Vesicles 2013, 2, 269. [Google Scholar] [CrossRef]
- Salomon, C.; Ryan, J.; Sobrevia, L.; Kobayashi, M.; Ashman, K.; Mitchell, M.; Rice, G.E. Exosomal Signaling during Hypoxia Mediates Microvascular Endothelial Cell Migration and Vasculogenesis. PLoS ONE 2013, 8, e68451. [Google Scholar] [CrossRef]
- Lai, R.C.; Tan, S.S.; Teh, B.J.; Sze, S.K.; Arslan, F.; De Kleijn, D.P.; Choo, A.; Lim, S.K. Proteolytic Potential of the MSC Exosome Proteome: Implications for an Exosome-Mediated Delivery of Therapeutic Proteasome. Int. J. Proteom. 2012, 2012, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramos, T.L.; Sanchez-Abarca, L.I.; Muntión, S.; Preciado, S.; Puig, N.; López-Ruano, G.; Hernández-Hernández, Á.; Redondo, A.; Ortega, R.; Rodríguez, C.; et al. MSC surface markers (CD44, CD73, and CD90) can identify human MSC-derived extracellular vesicles by conventional flow cytometry. Cell Commun. Signal. 2016, 14, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Balkom, B.W.M.; Gremmels, H.; Giebel, B.; Lim, S.K. Proteomic Signature of Mesenchymal Stromal Cell-Derived Small Extracellular Vesicles. Proteomics 2019, 19, e1800163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, B.; Shen, L.; Shi, H.; Pan, Z.; Wu, L.; Yan, Y.; Zhang, X.; Mao, F.; Qian, H.; Xu, W. Exosomes from Human Umbilical Cord Mesenchymal Stem Cells: Identification, Purification, and Biological Characteristics. Stem Cells Int. 2016, 2016, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Burrello, J.; Monticone, S.; Gai, C.; Gomez, Y.; Kholia, S.; Camussi, G. Stem Cell-Derived Extracellular Vesicles and Immune-Modulation. Front. Cell Dev. Boil. 2016, 4, 1815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ragni, E.; Banfi, F.; Barilani, M.; Cherubini, A.; Parazzi, V.; Larghi, P.; Dolo, V.; Bollati, V.; Lazzari, L. Extracellular Vesicle-Shuttled mRNA in Mesenchymal Stem Cell Communication. STEM CELLS 2017, 35, 1093–1105. [Google Scholar] [CrossRef] [Green Version]
- Bruno, S.; Tapparo, M.; Collino, F.; Chiabotto, G.; Deregibus, M.C.; Lindoso, R.S.; Neri, F.; Kholia, S.; Giunti, S.; Wen, S.; et al. Renal Regenerative Potential of Different Extracellular Vesicle Populations Derived from Bone Marrow Mesenchymal Stromal Cells. Tissue Eng. Part A 2017, 23, 1262–1273. [Google Scholar] [CrossRef]
- Haraszti, R.A.; Didiot, M.-C.; Sapp, E.; Leszyk, J.; Shaffer, S.A.; Rockwell, H.E.; Gao, F.; Narain, N.R.; DiFiglia, M.; Kiebish, M.A.; et al. High-resolution proteomic and lipidomic analysis of exosomes and microvesicles from different cell sources. J. Extracell. Vesicles 2016, 5, 2500. [Google Scholar] [CrossRef]
- Marzesco, A.-M. Prominin-1-Containing Membrane Vesicles: Origins, Formation, and Utility. Plant Promoters Transcr. Factors 2012, 777, 41–54. [Google Scholar] [CrossRef]
- Fargeas, C.A. Prominin-2 and Other Relatives of CD133. Plant Promoters Transcr. Factors 2012, 777, 25–40. [Google Scholar] [CrossRef]
- Phinney, D.; Di Giuseppe, M.; Njah, J.; Sala-Llinas, E.; Shiva, S.; Croix, C.M.S.; Stolz, N.B.; Watkins, S.C.; Di, Y.P.; Leikauf, G.; et al. Mesenchymal stem cells use extracellular vesicles to outsource mitophagy and shuttle microRNAs. Nat. Commun. 2015, 6, 8472. [Google Scholar] [CrossRef] [PubMed]
- Stahl, P.D.; Raposo, G. Extracellular Vesicles: Exosomes and Microvesicles, Integrators of Homeostasis. Physiology 2019, 34, 169–177. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, A.; Okada, R.; Nagao, K.; Kawamata, Y.; Hanyu, A.; Yoshimoto, S.; Takasugi, M.; Watanabe, S.; Kanemaki, M.T.; Obuse, C.; et al. Exosomes maintain cellular homeostasis by excreting harmful DNA from cells. Nat. Commun. 2017, 8, 15287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, B.; Wang, M.; Gong, A.; Zhang, X.; Wu, X.; Zhu, Y.; Shi, H.; Wu, L.; Zhu, W.; Qian, H.; et al. HucMSC-Exosome Mediated-Wnt4 Signaling Is Required for Cutaneous Wound Healing. STEM CELLS 2015, 33, 2158–2168. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.Y.; Lai, R.C.; Wong, W.; Dan, Y.Y.; Lim, S.K.; Ho, H.K. Mesenchymal stem cell-derived exosomes promote hepatic regeneration in drug-induced liver injury models. Stem Cell Res. Ther. 2014, 5, 76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, C.; Mitsialis, S.A.; Aslam, M.; Vitali, S.H.; Vergadi, E.; Konstantinou, G.; Sdrimas, K.; Fernandez-Gonzalez, A.; Kourembanas, S. Exosomes mediate the cytoprotective action of mesenchymal stromal cells on hypoxia-induced pulmonary hypertension. Circulation 2012, 126, 2601–2611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruno, S.; Grange, C.; Collino, F.; Deregibus, M.C.; Cantaluppi, V.; Biancone, L.; Tetta, C.; Camussi, G. Microvesicles Derived from Mesenchymal Stem Cells Enhance Survival in a Lethal Model of Acute Kidney Injury. PLoS ONE 2012, 7, e33115. [Google Scholar] [CrossRef]
- Ranghino, A.; Cantaluppi, V.; Grange, C.; Vitillo, L.; Fop, F.; Biancone, L.; Deregibus, M.; Tetta, C.; Segoloni, G.; Camussi, G. Endothelial Progenitor Cell-Derived Microvesicles Improve Neovascularization in a Murine Model of Hindlimb Ischemia. Int. J. Immunopathol. Pharmacol. 2012, 25, 75–85. [Google Scholar] [CrossRef] [Green Version]
