Mitochondrial Dysfunction, Oxidative Stress, and Neuroinflammation: Intertwined Roads to Neurodegeneration

 ,
,  ,
,  ,
, 

{kind=link}
{kind=link}
Abstract
:1. Introduction
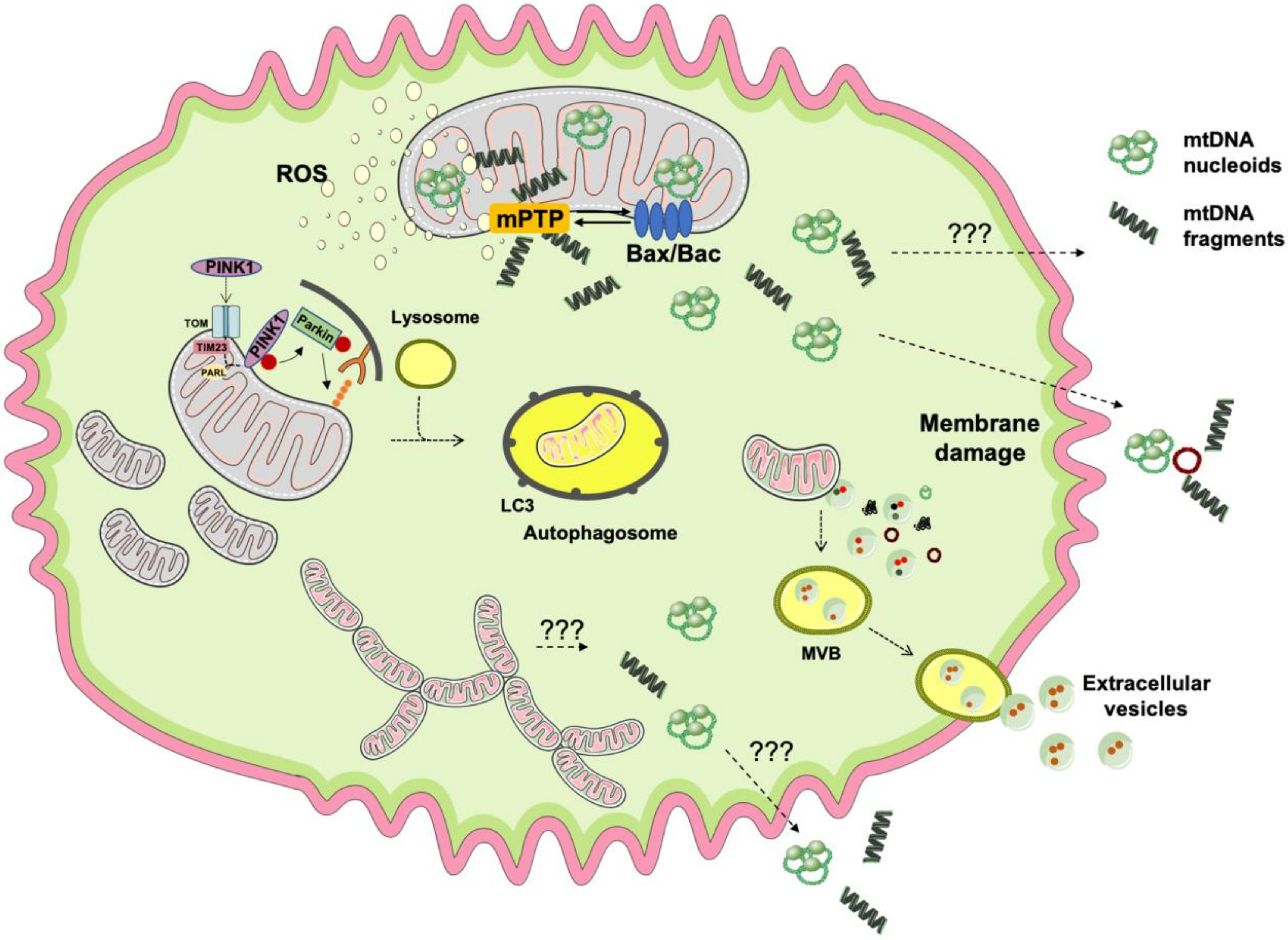
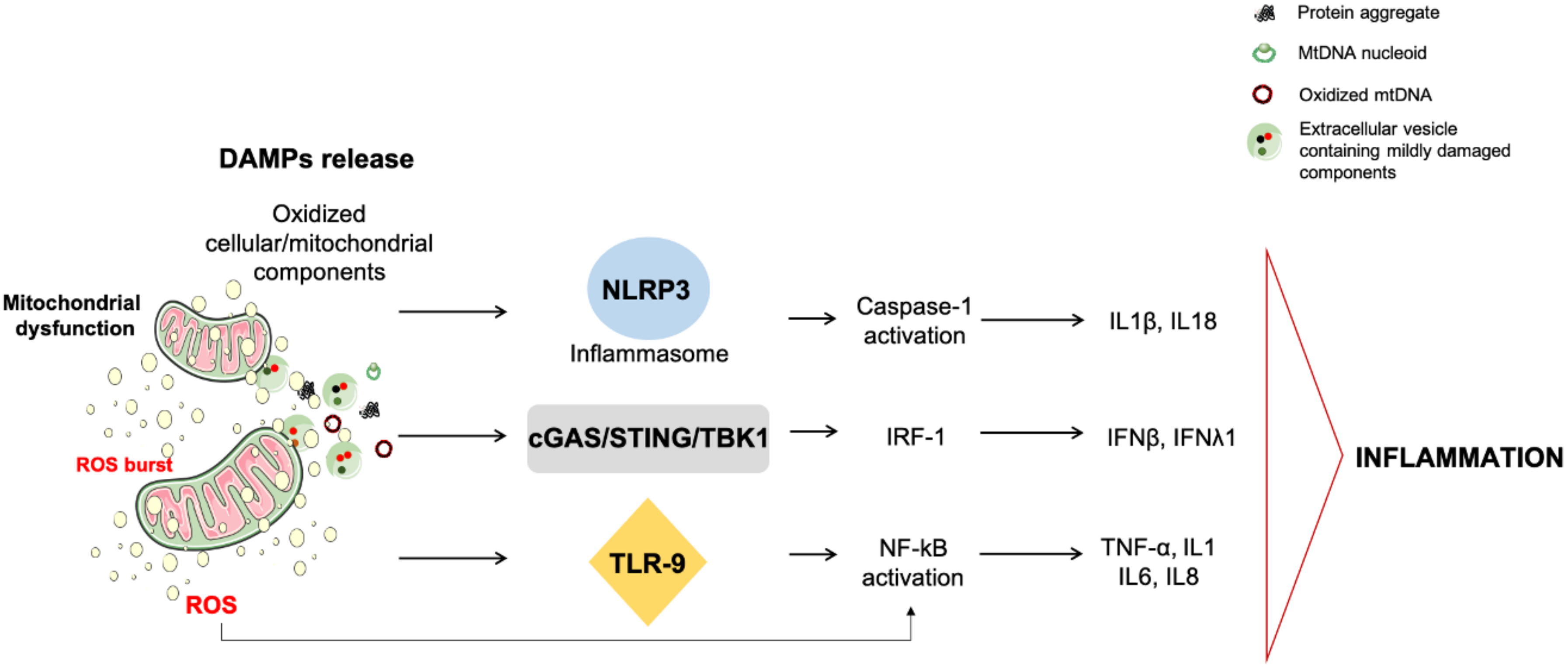
2. Mitochondrial Quality Control, mtDNA Release, and Inflammation
2.1. Failing Mitochondrial Quality Control and mtDNA Release
2.2. mtDNA Release and Inflammation
3. Alzheimer’s Disease
4. Parkinson’s Disease
5. Down Syndrome
6. Potential Therapeutics to Counteract Neuroinflammation
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Giasson, B.I.; Duda, J.E.; Murray, I.V.J.; Chen, Q.; Souza, J.M.; Hurtig, H.I.; Ischiropoulos, H.; Trojanowski, J.Q.; Lee, V.M.Y. Oxidative damage linked to neurodegeneration by selective α-synuclein nitration in synucleinopathy lesions. Science 2000, 290, 985–989. [Google Scholar] [CrossRef]
- Fischer, R.; Maier, O. Interrelation of oxidative stress and inflammation in neurodegenerative disease: Role of TNF. Oxid. Med. Cell. Longev. 2015, 2015, 610813. [Google Scholar] [CrossRef] [Green Version]
- Chausse, B.; Lewen, A.; Poschet, G.; Kann, O. Selective inhibition of mitochondrial respiratory complexes controls the transition of microglia into a neurotoxic phenotype in situ. Brain. Behav. Immun. 2020. online ahead of print. [Google Scholar] [CrossRef]
- Calvani, R.; Picca, A.; Marini, F.; Biancolillo, A.; Gervasoni, J.; Persichilli, S.; Primiano, A.; Coelho-Junior, H.J.; Bossola, M.; Urbani, A.; et al. A distinct pattern of circulating amino acids characterizes older persons with physical frailty and sarcopenia: Results from the BIOSPHERE Study. Nutrients 2018, 10, 1691. [Google Scholar] [CrossRef] [Green Version]
- DiSabato, D.J.; Quan, N.; Godbout, J.P. Neuroinflammation: The devil is in the details. J. Neurochem. 2016, 139, 136–153. [Google Scholar] [CrossRef] [Green Version]
- Yin, J.; Valin, K.L.; Dixon, M.L.; Leavenworth, J.W. The role of microglia and macrophages in CNS homeostasis, autoimmunity, and cancer. J. Immunol. Res. 2017, 2017, 5150678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Femminella, G.D.; Dani, M.; Wood, M.; Fan, Z.; Calsolaro, V.; Atkinson, R.; Edginton, T.; Hinz, R.; Brooks, D.J.; Edison, P. Microglial activation in early Alzheimer trajectory is associated with higher gray matter volume. Neurology 2019, 92, E1331–E1343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- George, S.; Rey, N.L.; Tyson, T.; Esquibel, C.; Meyerdirk, L.; Schulz, E.; Pierce, S.; Burmeister, A.R.; Madaj, Z.; Steiner, J.A.; et al. Microglia affect α-synuclein cell-to-cell transfer in a mouse model of Parkinson’s disease. Mol. Neurodegener. 2019, 14, 34. [Google Scholar] [CrossRef] [PubMed]
- Von Bernhardi, R.; Eugenín-von Bernhardi, L.; Eugenín, J. Microglial cell dysregulation in brain aging and neurodegeneration. Front. Aging Neurosci. 2015, 7, 124. [Google Scholar] [CrossRef] [Green Version]
- Koellhoffer, E.; McCullough, L.; Ritzel, R. Old maids: Aging and its impact on microglia function. Int. J. Mol. Sci. 2017, 18, 769. [Google Scholar] [CrossRef]
- Gulesserian, T.; Seidl, R.; Hardmeier, R.; Cairns, N.; Lubec, G. Superoxide dismutase SOD1, encoded on chromosome 21, but not SOD2 is overexpressed in brains of patients with Down syndrome. J. Investig. Med. 2001, 49, 41–46. [Google Scholar] [CrossRef] [PubMed]
- Perluigi, M.; Butterfield, D.A. Oxidative stress and Down syndrome: A route toward Alzheimer-like dementia. Curr. Gerontol. Geriatr. Res. 2012, 2012, 724904. [Google Scholar] [CrossRef] [Green Version]