- Barile, L.; Lionetti, V.; Cervio, E.; Matteucci, M.; Gherghiceanu, M.; Popescu, L.M.; Torre, T.; Siclari, F.; Moccetti, T.; Vassalli, G. Extracellular vesicles from human cardiac progenitor cells inhibit cardiomyocyte apoptosis and improve cardiac function after myocardial infarction. Cardiovasc. Res. 2014, 103, 530–541. [Google Scholar] [CrossRef]
- Saha, P.; Sharma, S.; Korutla, L.; Datla, S.R.; Shoja-Taheri, F.; Mishra, R.; Bigham, G.E.; Sarkar, M.; Morales, D.; Bittle, G.; et al. Circulating exosomes derived from transplanted progenitor cells aid the functional recovery of ischemic myocardium. Sci. Transl. Med. 2019, 11, eaau1168. [Google Scholar] [CrossRef]
- Rybinski, B.; Franco-Barraza, J.; Cukierman, E. The wound healing, chronic fibrosis, and cancer progression triad. Physiol. Genom. 2014, 46, 223–244. [Google Scholar] [CrossRef] [PubMed]
- Dvorak, H. Tumors: Wounds that do not heal-redux. Cancer Immunol. Res. 2015, 3, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schäfer, M.; Werner, S. Cancer as an overhealing wound: An old hypothesis revisited. Nat. Rev. Mol. Cell Boil. 2008, 9, 628–638. [Google Scholar] [CrossRef] [PubMed]
- Vermeulen, L.; Snippert, H.J. Stem cell dynamics in homeostasis and cancer of the intestine. Nat. Rev. Cancer 2014, 14, 468–480. [Google Scholar] [CrossRef]
- Gross, J.C.; Chaudhary, V.; Bartscherer, K.; Boutros, M. Active Wnt proteins are secreted on exosomes. Nat. Cell Biol. 2012, 14, 1036–1045. [Google Scholar] [CrossRef]
- Riazifar, M.; Pone, E.J.; Lötvall, J.; Zhao, W. Stem Cell Extracellular Vesicles: Extended Messages of Regeneration. Annu. Rev. Pharmacol. Toxicol. 2016, 57, 125–154. [Google Scholar] [CrossRef] [Green Version]
- Vakhshiteh, F.; Atyabi, F.; Ostad, S.N. Mesenchymal stem cell exosomes: A two-edged sword in cancer therapy. Int. J. Nanomed. 2019, 14, 2847–2859. [Google Scholar] [CrossRef] [Green Version]
- Moore, C.; Kosgodage, U.; Lange, S.; Inal, J. The emerging role of exosome and microvesicle- (EMV-) based cancer therapeutics and immunotherapy. Int. J. Cancer 2017, 141, 428–436. [Google Scholar] [CrossRef]
- Zhu, W.; Huang, L.; Li, Y.; Zhang, X.; Gu, J.; Yan, Y.; Xu, X.; Wang, M.; Qian, H.; Xu, W. Exosomes derived from human bone marrow mesenchymal stem cells promote tumor growth in vivo. Cancer Lett. 2012, 315, 28–37. [Google Scholar] [CrossRef]
- Qi, J.; Zhou, Y.; Jiao, Z.; Wang, X.; Zhao, Y.; Li, Y.; Chen, H.; Yang, L.; Zhu, H.; Li, Y. Exosomes Derived from Human Bone Marrow Mesenchymal Stem Cells Promote Tumor Growth Through Hedgehog Signaling Pathway. Cell. Physiol. Biochem. 2017, 42, 2242–2254. [Google Scholar] [CrossRef]
- Ren, W.; Hou, J.; Yang, C.; Zhang, H.; Wu, S.; Wu, Y.; Zhao, X.; Lu, C. Extracellular vesicles secreted by hypoxia pre-challenged mesenchymal stem cells promote non-small cell lung cancer cell growth and mobility as well as macrophage M2 polarization via miR-21-5p delivery. J. Exp. Clin. Cancer Res. 2019, 38, 62. [Google Scholar] [CrossRef] [PubMed]
- Ono, M.; Kosaka, N.; Tominaga, N.; Yoshioka, Y.; Takeshita, F.; Takahashi, R.-U.; Yoshida, M.; Tsuda, H.; Tamura, K.; Ochiya, T. Exosomes from bone marrow mesenchymal stem cells contain a microRNA that promotes dormancy in metastatic breast cancer cells. Sci. Signal. 2014, 7, ra63. [Google Scholar] [CrossRef] [PubMed]
- Shi, S.; Zhang, Q.; Xia, Y.; You, B.; Shan, Y.; Bao, L.; Li, L.; You, Y.; Gu, Z. Mesenchymal stem cell-derived exosomes facilitate nasopharyngeal carcinoma progression. Am. J. Cancer Res. 2016, 6, 459–472. [Google Scholar] [PubMed]
- Mao, J.; Lian, Z.; Zhanb, B.; Yang, H.; Li, H.; Fu, H.; Zhang, X.; Yan, Y.; Xu, W.; Qian, H. UBR2 Enriched in p53 Deficient Mouse Bone Marrow Mesenchymal Stem Cell-Exosome Promoted Gastric Cancer Progression via Wnt/beta-Catenin Pathway. Stem Cells 2017, 35, 2267–2279. [Google Scholar] [CrossRef] [Green Version]
- Ma, M.; Chen, S.; Liu, Z.; Xie, H.; Deng, H.; Shang, S.; Wang, X.; Xia, M.; Zuo, C. miRNA-221 of exosomes originating from bone marrow mesenchymal stem cells promotes oncogenic activity in gastric cancer. OncoTargets Ther. 2017, 10, 4161–4171. [Google Scholar] [CrossRef] [Green Version]
- Lin, R.; Wang, S.; Zhao, R.C. Exosomes from human adipose-derived mesenchymal stem cells promote migration through Wnt signaling pathway in a breast cancer cell model. Mol. Cell. Biochem. 2013, 383, 13–20. [Google Scholar] [CrossRef]
- Ji, R.; Zhang, B.; Zhang, X.; Xue, J.; Yuan, X.; Yan, Y.; Wang, M.; Zhu, W.; Qian, H.; Xu, W. Exosomes derived from human mesenchymal stem cells confer drug resistance in gastric cancer. Cell Cycle 2015, 14, 2473–2483. [Google Scholar] [CrossRef] [Green Version]
- Dong, L.; Pu, Y.; Zhang, L.; Qi, Q.; Xu, L.; Li, W.; Wei, C.; Wang, X.; Zhou, S.; Zhu, J.; et al. Human umbilical cord mesenchymal stem cell-derived extracellular vesicles promote lung adenocarcinoma growth by transferring miR-410. Cell Death Dis. 2018, 9, 218. [Google Scholar] [CrossRef]
- Yang, Y.; Bucan, V.; Baehre, H.; Von Der Ohe, J.; Otte, A.; Hass, R. Acquisition of new tumor cell properties by MSC-derived exosomes. Int. J. Oncol. 2015, 47, 244–252. [Google Scholar] [CrossRef] [Green Version]