- Butterfield, D.A.; Perluigi, M. Down syndrome: From development to adult life to Alzheimer disease. Free Radic. Biol. Med. 2018, 114, 1–2. [Google Scholar] [CrossRef] [PubMed]
- Barja, G. Updating the mitochondrial free radical theory of aging: An integrated view, key aspects, and confounding concepts. Antioxid. Redox Signal. 2013, 19, 1420–1445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Y.; Chen, M.; Jiang, J. Mitochondrial dysfunction in neurodegenerative diseases and drug targets via apoptotic signaling. Mitochondrion 2019, 49, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Grazioli, S.; Pugin, J. Mitochondrial damage-associated molecular patterns: From inflammatory signaling to human diseases. Front. Immunol. 2018, 9, 832. [Google Scholar] [CrossRef]
- Picca, A.; Lezza, A.M.S.; Leeuwenburgh, C.; Pesce, V.; Calvani, R.; Landi, F.; Bernabei, R.; Marzetti, E. Fueling inflamm-aging through mitochondrial dysfunction: Mechanisms and molecular targets. Int. J. Mol. Sci. 2017, 18, 933. [Google Scholar] [CrossRef]
- Pérez-Treviño, P.; Velásquez, M.; García, N. Mechanisms of mitochondrial DNA escape and its relationship with different metabolic diseases. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165761. [Google Scholar] [CrossRef]
- Picca, A.; Guerra, F.; Calvani, R.; Coelho-Junior, H.J.; Bossola, M.; Landi, F.; Bernabei, R.; Bucci, C.; Marzetti, E. Generation and release of mitochondrial-derived vesicles in health, aging and disease. J. Clin. Med. 2020, 9, 1440. [Google Scholar] [CrossRef]
- Picca, A.; Guerra, F.; Calvani, R.; Bucci, C.; Lo Monaco, M.R.; Bentivoglio, A.R.; Coelho-Júnior, H.J.; Landi, F.; Bernabei, R.; Marzetti, E. Mitochondrial dysfunction and aging: Insights from the analysis of extracellular vesicles. Int. J. Mol. Sci. 2019, 20, 805. [Google Scholar] [CrossRef] [Green Version]
- Al Amir Dache, Z.; Otandault, A.; Tanos, R.; Pastor, B.; Meddeb, R.; Sanchez, C.; Arena, G.; Lasorsa, L.; Bennett, A.; Grange, T.; et al. Blood contains circulating cell-free respiratory competent mitochondria. FASEB J. 2020, 34, 3616–3630. [Google Scholar] [CrossRef] [Green Version]
- Picca, A.; Calvani, R.; Coelho-Junior, H.J.; Landi, F.; Bernabei, R.; Marzetti, E. Inter-organelle membrane contact sites and mitochondrial quality control during aging: A geroscience view. Cells 2020, 9, 598. [Google Scholar] [CrossRef] [Green Version]
- Toyoshima, K.; Nakamura, M.; Adachi, Y.; Imaizumi, A.; Hakamada, T.; Abe, Y.; Kaneko, E.; Takahashi, S.; Shimokado, K. Increased plasma proline concentrations are associated with sarcopenia in the elderly. PLoS One 2017, 12, e0185206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hou, Y.; Dan, X.; Babbar, M.; Wei, Y.; Hasselbalch, S.G.; Croteau, D.L.; Bohr, V.A. Ageing as a risk factor for neurodegenerative disease. Nat. Rev. Neurol. 2019, 15, 565–581. [Google Scholar] [CrossRef]
- Picard, M.; McManus, M.J.; Csordás, G.; Várnai, P.; Dorn, G.W.; Williams, D.; Hajnóczky, G.; Wallace, D.C. Trans-mitochondrial coordination of cristae at regulated membrane junctions. Nat. Commun. 2015, 6, 6259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Twig, G.; Elorza, A.; Molina, A.J.A.; Mohamed, H.; Wikstrom, J.D.; Walzer, G.; Stiles, L.; Haigh, S.E.; Katz, S.; Las, G.; et al. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008, 27, 433–446. [Google Scholar] [CrossRef] [Green Version]