- Du, T.; Ju, G.; Wu, S.; Cheng, Z.; Cheng, J.; Zou, X.; Zhang, G.; Miao, S.; Liu, G.; Zhu, Y. Microvesicles Derived from Human Wharton’s Jelly Mesenchymal Stem Cells Promote Human Renal Cancer Cell Growth and Aggressiveness through Induction of Hepatocyte Growth Factor. PLoS ONE 2014, 9, e96836. [Google Scholar] [CrossRef] [Green Version]
- Pascucci, L.; Coccè, V.; Bonomi, A.; Ami, D.; Ceccarelli, P.; Ciusani, E.; Viganò, L.; Locatelli, A.; Sisto, F.; Doglia, S.M.; et al. Paclitaxel is incorporated by mesenchymal stromal cells and released in exosomes that inhibit in vitro tumor growth: A new approach for drug delivery. J. Control. Release 2014, 192, 262–270. [Google Scholar] [CrossRef] [PubMed]
- Kalimuthu, S.; Gangadaran, P.; Rajendran, R.L.; Zhu, L.; Oh, J.M.; Lee, H.W.; Gopal, A.; Baek, S.H.; Jeong, S.Y.; Lee, S.-W.; et al. A New Approach for Loading Anticancer Drugs Into Mesenchymal Stem Cell-Derived Exosome Mimetics for Cancer Therapy. Front. Pharmacol. 2018, 9, 1116. [Google Scholar] [CrossRef] [PubMed]
- Munoz, J.L.; A Bliss, S.; Greco, S.J.; Ramkissoon, S.H.; Ligon, K.L.; Rameshwar, P. Delivery of Functional Anti-miR-9 by Mesenchymal Stem Cell-derived Exosomes to Glioblastoma Multiforme Cells Conferred Chemosensitivity. Mol. Ther. Nucleic Acids 2013, 2, e126. [Google Scholar] [CrossRef]
- Lee, H.-K.; Finniss, S.; Cazacu, S.; Bucris, E.; Ziv-Av, A.; Xiang, C.; Bobbitt, K.; Rempel, S.A.; Hasselbach, L.; Mikkelsen, T.; et al. Mesenchymal stem cells deliver synthetic microRNA mimics to glioma cells and glioma stem cells and inhibit their cell migration and self-renewal. Oncotarget 2013, 4, 346–361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimbo, K.; Miyaki, S.; Ishitobi, H.; Kato, Y.; Kubo, T.; Shimose, S.; Ochi, M. Exosome-formed synthetic microRNA-143 is transferred to osteosarcoma cells and inhibits their migration. Biochem. Biophys. Res. Commun. 2014, 445, 381–387. [Google Scholar] [CrossRef]
- Katakowski, M.; Buller, B.; Zheng, X.; Lu, Y.; Rogers, T.; Osobamiro, O.; Shu, W.; Jiang, F.; Chopp, M. Exosomes from marrow stromal cells expressing miR-146b inhibit glioma growth. Cancer Lett. 2013, 335, 201–204. [Google Scholar] [CrossRef] [Green Version]
- Bliss, S.A.; Sinha, G.; Sandiford, O.A.; Williams, L.M.; Engelberth, D.J.; Guiro, K.; Isenalumhe, L.L.; Greco, S.J.; Ayer, S.; Bryan, M.; et al. Mesenchymal Stem Cell-Derived Exosomes Stimulate Cycling Quiescence and Early Breast Cancer Dormancy in Bone Marrow. Cancer Res. 2016, 76, 5832–5844. [Google Scholar] [CrossRef] [Green Version]
- Naseri, Z.; Oskuee, R.K.; Jaafari, M.R.; Moghadam, M.F. Exosome-mediated delivery of functionally active miRNA-142-3p inhibitor reduces tumorigenicity of breast cancer in vitro and in vivo. Int. J. Nanomed. 2018, 13, 7727–7747. [Google Scholar] [CrossRef] [Green Version]
- O’Brien, K.P.; Khan, S.; Gilligan, K.; Zafar, H.; Lalor, P.; Glynn, C.; O’Flatharta, C.; Ingoldsby, H.; Dockery, P.; De Bhulbh, A.; et al. Employing mesenchymal stem cells to support tumor-targeted delivery of extracellular vesicle (EV)-encapsulated microRNA-379. Oncogene 2018, 37, 2137–2149. [Google Scholar] [CrossRef] [Green Version]
- Lang, F.M.; Hossain, A.; Gumin, J.; Momin, E.N.; Shimizu, Y.; Ledbetter, D.; Shahar, T.; Yamashita, S.; Kerrigan, B.P.; Fueyo, J.; et al. Mesenchymal stem cells as natural biofactories for exosomes carrying miR-124a in the treatment of gliomas. Neuro-Oncology 2017, 20, 380–390. [Google Scholar] [CrossRef] [Green Version]
- Takahara, K.; Ii, M.; Inamoto, T.; Nakagawa, T.; Ibuki, N.; Yoshikawa, Y.; Tsujino, T.; Uchimoto, T.; Saito, K.; Takai, T.; et al. microRNA-145 Mediates the Inhibitory Effect of Adipose Tissue-Derived Stromal Cells on Prostate Cancer. Stem Cells Dev. 2016, 25, 1290–1298. [Google Scholar] [CrossRef] [PubMed]
- Reza, A.M.M.T.; Choi, Y.-J.; Yasuda, H.; Kim, J.-H. Human adipose mesenchymal stem cell-derived exosomal-miRNAs are critical factors for inducing anti-proliferation signalling to A2780 and SKOV-3 ovarian cancer cells. Sci. Rep. 2016, 6, 38498. [Google Scholar] [CrossRef] [PubMed]
- Ko, S.-F.; Yip, H.-K.; Zhen, Y.-Y.; Lee, C.-C.; Lee, C.-C.; Huang, C.-C.; Ng, S.-H.; Lin, J.-W. Adipose-Derived Mesenchymal Stem Cell Exosomes Suppress Hepatocellular Carcinoma Growth in a Rat Model: Apparent Diffusion Coefficient, Natural Killer T-Cell Responses, and Histopathological Features. Stem Cells Int. 2015, 2015, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lou, G.; Song, X.; Yang, F.; Wu, S.; Wang, J.; Chen, Z.; Liu, Y. Exosomes derived from miR-122-modified adipose tissue-derived MSCs increase chemosensitivity of hepatocellular carcinoma. J. Hematol. Oncol. 2015, 8, 122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, L.; Liu, Y.; Qu, Y.; Liu, L.; Li, H. Exosomes Derived From MicroRNA-148b-3p-Overexpressing Human Umbilical Cord Mesenchymal Stem Cells Restrain Breast Cancer Progression. Front. Oncol. 2019, 9, 1076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, S.; Ju, G.-Q.; Du, T.; Zhu, Y.-J.; Liu, G.-H. Microvesicles Derived from Human Umbilical Cord Wharton’s Jelly Mesenchymal Stem Cells Attenuate Bladder Tumor Cell Growth In Vitro and In Vivo. PLoS ONE 2013, 8, e61366. [Google Scholar] [CrossRef]