- Vincent, A.E.; Turnbull, D.M.; Eisner, V.; Hajnóczky, G.; Picard, M. Mitochondrial nanotunnels. Trends Cell Biol. 2017, 27, 787–799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rasmussen, M.L.; Robertson, G.L.; Gama, V. Break on through: Golgi-derived vesicles aid in mitochondrial fission. Cell Metab. 2020, 31, 1047–1049. [Google Scholar] [CrossRef]
- Meldolesi, J. Exosomes and ectosomes in intercellular communication. Curr. Biol. 2018, 28, R435–R444. [Google Scholar] [CrossRef] [Green Version]
- Picca, A.; Lezza, A.M.S.; Leeuwenburgh, C.; Pesce, V.; Calvani, R.; Bossola, M.; Manes-Gravina, E.; Landi, F.; Bernabei, R.; Marzetti, E. Circulating mitochondrial DNA at the crossroads of mitochondrial dysfunction and inflammation during aging and muscle wasting disorders. Rejuvenation Res. 2018, 21, 350–359. [Google Scholar] [CrossRef] [PubMed]
- Blanch, M.; Mosquera, J.L.; Ansoleaga, B.; Ferrer, I.; Barrachina, M. Altered mitochondrial DNA methylation pattern in Alzheimer disease-related pathology and in Parkinson disease. Am. J. Pathol. 2016, 186, 385–397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merkwirth, C.; Jovaisaite, V.; Durieux, J.; Matilainen, O.; Jordan, S.D.; Quiros, P.M.; Steffen, K.K.; Williams, E.G.; Mouchiroud, L.; Tronnes, S.U.; et al. Two conserved histone demethylases regulate mitochondrial stress-induced longevity. Cell 2016, 165, 1209–1223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, Y.; Garcia, G.; Bian, Q.; Steffen, K.K.; Joe, L.; Wolff, S.; Meyer, B.J.; Dillin, A. Mitochondrial stress induces chromatin reorganization to promote longevity and UPRmt. Cell 2016, 165, 1197–1208. [Google Scholar] [CrossRef] [PubMed]
- Caielli, S.; Athale, S.; Domic, B.; Murat, E.; Chandra, M.; Banchereau, R.; Baisch, J.; Phelps, K.; Clayton, S.; Gong, M.; et al. Oxidized mitochondrial nucleoids released by neutrophils drive type I interferon production in human lupus. J. Exp. Med. 2016, 213, 697–713. [Google Scholar] [CrossRef] [PubMed]
- Bae, J.H.; Jo, S.I.; Kim, S.J.; Lee, J.M.; Jeong, J.H.; Kang, J.S.; Cho, N.-J.; Kim, S.S.; Lee, E.Y.; Moon, J.-S. Circulating cell-free mtDNA contributes to AIM2 inflammasome-mediated chronic inflammation in patients with type 2 diabetes. Cells 2019, 8, 328. [Google Scholar] [CrossRef] [Green Version]
- Silzer, T.; Barber, R.; Sun, J.; Pathak, G.; Johnson, L.; O’Bryant, S.; Phillips, N. Circulating mitochondrial DNA: New indices of type 2 diabetes-related cognitive impairment in Mexican Americans. PLoS One 2019, 14, e0213527. [Google Scholar] [CrossRef] [PubMed]
- Riley, J.S.; Quarato, G.; Cloix, C.; Lopez, J.; O’Prey, J.; Pearson, M.; Chapman, J.; Sesaki, H.; Carlin, L.M.; Passos, J.F.; et al. Mitochondrial inner membrane permeabilisation enables mtDNA release during apoptosis. EMBO J. 2018, 37, e99238. [Google Scholar] [CrossRef]
- McArthur, K.; Whitehead, L.W.; Heddleston, J.M.; Li, L.; Padman, B.S.; Oorschot, V.; Geoghegan, N.D.; Chappaz, S.; Davidson, S.; Chin, H.S.; et al. BAK/BAX macropores facilitate mitochondrial herniation and mtDNA efflux during apoptosis. Science 2018, 359, eaao6047. [Google Scholar] [CrossRef] [Green Version]
- Oka, T.; Hikoso, S.; Yamaguchi, O.; Taneike, M.; Takeda, T.; Tamai, T.; Oyabu, J.; Murakawa, T.; Nakayama, H.; Nishida, K.; et al. Mitochondrial DNA that escapes from autophagy causes inflammation and heart failure. Nature 2012, 485, 251–255. [Google Scholar] [CrossRef]
- Afrifa, J.; Zhao, T.; Yu, J. Circulating mitochondria DNA, a non-invasive cancer diagnostic biomarker candidate. Mitochondrion 2019, 47, 238–243. [Google Scholar] [CrossRef]
- Sansone, P.; Savini, C.; Kurelac, I.; Chang, Q.; Amato, L.B.; Strillacci, A.; Stepanova, A.; Iommarini, L.; Mastroleo, C.; Daly, L.; et al. Packaging and transfer of mitochondrial DNA via exosomes regulate escape from dormancy in hormonal therapy-resistant breast cancer. Proc. Natl. Acad. Sci. USA 2017, 114, E9066–E9075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- García, N.; Chávez, E. Mitochondrial DNA fragments released through the permeability transition pore correspond to specific gene size. Life Sci. 2007, 81, 1160–1166. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Gupta, R.; Blanco, L.P.; Yang, S.; Shteinfer-Kuzmine, A.; Wang, K.; Zhu, J.; Yoon, H.E.; Wang, X.; Kerkhofs, M.; et al. VDAC oligomers form mitochondrial pores to release mtDNA fragments and promote lupus-like disease. Science 2019, 366, 1531–1536. [Google Scholar] [CrossRef] [PubMed]
- López-Armada, M.J.; Riveiro-Naveira, R.R.; Vaamonde-García, C.; Valcárcel-Ares, M.N. Mitochondrial dysfunction and the inflammatory response. Mitochondrion 2013, 13, 106–118. [Google Scholar] [CrossRef]
- Maass, D.L.; White, J.; Sanders, B.; Horton, J.W. Role of cytosolic vs. mitochondrial Ca2+ accumulation in burn injury-related myocardial inflammation and function. Am. J. Physiol. Heart Circ. Physiol. 2005, 288, H744–H751. [Google Scholar] [CrossRef] [Green Version]
- Picca, A.; Mankowski, R.T.; Kamenov, G.; Anton, S.D.; Manini, T.M.; Buford, T.W.; Saini, S.K.; Calvani, R.; Landi, F.; Bernabei, R.; et al. Advanced age is associated with iron dyshomeostasis and mitochondrial DNA damage in human skeletal muscle. Cells 2019, 8, 1525. [Google Scholar] [CrossRef] [Green Version]
- Schreck, R.; Rieber, P.; Baeuerle, P.A. Reactive oxygen intermediates as apparently widely used messengers in the activation of the NF-kappa B transcription factor and HIV-1. EMBO J. 1991, 10, 2247–2258. [Google Scholar] [CrossRef]
- Ma, K.; Chen, G.; Li, W.; Kepp, O.; Zhu, Y.; Chen, Q. Mitophagy, mitochondrial homeostasis, and cell fate. Front. Cell Dev. Biol. 2020, 8, 467. [Google Scholar] [CrossRef]
- Liu, R.; Xu, F.; Bi, S.; Zhao, X.; Jia, B.; Cen, Y. Mitochondrial DNA-induced inflammatory responses and lung injury in thermal injury murine model: Protective effect of cyclosporine-A. J. Burn Care Res. 2019, 40, 355–360. [Google Scholar] [CrossRef]
- Xiao, Z.; Jia, B.; Zhao, X.; Bi, S.; Meng, W. Attenuation of lipopolysaccharide-induced acute lung injury by cyclosporine-a via suppression of mitochondrial DNA. Med. Sci. Monit. 2018, 24, 7682–7688. [Google Scholar] [CrossRef]
- Lin, J.-Y.; Jing, R.; Lin, F.; Ge, W.-Y.; Dai, H.-J.; Pan, L. High tidal volume induces mitochondria damage and releases mitochondrial DNA to aggravate the ventilator-induced lung injury. Front. Immunol. 2018, 9, 1477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sliter, D.A.; Martinez, J.; Hao, L.; Chen, X.; Sun, N.; Fischer, T.D.; Burman, J.L.; Li, Y.; Zhang, Z.; Narendra, D.P.; et al. Parkin and PINK1 mitigate STING-induced inflammation. Nature 2018, 561, 258–262. [Google Scholar] [CrossRef] [PubMed]
- Atamaniuk, J.; Kopecky, C.; Skoupy, S.; Säemann, M.D.; Weichhart, T. Apoptotic cell-free DNA promotes inflammation in haemodialysis patients. Nephrol. Dial. Transplant. 2012, 27, 902–905. [Google Scholar] [CrossRef] [Green Version]