- Melzer, C.; Rehn, V.; Yang, Y.; Bähre, H.; Von Der Ohe, J.; Hass, R. Taxol-Loaded MSC-Derived Exosomes Provide a Therapeutic Vehicle to Target Metastatic Breast Cancer and Other Carcinoma Cells. Cancers 2019, 11, 798. [Google Scholar] [CrossRef] [Green Version]
- Sharif, S.; Ghahremani, M.H.; Soleimani, M. Delivery of Exogenous miR-124 to Glioblastoma Multiform Cells by Wharton’s Jelly Mesenchymal Stem Cells Decreases Cell Proliferation and Migration, and Confers Chemosensitivity. Stem Cell Rev. Rep. 2017, 14, 236–246. [Google Scholar] [CrossRef]
- Fonsato, V.; Collino, F.; Herrera, M.B.; Cavallari, C.; Deregibus, M.C.; Cisterna, B.; Bruno, S.; Romagnoli, R.; Salizzoni, M.; Tetta, C.; et al. Human Liver Stem Cell-Derived Microvesicles Inhibit Hepatoma Growth in SCID Mice by Delivering Antitumor MicroRNAs. STEM CELLS 2012, 30, 1985–1998. [Google Scholar] [CrossRef] [Green Version]
- Lopatina, T.; Grange, C.; Fonsato, V.; Tapparo, M.; Brossa, A.; Fallo, S.; Pitino, A.; Herrera-Sanchez, M.B.; Kholia, S.; Camussi, G.; et al. Extracellular vesicles from human liver stem cells inhibit tumor angiogenesis. Int. J. Cancer 2018, 144, 322–333. [Google Scholar] [CrossRef] [Green Version]
- Brossa, A.; Fonsato, V.; Grange, C.; Tritta, S.; Tapparo, M.; Calvetti, R.; Cedrino, M.; Fallo, S.; Gontero, P.; Camussi, G.; et al. Extracellular vesicles from human liver stem cells inhibit renal cancer stem cell-derived tumor growth in vitro and in vivo. Int. J. Cancer 2020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greco, K.A.; Franzen, C.; Foreman, K.E.; Flanigan, R.C.; Kuo, P.; Gupta, G. PLK-1 Silencing in Bladder Cancer by siRNA Delivered With Exosomes. Urology 2016, 91, 241.e1–241.e7. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Z.; Kolluri, K.K.; Gowers, K.H.; Janes, S.M. TRAIL delivery by MSC-derived extracellular vesicles is an effective anti-cancer therapy. J. Extracell. Vesicles 2017, 6, 1265291. [Google Scholar] [CrossRef] [PubMed]
- Rosenberger, L.; Ezquer, M.; Lillo-Vera, F.; Pedraza, P.L.; Ortúzar, M.I.; González, P.L.; Figueroa-Valdés, A.I.; Cuenca, J.; Ezquer, F.; Khoury, M.; et al. Stem cell exosomes inhibit angiogenesis and tumor growth of oral squamous cell carcinoma. Sci. Rep. 2019, 9, 663. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Li, N.; Song, K.; Wei, J.; Yao, S.; Li, Z.; Su, X.; Ju, X.; Chao, L.; Deng, X.; et al. Exosomes derived from human umbilical cord mesenchymal stem cells protect against cisplatin-induced ovarian granulosa cell stress and apoptosis in vitro. Sci. Rep. 2017, 7, 2552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- García-Manrique, P.; Matos, M.; Gutierrez, G.; Pazos, C.; Blanco-López, M.C. Therapeutic biomaterials based on extracellular vesicles: Classification of bio-engineering and mimetic preparation routes. J. Extracell. Vesicles 2018, 7, 1422676. [Google Scholar] [CrossRef] [Green Version]
- You, B.; Xu, W.; Zhang, B. Engineering exosomes: A new direction for anti-cancer treatment. Am. J. Cancer Res 2018, 8, 1332–1342. [Google Scholar]
- Fuhrmann, G.; Serio, A.; Mazo, M.M.; Nair, R.; Stevens, M.M. Active loading into extracellular vesicles significantly improves the cellular uptake and photodynamic effect of porphyrins. J. Control. Release 2015, 205, 35–44. [Google Scholar] [CrossRef]
- Sutaria, D.; Badawi, M.; Phelps, M.A.; Schmittgen, T.D. Achieving the Promise of Therapeutic Extracellular Vesicles: The Devil is in Details of Therapeutic Loading. Pharm. Res. 2017, 34, 1053–1066. [Google Scholar] [CrossRef] [Green Version]
- Antimisiaris, S.G.; Mourtas, S.; Marazioti, A. Exosomes and Exosome-Inspired Vesicles for Targeted Drug Delivery. Pharmaceutics 2018, 10, 218. [Google Scholar] [CrossRef] [Green Version]
- Haney, M.J.; Klyachko, N.L.; Zhao, Y.; Gupta, R.; Plotnikova, E.G.; He, Z.; Patel, T.; Piroyan, A.; Sokolsky, M.; Kabanov, A.V.; et al. Exosomes as drug delivery vehicles for Parkinson’s disease therapy. J. Control. Release 2015, 207, 18–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Micheau, O.; Shirley, S.; Dufour, F. Death receptors as targets in cancer. Br. J. Pharmacol. 2013, 169, 1723–1744. [Google Scholar] [CrossRef] [PubMed]
- Yim, N.; Ryu, S.-W.; Choi, K.; Lee, K.R.; Lee, S.; Choi, H.; Kim, J.; Shaker, M.R.; Sun, W.; Park, J.-H.; et al. Exosome engineering for efficient intracellular delivery of soluble proteins using optically reversible protein–protein interaction module. Nat. Commun. 2016, 7, 12277. [Google Scholar] [CrossRef] [PubMed]
- Mateescu, B.; Kowal, E.; Van Balkom, B.W.M.; Bartel, S.; Bhattacharyya, S.N.; Buzás, E.I.; Buck, A.H.; De Candia, P.; Chow, W.-N.; Das, S.; et al. Obstacles and opportunities in the functional analysis of extracellular vesicle RNA – an ISEV position paper. J. Extracell. Vesicles 2017, 6, 1286095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vasconcelos, M.H.; Caires, H.R.; Ābols, A.; Xavier, C.P.; Linē, A. Extracellular vesicles as a novel source of biomarkers in liquid biopsies for monitoring cancer progression and drug resistance. Drug Resist. Updat. 2019, 47, 100647. [Google Scholar] [CrossRef] [PubMed]