- Bernardi, P. Why F-ATP synthase remains a strong candidate as the mitochondrial permeability transition pore. Front. Physiol. 2018, 9, 1543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giorgio, V.; Guo, L.; Bassot, C.; Petronilli, V.; Bernardi, P. Calcium and regulation of the mitochondrial permeability transition. Cell Calcium 2018, 70, 56–63. [Google Scholar] [CrossRef]
- García, N.; García, J.J.; Correa, F.; Chávez, E. The permeability transition pore as a pathway for the release of mitochondrial DNA. Life Sci. 2005, 76, 2873–2880. [Google Scholar] [CrossRef] [PubMed]
- Kim, I.; Rodriguez-Enriquez, S.; Lemasters, J.J. Selective degradation of mitochondria by mitophagy. Arch. Biochem. Biophys. 2007, 462, 245–253. [Google Scholar] [CrossRef] [Green Version]
- Gottlieb, R.A.; Mentzer, R.M., Jr.; Linton, P.-J. Impaired mitophagy at the heart of injury. Autophagy 2011, 7, 1573–1574. [Google Scholar] [CrossRef] [Green Version]
- Evans, C.J.; Aguilera, R.J. DNase II: Genes, enzymes and function. Gene 2003, 322, 1–15. [Google Scholar] [CrossRef]
- Ding, Z.; Liu, S.; Wang, X.; Khaidakov, M.; Dai, Y.; Mehta, J.L. Oxidant stress in mitochondrial DNA damage, autophagy and inflammation in atherosclerosis. Sci. Rep. 2013, 3, 1077. [Google Scholar] [CrossRef] [Green Version]
- Ding, Z.; Liu, S.; Wang, X.; Dai, Y.; Khaidakov, M.; Deng, X.; Fan, Y.; Xiang, D.; Mehta, J.L. LOX-1, mtDNA damage, and NLRP3 inflammasome activation in macrophages: Implications in atherogenesis. Cardiovasc. Res. 2014, 103, 619–628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakahira, K.; Haspel, J.A.; Rathinam, V.A.K.; Lee, S.-J.; Dolinay, T.; Lam, H.C.; Englert, J.A.; Rabinovitch, M.; Cernadas, M.; Kim, H.P.; et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol. 2011, 12, 222–230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiers, W.; Beyaert, R.; Declercq, W.; Vandenabeele, P. More than one way to die: Apoptosis, necrosis and reactive oxygen damage. Oncogene 1999, 18, 7719–7730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vianello, A.; Casolo, V.; Petrussa, E.; Peresson, C.; Patui, S.; Bertolini, A.; Passamonti, S.; Braidot, E.; Zancani, M. The mitochondrial permeability transition pore (PTP) — An example of multiple molecular exaptation? Biochim. Biophys. Acta Bioenerg. 2012, 1817, 2072–2086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ader, N.R.; Hoffmann, P.C.; Ganeva, I.; Borgeaud, A.C.; Wang, C.; Youle, R.J.; Kukulski, W. Molecular and topological reorganizations in mitochondrial architecture interplay during bax-mediated steps of apoptosis. Elife 2019, 8, e40712. [Google Scholar] [CrossRef]
- Rodríguez-Nuevo, A.; Díaz-Ramos, A.; Noguera, E.; Díaz-Sáez, F.; Duran, X.; Muñoz, J.P.; Romero, M.; Plana, N.; Sebastián, D.; Tezze, C.; et al. Mitochondrial DNA and TLR9 drive muscle inflammation upon Opa1 deficiency. EMBO J. 2018, 37, e96553. [Google Scholar] [CrossRef]
- Soubannier, V.; McLelland, G.-L.; Zunino, R.; Braschi, E.; Rippstein, P.; Fon, E.A.; McBride, H.M. A vesicular transport pathway shuttles cargo from mitochondria to lysosomes. Curr. Biol. 2012, 22, 135–141. [Google Scholar] [CrossRef] [Green Version]
- Sugiura, A.; McLelland, G.-L.; Fon, E.A.; McBride, H.M. A new pathway for mitochondrial quality control: Mitochondrial-derived vesicles. EMBO J. 2014, 33, 2142–2156. [Google Scholar] [CrossRef] [Green Version]
- Desdín-Micó, G.; Mittelbrunn, M. Role of exosomes in the protection of cellular homeostasis. Cell Adh. Migr. 2017, 11, 127–134. [Google Scholar] [CrossRef] [Green Version]
- Cardon, L.R.; Burge, C.; Clayton, D.A.; Karlin, S. Pervasive CpG suppression in animal mitochondrial genomes. Proc. Natl. Acad. Sci. USA 1994, 91, 3799–3803. [Google Scholar] [CrossRef] [Green Version]
- Pollack, Y.; Kasir, J.; Shemer, R.; Metzger, S.; Szyf, M. Methylation pattern of mouse mitochondrial DNA. Nucleic Acids Res. 1984, 12, 4811–4824. [Google Scholar] [CrossRef] [Green Version]
- Collins, L.V.; Hajizadeh, S.; Holme, E.; Jonsson, I.-M.; Tarkowski, A. Endogenously oxidized mitochondrial DNA induces in vivo and in vitro inflammatory responses. J. Leukoc. Biol. 2004, 75, 995–1000. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.; Chiu, Y.H.; Chen, Z.J. The cGAS-cGAMP-STING pathway of cytosolic DNA sensing and signaling. Mol. Cell 2014, 54, 289–296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeuchi, O.; Akira, S. Pattern recognition receptors and inflammation. Cell 2010, 140, 805–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Q.; Raoof, M.; Chen, Y.; Sumi, Y.; Sursal, T.; Junger, W.; Brohi, K.; Itagaki, K.; Hauser, C.J. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 2010, 464, 104–107. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Itagaki, K.; Hauser, C.J. Mitochondrial DNA is released by shock and activates neutrophils via P38 map kinase. Shock 2010, 34, 55–59. [Google Scholar] [CrossRef]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011, 469, 221–225. [Google Scholar] [CrossRef]
- Shimada, K.; Crother, T.R.; Karlin, J.; Dagvadorj, J.; Chiba, N.; Chen, S.; Ramanujan, V.K.; Wolf, A.J.; Vergnes, L.; Ojcius, D.M.; et al. Oxidized Mitochondrial DNA Activates the NLRP3 Inflammasome during Apoptosis. Immunity 2012, 36, 401–414. [Google Scholar] [CrossRef] [Green Version]
- White, M.J.; McArthur, K.; Metcalf, D.; Lane, R.M.; Cambier, J.C.; Herold, M.J.; Van Delft, M.F.; Bedoui, S.; Lessene, G.; Ritchie, M.E.; et al. Apoptotic caspases suppress mtDNA-induced STING-mediated type i IFN production. Cell 2014, 159, 1549–1562. [Google Scholar] [CrossRef] [Green Version]
- Rongvaux, A.; Jackson, R.; Harman, C.C.D.; Li, T.; West, A.P.; De Zoete, M.R.; Wu, Y.; Yordy, B.; Lakhani, S.A.; Kuan, C.Y.; et al. Apoptotic caspases prevent the induction of type I interferons by mitochondrial DNA. Cell 2014, 159, 1563–1577. [Google Scholar] [CrossRef] [Green Version]
- West, A.P.; Khoury-Hanold, W.; Staron, M.; Tal, M.C.; Pineda, C.M.; Lang, S.M.; Bestwick, M.; Duguay, B.A.; Raimundo, N.; MacDuff, D.A.; et al. Mitochondrial DNA stress primes the antiviral innate immune response. Nature 2015, 520, 553–557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinon, F.; Burns, K.; Tschopp, J. The Inflammasome: A molecular platform triggering activation of inflammatory caspases and processing of proIL-β. Mol. Cell 2002, 10, 417–426. [Google Scholar] [CrossRef]
- Mangan, M.S.J.; Olhava, E.J.; Roush, W.R.; Seidel, H.M.; Glick, G.D.; Latz, E. Targeting the NLRP3 inflammasome in inflammatory diseases. Nat. Rev. Drug Discov. 2018, 17, 688. [Google Scholar] [CrossRef] [Green Version]
- Strowig, T.; Henao-Mejia, J.; Elinav, E.; Flavell, R. Inflammasomes in health and disease. Nature 2012, 481, 278–286. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Z.; Liang, S.; Sanchez-Lopez, E.; He, F.; Shalapour, S.; Lin, X.; Wong, J.; Ding, S.; Seki, E.; Schnabl, B.; et al. New mitochondrial DNA synthesis enables NLRP3 inflammasome activation. Nature 2018, 560, 198–203. [Google Scholar] [CrossRef]
- Masters, C.L.; Bateman, R.; Blennow, K.; Rowe, C.C.; Sperling, R.A.; Cummings, J.L. Alzheimer’s disease. Nat. Rev. Dis. Prim. 2015, 1, 15056. [Google Scholar] [CrossRef] [PubMed]