- Ullah, M.; Qiao, Y.; Concepcion, W.; Thakor, A.S. Stem cell-derived extracellular vesicles: Role in oncogenic processes, bioengineering potential, and technical challenges. Stem Cell Res. Ther. 2019, 10, 347. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.; Su, C. Design strategies and application progress of therapeutic exosomes. Theranostics 2019, 9, 1015–1028. [Google Scholar] [CrossRef]
- Murphy, D.E.; De Jong, O.G.; Brouwer, M.; Wood, M.J.; Lavieu, G.; Schiffelers, R.M.; Vader, P. Extracellular vesicle-based therapeutics: Natural versus engineered targeting and trafficking. Exp. Mol. Med. 2019, 51, 1–12. [Google Scholar] [CrossRef]
- Sato, Y.T.; Umezaki, K.; Sawada, S.; Mukai, S.-A.; Sasaki, Y.; Harada, N.; Shiku, H.; Akiyoshi, K. Engineering hybrid exosomes by membrane fusion with liposomes. Sci. Rep. 2016, 6, 21933. [Google Scholar] [CrossRef] [Green Version]
- Pachler, K.; Lener, T.; Streif, D.; Dunai, Z.A.; Desgeorges, A.; Feichtner, M.; Öller, M.; Schallmoser, K.; Rohde, E.; Gimona, M. A Good Manufacturing Practice–grade standard protocol for exclusively human mesenchymal stromal cell–derived extracellular vesicles. Cytotherapy 2017, 19, 458–472. [Google Scholar] [CrossRef] [Green Version]
- Lai, R.C.; Yeo, R.W.Y.; Padmanabhan, J.; Choo, A.; De Kleijn, D.P.V.; Lim, S.K. Isolation and Characterization of Exosome from Human Embryonic Stem Cell-Derived C-Myc-Immortalized Mesenchymal Stem Cells. Methods Mol. Biol. 2016, 1416, 477–494. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Kim, T.M. Generation of mesenchymal stem-like cells for producing extracellular vesicles. World J. Stem Cells 2019, 11, 270–280. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Bonacquisti, E.E.; Brown, A.D.; Nguyen, J. Boosting the Biogenesis and Secretion of Mesenchymal Stem Cell-Derived Exosomes. Cells 2020, 9, 660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phan, J.; Kumar, P.; Hao, D.; Gao, K.; Farmer, D.L.; Wang, A. Engineering mesenchymal stem cells to improve their exosome efficacy and yield for cell-free therapy. J. Extracell. Vesicles 2018, 7, 1522236. [Google Scholar] [CrossRef]
- Aboody, K.S.; Brown, A.; Rainov, N.G.; Bower, K.A.; Liu, S.; Yang, W.; Small, J.E.; Herrlinger, U.; Ourednik, V.; Black, P.M.; et al. Neural stem cells display extensive tropism for pathology in adult brain: Evidence from intracranial gliomas. Proc. Natl. Acad. Sci. USA 2000, 97, 12846–12851. [Google Scholar] [CrossRef] [Green Version]
- Bernardo, M.E.; Fibbe, W.E. Mesenchymal Stromal Cells: Sensors and Switchers of Inflammation. Cell Stem Cell 2013, 13, 392–402. [Google Scholar] [CrossRef] [Green Version]
- Qiao, L.; Xu, Z.; Zhao, T.; Zhao, Z.; Shi, M.; Zhao, R.C.; Ye, L.; Zhang, X. Suppression of tumorigenesis by human mesenchymal stem cells in a hepatoma model. Cell Res. 2008, 18, 500–507. [Google Scholar] [CrossRef] [Green Version]
- Momin, E.N.; Vela, G.; Zaidi, H.A.; Quiñones-Hinojosa, A. The Oncogenic Potential of Mesenchymal Stem Cells in the Treatment of Cancer: Directions for Future Research. Curr. Immunol. Rev. 2010, 6, 137–148. [Google Scholar] [CrossRef]
- A Park, S.; Ryu, C.H.; Kim, S.M.; Lim, J.Y.; Park, S.I.; Jeong, C.H.; Jun, J.A.; Oh, J.H.; Park, S.H.; Oh, W.; et al. CXCR4-transfected human umbilical cord blood-derived mesenchymal stem cells exhibit enhanced migratory capacity toward gliomas. Int. J. Oncol. 2011, 38, 97–103. [Google Scholar]
- Friedman, R.; Betancur, M.; Boissel, L.; Tuncer, H.; Cetrulo, C.; Klingemann, H. Umbilical Cord Mesenchymal Stem Cells: Adjuvants for Human Cell Transplantation. Boil. Blood Marrow Transplant. 2007, 13, 1477–1486. [Google Scholar] [CrossRef] [Green Version]
- Qiao, L.; Xu, Z.-L.; Zhao, T.-J.; Ye, L.-H.; Zhang, X.-D. Dkk-1 secreted by mesenchymal stem cells inhibits growth of breast cancer cells via depression of Wnt signalling. Cancer Lett. 2008, 269, 67–77. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Sun, Z.; Han, Q.; Liao, L.; Wang, J.; Bian, C.; Li, J.; Yan, X.; Liu, Y.; Shao, C.; et al. Human mesenchymal stem cells inhibit cancer cell proliferation by secreting DKK-1. Leukemia 2009, 23, 925–933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serakinci, N.; Tulay, P.; Kalkan, R. Role of Mesenchymal Stem Cells in Cancer Development and Their Use in Cancer Therapy. Single Mol. Single Cell Seq. 2017, 45–62. [Google Scholar] [CrossRef]
- Huang, W.-H.; Chang, M.-C.; Tsai, K.-S.; Hung, M.-C.; Chen, H.-L.; Hung, S.-C. Mesenchymal stem cells promote growth and angiogenesis of tumors in mice. Oncogene 2012, 32, 4343–4354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathew, E.; Brannon, A.L.; Del Vecchio, A.; Garcia, P.E.; Penny, M.K.; Kane, K.T.; Vinta, A.; Buckanovich, R.J.; Di Magliano, M.P. Mesenchymal Stem Cells Promote Pancreatic Tumor Growth by Inducing Alternative Polarization of Macrophages. Neoplasia 2016, 18, 142–151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheng, H.; Wang, Y.; Jin, Y.; Zhang, Q.; Zhang, Y.; Wang, L.; Shen, B.; Yin, S.; Liu, W.; Cui, L.; et al. A critical role of IFNγ in priming MSC-mediated suppression of T cell proliferation through up-regulation of B7-H1. Cell Res. 2008, 18, 846–857. [Google Scholar] [CrossRef] [PubMed]