- De Strooper, B.; Iwatsubo, T.; Wolfe, M.S. Presenilins and γ-secretase: Structure, function, and role in Alzheimer disease. Cold Spring Harb. Perspect. Med. 2012, 2, a006304. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.Y.; Tan, M.S.; Yu, J.T.; Tan, L. Role of pro-inflammatory cytokines released from microglia in Alzheimer’s disease. Ann. Transl. Med. 2015, 3, 136. [Google Scholar] [CrossRef]
- Kim, S.-M.; Song, J.; Kim, S.; Han, C.; Park, M.H.; Koh, Y.; Jo, S.A.; Kim, Y.-Y. Identification of peripheral inflammatory markers between normal control and Alzheimer’s disease. BMC Neurol. 2011, 11, 51. [Google Scholar] [CrossRef] [Green Version]
- Neumann, H.; Kotter, M.R.; Franklin, R.J.M. Debris clearance by microglia: An essential link between degeneration and regeneration. Brain 2009, 132, 288–295. [Google Scholar] [CrossRef]
- D’Andrea, M.R.; Nagele, R.G.; Wang, H.Y.; Peterson, P.A.; Lee, D.H.S. Evidence that neurones accumulating amyloid can undergo lysis to form amyloid plaques in Alzheimer’s disease. Histopathology 2001, 38, 120–134. [Google Scholar] [CrossRef] [PubMed]
- Willén, K.; Edgar, J.R.; Hasegawa, T.; Tanaka, N.; Futter, C.E.; Gouras, G.K. Aβ accumulation causes MVB enlargement and is modelled by dominant negative VPS4A. Mol. Neurodegener. 2017, 12, 61. [Google Scholar] [CrossRef]
- Tang, B.L. Neuronal protein trafficking associated with Alzheimer disease: From APP and BACE1 to glutamate receptors. Cell Adhes. Migr. 2009, 3, 118–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Q.Y.; Tan, M.S.; Yu, J.T.; Tan, L. The role of retromer in Alzheimer’s disease. Mol. Neurobiol. 2016, 53, 4201–4209. [Google Scholar] [CrossRef]
- Coskun, P.E.; Beal, M.F.; Wallace, D.C. Alzheimer’s brains harbor somatic mtDNA control-region mutations that suppress mitochondrial transcription and replication. Proc. Natl. Acad. Sci. USA 2004, 101, 10726–10731. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez-Santiago, B.; Casademont, J.; Nunes, V. Is mitochondrial DNA depletion involved in Alzheimer’s disease? Eur. J. Hum. Genet. 2001, 9, 279–285. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.; Keogh, M.J.; Wilson, I.; Coxhead, J.; Ryan, S.; Rollinson, S.; Griffin, H.; Kurzawa-Akanbi, M.; Santibanez-Koref, M.; Talbot, K.; et al. Mitochondrial DNA point mutations and relative copy number in 1363 disease and control human brains. Acta Neuropathol. Commun. 2017, 5, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, M.A.; Harris, P.L.R.; Sayre, L.M.; Perry, G. Iron accumulation in Alzheimer disease is a source of redox-generated free radicals. Proc. Natl. Acad. Sci. USA 1997, 94, 9866–9868. [Google Scholar] [CrossRef] [Green Version]
- De La Monte, S.M.; Wands, J.R. Molecular indices of oxidative stress and mitochondrial dysfunction occur early and often progress with severity of Alzheimer’s disease. J. Alzheimers Dis. 2006, 9, 167–181. [Google Scholar] [CrossRef]
- Nunomura, A.; Perry, G.; Aliev, G.; Hirai, K.; Takeda, A.; Balraj, E.K.; Jones, P.K.; Ghanbari, H.; Wataya, T.; Shimohama, S.; et al. Oxidative damage is the earliest event in Alzheimer disease. J. Neuropathol. Exp. Neurol. 2001, 60, 759–767. [Google Scholar] [CrossRef] [Green Version]