- Eliopoulos, N.; Stagg, J.; Lejeune, L.; Pommey, S.; Galipeau, J. Allogeneic marrow stromal cells are immune rejected by MHC class I– and class II–mismatched recipient mice. Blood 2005, 106, 4057–4065. [Google Scholar] [CrossRef]
- Liu, Z.; Tang, Y.; Lü, S.; Zhou, J.; Du, Z.; Duan, C.; Li, Z.; Wang, C. The tumourigenicity of iPS cells and their differentiated derivates. J. Cell. Mol. Med. 2013, 17, 782–791. [Google Scholar] [CrossRef]
- Malecki, M. ’Above all, do no harm’: Safeguarding pluripotent stem cell therapy against iatrogenic tumorigenesis. Stem Cell Res. Ther. 2014, 5, 73. [Google Scholar] [CrossRef] [Green Version]
- Aboody, K.; Najbauer, J.; Metz, M.Z.; D’Apuzzo, M.; Gutova, M.; Annala, A.; Synold, T.W.; Couture, L.A.; Blanchard, S.; Moats, R.A.; et al. Neural Stem Cell-Mediated Enzyme/Prodrug Therapy for Glioma: Preclinical Studies. Sci. Transl. Med. 2013, 5, 184ra59. [Google Scholar] [CrossRef] [Green Version]
- Røsland, G.V.; Svendsen, A.; Torsvik, A.; Sobala, E.; Mc Cormack, E.; Immervoll, H.; Mysliwietz, J.; Tonn, J.-C.; Goldbrunner, R.; Lønning, P.E.; et al. Long-term Cultures of Bone Marrow–Derived Human Mesenchymal Stem Cells Frequently Undergo Spontaneous Malignant Transformation. Cancer Res. 2009, 69, 5331–5339. [Google Scholar] [CrossRef] [Green Version]
- Rubio, D.; García, S.; Paz, M.F.; De La Cueva, T.; Fernández, L.A.L.; Lloyd, A.C.; García-Castro, J.; Bernad, A. Molecular Characterization of Spontaneous Mesenchymal Stem Cell Transformation. PLoS ONE 2008, 3, e1398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gholamrezanezhad, A.; Mirpour, S.; Bagheri, M.; Mohamadnejad, M.; Alimoghaddam, K.; Abdolahzadeh, L.; Saghari, M.; Malekzadeh, R. In vivo tracking of 111In-oxine labeled mesenchymal stem cells following infusion in patients with advanced cirrhosis. Nucl. Med. Boil. 2011, 38, 961–967. [Google Scholar] [CrossRef] [PubMed]
- Krueger, T.E.G.; Thorek, D.L.J.; Denmeade, S.R.; Isaacs, J.T.; Brennen, W.N. Concise Review: Mesenchymal Stem Cell-Based Drug Delivery: The Good, the Bad, the Ugly, and the Promise. STEM CELLS Transl. Med. 2018, 7, 651–663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, L.; Tang, J.; Nishi, K.; Yan, C.; Dinh, P.-U.; Cores, J.; Kudo, T.; Zhang, J.; Li, T.-S.; Cheng, K. Fabrication of Synthetic Mesenchymal Stem Cells for the Treatment of Acute Myocardial Infarction in Mice. Circ. Res. 2017, 120, 1768–1775. [Google Scholar] [CrossRef] [Green Version]
- Walker, S.; Busatto, S.; Pham, A.; Tian, M.; Suh, A.; Carson, K.; Quintero, A.; Lafrence, M.; Malik, H.; Santana, M.X.; et al. Extracellular vesicle-based drug delivery systems for cancer treatment. Theranostics 2019, 9, 8001–8017. [Google Scholar] [CrossRef]
- Rani, S.; Ryan, A.; Griffin, M.D.; Ritter, T. Mesenchymal Stem Cell-derived Extracellular Vesicles: Toward Cell-free Therapeutic Applications. Mol. Ther. 2015, 23, 812–823. [Google Scholar] [CrossRef] [Green Version]
- Smyth, T.J.; Redzic, J.S.; Graner, M.W.; Anchordoquy, T.J. Examination of the specificity of tumor cell derived exosomes with tumor cells in vitro. Biochim. Biophys. Acta (BBA) - Bioenerg. 2014, 1838, 2954–2965. [Google Scholar] [CrossRef] [Green Version]
- Konoshenko, M.Y.; Lekchnov, E.A.; Vlassov, A.V.; Laktionov, P.P. Isolation of Extracellular Vesicles: General Methodologies and Latest Trends. BioMed Res. Int. 2018, 2018, 8545347-27. [Google Scholar] [CrossRef]
- Thippabhotla, S.; Zhong, C.; He, M. 3D cell culture stimulates the secretion of in vivo like extracellular vesicles. Sci. Rep. 2019, 9, 13012-14. [Google Scholar] [CrossRef] [Green Version]
- Lener, T.; Gimona, M.; Aigner, L.; Börger, V.; Buzas, E.; Camussi, G.; Chaput, N.; Chatterjee, D.; Court, F.A.; Del Portillo, H.A.; et al. Applying extracellular vesicles based therapeutics in clinical trials – an ISEV position paper. J. Extracell. Vesicles 2015, 4, 30087. [Google Scholar] [CrossRef] [PubMed]
- Kamerkar, S.; LeBleu, V.S.; Sugimoto, H.; Yang, S.; Ruivo, C.; Melo, S.A.; Lee, J.J.; Kalluri, R. Exosomes facilitate therapeutic targeting of oncogenic KRAS in pancreatic cancer. Nature 2017, 546, 498–503. [Google Scholar] [CrossRef] [PubMed]
- Gao, F.; Chiu, S.M.; Motan, D.A.L.; Zhang, Z.; Chen, L.; Ji, H.-L.; Tse, H.-F.; Fu, Q.-L.; Lian, Q. Mesenchymal stem cells and immunomodulation: Current status and future prospects. Cell Death Dis. 2016, 7, e2062. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomzikova, M.O.; James, V.; Rizvanov, A.A. Therapeutic Application of Mesenchymal Stem Cells Derived Extracellular Vesicles for Immunomodulation. Front. Immunol. 2019, 10, 2663. [Google Scholar] [CrossRef] [PubMed]
- Robbins, P.D.; Morelli, A.E. Regulation of immune responses by extracellular vesicles. Nat. Rev. Immunol. 2014, 14, 195–208. [Google Scholar] [CrossRef] [Green Version]
- Daassi, D.; Mahoney, K.M.; Freeman, G.J. The importance of exosomal PDL1 in tumour immune evasion. Nat. Rev. Immunol. 2020, 20, 209–215. [Google Scholar] [CrossRef]
- Biswas, S.; Mandal, G.; Chowdhury, S.R.; Purohit, S.; Payne, K.K.; Anadon, C.; Gupta, A.; Swanson, P.; Yu, X.; Conejo-Garcia, J.R.; et al. Exosomes Produced by Mesenchymal Stem Cells Drive Differentiation of Myeloid Cells into Immunosuppressive M2-Polarized Macrophages in Breast Cancer. J. Immunol. 2019, 203, 3447–3460. [Google Scholar] [CrossRef]
- Cattaneo, C.M.; Dijkstra, K.K.; Fanchi, L.F.; Kelderman, S.; Kaing, S.; van Rooij, N.; van den Brink, S.; Schumacher, T.N.; Voest, E.E. Tumor organoid-T-cell co-culture systems. Nat. Protoc. 2020, 15, 15–39. [Google Scholar] [CrossRef]
- Galleu, A.; Riffo-Vasquez, Y.; Trento, C.; Lomas, C.; Dolcetti, L.; Cheung, T.S.; Von Bonin, M.; Barbieri, L.; Halai, K.; Ward, S.; et al. Apoptosis in mesenchymal stromal cells induces in vivo recipient-mediated immunomodulation. Sci. Transl. Med. 2017, 9, eaam7828. [Google Scholar] [CrossRef] [Green Version]
- De Witte, S.F.; Luk, F.; Parraga, J.M.S.; Gargesha, M.; Merino, A.; Korevaar, S.S.; Shankar, A.S.; O’Flynn, L.; Elliman, S.J.; Roy, D.; et al. Immunomodulation By Therapeutic Mesenchymal Stromal Cells (MSC) Is Triggered Through Phagocytosis of MSC By Monocytic Cells. STEM CELLS 2018, 36, 602–615. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Kasagi, S.; Chia, C.; Zhang, D.; Tu, E.; Wu, R.; Zanvit, P.; Goldberg, N.; Jin, W.; Chen, W. Extracellular Vesicles from Apoptotic Cells Promote TGFbeta Production in Macrophages and Suppress Experimental Colitis. Sci. Rep 2019, 9, 5875. [Google Scholar]
- Matusek, T.; Wendler, F.; Polès, S.; Pizette, S.; D’Angelo, G.; Fürthauer, M.; Thérond, P.P. The ESCRT machinery regulates the secretion and long-range activity of Hedgehog. Nature 2014, 516, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Gangadaran, P.; Hong, C.M.; Ahn, B.-C. An Update on in Vivo Imaging of Extracellular Vesicles as Drug Delivery Vehicles. Front. Pharmacol. 2018, 9, 169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verweij, F.J.; Revenu, C.; Arras, G.; Dingli, F.; Loew, D.; Pegtel, D.M.; Follain, G.; Allio, G.; Goetz, J.G.; Zimmermann, P.; et al. Live Tracking of Inter-organ Communication by Endogenous Exosomes In Vivo. Dev. Cell 2019, 48, 573–589.e4. [Google Scholar] [CrossRef] [Green Version]
- Lancaster, M.A.; Huch, M. Disease modelling in human organoids. Dis. Model. Mech. 2019, 12, dmm039347. [Google Scholar] [CrossRef] [Green Version]


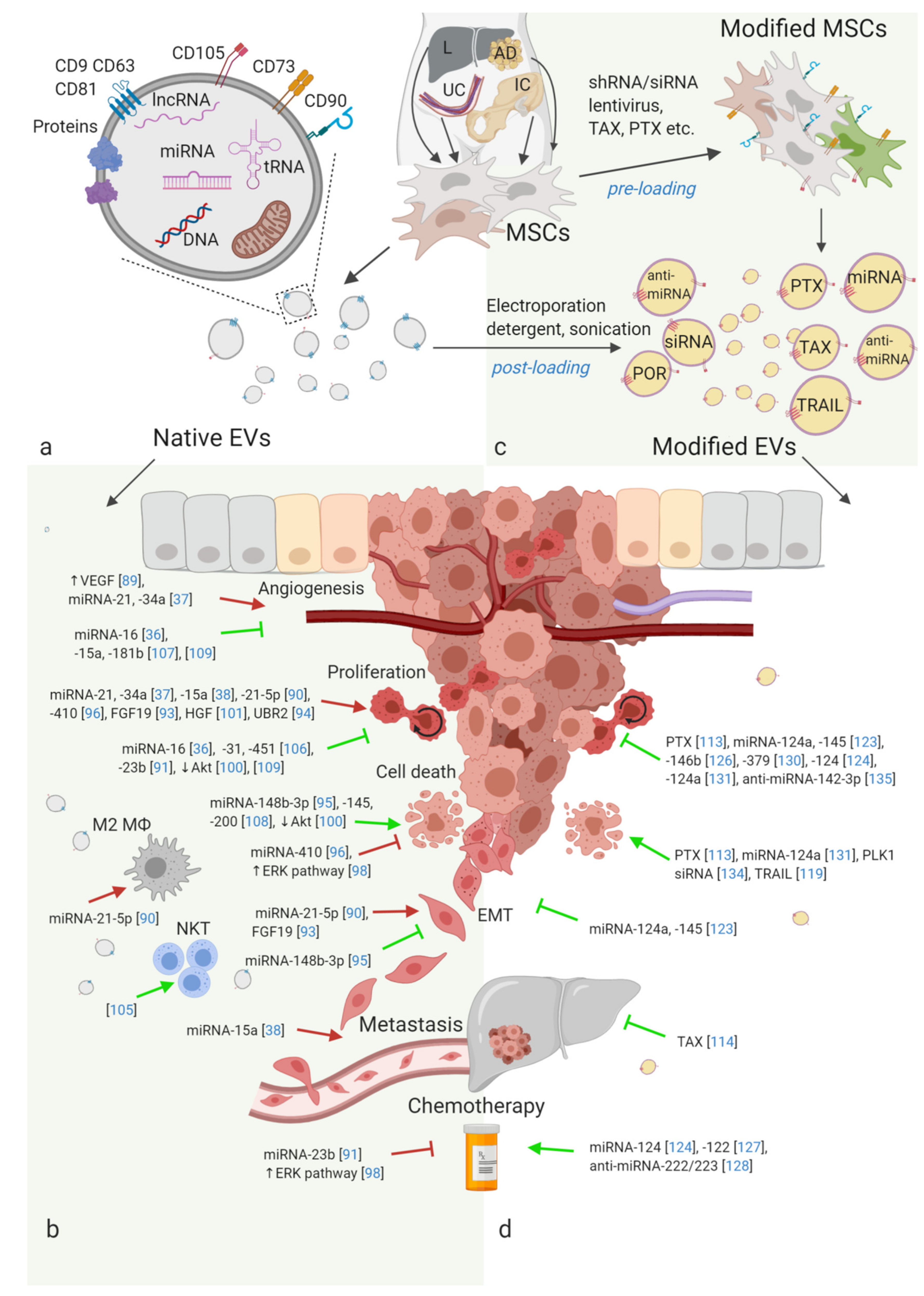{kind=link}
| EV Source | Target | Cargo/Mechanism | Method | Effect |
|---|---|---|---|---|
| BM-MSC | ||||
| Human primary | GC, colon cancer cells | ↑VEGF through ERK1/2 | In vivo | ↑growth & angiogenesis [89] |
| MM patients primary | multiple myeloma cells | miRNA15a, oncogenic proteins | In vivo | ↑homing to BM & growth [38] |
| Human primary | breast cancer cells | miRNA21, miRNA34a | In vivo | ↑growth & angiogenesis [37] |
| Human primary | osteosarcoma, GC cells | HH pathway activation | In vitro | ↑growth & migration [90] |
| Human commercial | lung cancer cells A549 and H23 | hypoxia-induced miRNA-21-5p | In vitro In vivo | ↑proliferation, survival, invasiveness, EMT ↓apoptosis, macrophage M2 polarisation [91] |
| Human commercial | metastatic breast cancer cells | suppression of MARCKS by miRNA23b | In vivo, clinical sample analysis | ↑cancer cell dormancy and ↓sensitivity to docetaxel [92] |
| Human primary | nasopharyngeal carcinoma cells | FGF19 activated FGFR4-dependent ERK cascade | In vivo | ↑EMT, ↑tumour incidence & growth [93] |
| p53-/- knockout mouse primary | p53+/+ primary mouse BM-MSC and MFC cells | UBR2 protein and mRNA | In vitro | ↑proliferation, migration, expression of stemness related genes [94] |
| Human primary | GC cells | miRNA-221 pre-loaded | In vitro | ↑proliferation, migration, invasion, and adhesion [95] |
| AD-MSC | ||||
| Human primary | breast cancer cells | Wnt pathway activation | In vitro | ↑ migration [96] |
| UC-MSC | ||||
| Human primary | GC cells | ↑ CaMKs -Raf/MEK/ERK pathway | In vivo | ↑ resistance to 5-fluorouracil ↓apoptosis [97] |
| Human primary | lung adenocarcinoma cells | miRNA-410 transfer, ↓PTEN expression | In vivo, in silico prediction | ↑growth, ↓apoptosis [98] |
| Human primary | breast & ovarian cancer cells | enzyme transfer | In vitro | ↑ cancer cell heterogeneity [99] |
| Wharton’s jelly primary | renal cancer cells | ↑HGF, ↑ ERK1/2 and AKT pathways | In vivo | ↑ tumourigenesis & tumour growth [100] |
| EV Source | Target | Cargo/Mechanism/ Modification | Method | Effect/Reference |
|---|---|---|---|---|
| BM-MSC | ||||
| Human commercial | Kaposi sarcoma, ovarian cancer, hepatoma cells | N/A | In vivo | ↓growth, ↑apoptosis [50] |
| Mouse commercial | breast cancer cells | miRNA-16, ↓VEGF | In vivo | ↓growth & angiogenesis [36] |
| Human primary | multiple myeloma cells | lack of miRNA-15a & oncogenic protein transfer | In vivo | ↓homing to BM & growth [38] |
| Mouse commercial | human pancreatic cells CFPAC-1 | PTX pre-loaded EVs | In vitro | ↓proliferation [101] |
| Human commercial | breast cancer cells MDA-MB-231 | PTX pre-loaded EMs | In vitroIn vivo | ↓viability ↓tumour growth [102] |
| Human primary | human commercial and primary GBM cells | Cy5-tagged anti-miRNA-9 pre-loaded EVs | In vitro | ↓chemoresistance to TMZ [103] |
| Human commercial | human-derived glioma cells and GSC | Cy3-miRNA-124a & miRNA-145 pre-loaded EVs | In vitro In vivo | ↓migration of glioma cells and the self-renewal of GSCs [104] |
| Human commercial | human osteosarcoma cells 143B | miRNA-143 pre-loaded EVs | In vitro | ↓migration [105] |
| Rat primary | rat model of primary brain tumour | miRNA-146b pre-loaded EVs | In vivo | ↓glioma xenograft growth [106] |
| Human primary | Breast cancer cells MDA-MB-231 and T47D | Anti-miRNA-222/223 pre-loaded EVs | In vivo | ↑carboplatin-based therapy efficiency [107] |
| Mouse primary | mouse breast cancer cells 4T1 and TUBO | LNA-anti-miRNA-142-3p post-loaded EVs | In vitro In vivo | ↑APC and P2X7R expression [108] |
| Primary human | T47D and HCC-1954 (HCC) breast cancer cells | miRNA-379 pre-loaded EVs | In vitroIn vivo | ↓COX-2 ↓tumour formation and growth rate [109] |
| Human commercial | 5 GSC primary cells | miRNA-124a pre-loaded EVs | In vitro In vivo | ↓ FOXA2, viability, clonogenicity, ↑survival [110] |
| AD-MSC | ||||
| Human commercial | metastatic prostate cancer cells | miRNA-145 | In vitro | ↓growth, ↑apoptosis [111] |
| Human primary | ovarian cancer cells | miRNA mediated ↓BCL2 | In vitro | ↓growth & migration, ↑apoptosis [112] |
| Rat primary | Hepatocellular carcinoma animal model | NKT-cell anti-tumour response | In vivo | Improved tumour grading, ↑NKT-cells [113] |
| Human commercial | human-derived glioma cells and GSC | Cy3-miRNA-124a & miRNA-145 pre-loaded EVs | In vitro In vivo | ↓migration of glioma cells and the self-renewal of GSCs [104] |
| Human primary | human liver cancer cell line HepG2 | miRNA-122 pre-loaded EVs | In vitro In vivo | ↑anti-tumour efficacy of sorafenib [114] |
| UC-MSC | ||||
| Human primary | Breast cancer lines | miRNA-148b-3p by regulating TRIM59 expression | In vivo | ↓EMT, tumour growth, ↑apoptosis [115] |
| Wharton’s jelly primary | bladder carcinoma | inhibition of Akt pathway, cleaved Caspase3 induction | In vivo | ↓growth, ↑apoptosis [116] |
| Human primary | cancer cell lines: A549 SK-OV-3 MDA-hyb1 | Taxol pre-loaded EVs | In vitro In vivo | ↑cytotoxicity ↓subcutaneous primary tumours & metastasis [117] |
| Human commercial | human-derived glioma cells and GSC | Cy3-miRNA-124a & miRNA-145 pre-loaded EVs | In vitro In vivo | ↓migration of glioma cells and the self-renewal of GSCs [104] |
| Wharton’s jelly primary | GBM cells U87 | miRNA-124 pre-loaded EVs | In vitro | ↓CDK6 ↑Chemosensitivity to temozolomide [118] |
| Other ASCs | ||||
| Human liver MSCs primary/ commercial | Hepatoma [119], tumour-derived endothelial cells [120], renal CSC [121] | miRNA-31, 451 [119] miRNA-15a, 181b, 320c, 874 [120] miRNA-145, 200 [121] | In vivo | ↓tumour growth [119] ↓angiogenesis [120,121], ↑apoptosis [121] delayed metastasis [121] |
| Human placenta-derived MSC commercial | human-derived glioma cells and GSC | Cy3-miRNA-124a & miRNA-145 pre-loaded EVs | In vitro In vivo | ↓migration of glioma cells and the self-renewal of GSCs [104] |
| MSC with unspecified origin | bladder cancer cells UMUC3 & SW780 | PLK-1 siRNA post-loaded EVs | In vitro | ↓PLK-1 expression ↑apoptosis and necrosis [122] |
| Human MSC with an unspecified origin | 11 different cancer cells | TRAIL pre-loaded EVs | In vitro | ↑apoptosis in 11 cancer cell lines including TRAIL-resistant cells [123] |
| Human MenSC primary | chemically-induced OSCC | N/A | In vivo | ↓ tumour growth, ↓angiogenesis [124] |
| Advantages | Disadvantages |
|---|---|
| 1. Less mechanical entrapment in tissues 2. Paracrine function 3. Tumour homing similar to MSCs 4. Good safety profile 5. Modification options 6. Easier and less costly to handle 7. Yield can be easier increased compared to MSCs | 1. Lack of standardised production, modification, and characterisation 2. Modifications can cause immunogenicity or toxicity risk 3. Potential of tumour promoting effects similar to MSCs 4. Lack of information on tumour selectivity depending on the EV source |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Parfejevs, V.; Sagini, K.; Buss, A.; Sobolevska, K.; Llorente, A.; Riekstina, U.; Abols, A. Adult Stem Cell-Derived Extracellular Vesicles in Cancer Treatment: Opportunities and Challenges. Cells 2020, 9, 1171. https://doi.org/10.3390/cells9051171
Parfejevs V, Sagini K, Buss A, Sobolevska K, Llorente A, Riekstina U, Abols A. Adult Stem Cell-Derived Extracellular Vesicles in Cancer Treatment: Opportunities and Challenges. Cells. 2020; 9(5):1171. https://doi.org/10.3390/cells9051171
Chicago/Turabian StyleParfejevs, Vadims, Krizia Sagini, Arturs Buss, Kristine Sobolevska, Alicia Llorente, Una Riekstina, and Arturs Abols. 2020. "Adult Stem Cell-Derived Extracellular Vesicles in Cancer Treatment: Opportunities and Challenges" Cells 9, no. 5: 1171. https://doi.org/10.3390/cells9051171
APA StyleParfejevs, V., Sagini, K., Buss, A., Sobolevska, K., Llorente, A., Riekstina, U., & Abols, A. (2020). Adult Stem Cell-Derived Extracellular Vesicles in Cancer Treatment: Opportunities and Challenges. Cells, 9(5), 1171. https://doi.org/10.3390/cells9051171