- Devi, L.; Prabhu, B.M.; Galati, D.F.; Avadhani, N.G.; Anandatheerthavarada, H.K. Accumulation of amyloid precursor protein in the mitochondrial import channels of human Alzheimer’s disease brain is associated with mitochondrial dysfunction. J. Neurosci. 2006, 26, 9057–9068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eckert, A.; Schulz, K.L.; Rhein, V.; Götz, J. Convergence of amyloid-β and tau pathologies on mitochondria in vivo. Mol. Neurobiology 2010, 41, 107–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montagne, A.; Nation, D.A.; Sagare, A.P.; Barisano, G.; Sweeney, M.D.; Chakhoyan, A.; Pachicano, M.; Joe, E.; Nelson, A.R.; D’Orazio, L.M.; et al. APOE4 leads to blood–brain barrier dysfunction predicting cognitive decline. Nature 2020, 581, 71–76. [Google Scholar] [CrossRef] [PubMed]
- Mahley, R.W.; Weisgraber, K.H.; Huang, Y. Apolipoprotein E4: A causative factor and therapeutic target in neuropathology, including Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2006, 103, 5644–5651. [Google Scholar] [CrossRef] [Green Version]
- Kerr, J.S.; Adriaanse, B.A.; Greig, N.H.; Mattson, M.P.; Cader, M.Z.; Bohr, V.A.; Fang, E.F. Mitophagy and Alzheimer’s disease: Cellular and molecular mechanisms. Trends Neurosci. 2017, 40, 151–166. [Google Scholar] [CrossRef] [Green Version]
- Fang, E.F.; Hou, Y.; Palikaras, K.; Adriaanse, B.A.; Kerr, J.S.; Yang, B.; Lautrup, S.; Hasan-Olive, M.M.; Caponio, D.; Dan, X.; et al. Mitophagy inhibits amyloid-β and tau pathology and reverses cognitive deficits in models of Alzheimer’s disease. Nat. Neurosci. 2019, 22, 401–412. [Google Scholar] [CrossRef]
- Du, F.; Yu, Q.; Yan, S.; Hu, G.; Lue, L.F.; Walker, D.G.; Wu, L.; Yan, S.F.; Tieu, K.; Yan, S.S. PINK1 signalling rescues amyloid pathology and mitochondrial dysfunction in Alzheimer’s disease. Brain 2017, 140, 3233–3251. [Google Scholar] [CrossRef] [Green Version]
- Sorrentino, V.; Romani, M.; Mouchiroud, L.; Beck, J.S.; Zhang, H.; D’Amico, D.; Moullan, N.; Potenza, F.; Schmid, A.W.; Rietsch, S.; et al. Enhancing mitochondrial proteostasis reduces amyloid-β proteotoxicity. Nature 2017, 552, 187–193. [Google Scholar] [CrossRef]
- Melber, A.; Haynes, C.M. UPRmt regulation and output: A stress response mediated by mitochondrial-nuclear communication. Cell Res. 2018, 28, 281–295. [Google Scholar] [CrossRef]
- Wang, Z.T.; Lu, M.H.; Zhang, Y.; Ji, W.L.; Lei, L.; Wang, W.; Fang, L.P.; Wang, L.W.; Yu, F.; Wang, J.; et al. Disrupted-in-schizophrenia-1 protects synaptic plasticity in a transgenic mouse model of Alzheimer’s disease as a mitophagy receptor. Aging Cell 2019, 18, e12860. [Google Scholar] [CrossRef]
- Fang, E.F. Mitophagy and NAD+ inhibit Alzheimer disease. Autophagy 2019, 15, 1112–1114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Picca, A.; Guerra, F.; Calvani, R.; Bucci, C.; Lo Monaco, M.R.; Bentivoglio, A.R.; Landi, F.; Bernabei, R.; Marzetti, E. Mitochondrial-derived vesicles as candidate biomarkers in Parkinson’s disease: Rationale, design and methods of the EXosomes in PArkiNson Disease (EXPAND) study. Int. J. Mol. Sci. 2019, 20, 2373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Picca, A.; Guerra, F.; Calvani, R.; Marini, F.; Biancolillo, A.; Landi, G.; Beli, R.; Landi, F.; Bernabei, R.; Bentivoglio, A.; et al. Mitochondrial signatures in circulating extracellular vesicles of older adults with Parkinson’s disease: Results from the EXosomes in PArkiNson’s Disease (EXPAND) study. J. Clin. Med. 2020, 9, 504. [Google Scholar] [CrossRef] [Green Version]
- Holmes, C.; Butchart, J. Systemic inflammation and Alzheimer’s disease. Biochem. Soc. Trans. 2011, 39, 898–901. [Google Scholar] [CrossRef] [PubMed]
- Olsson, B.; Lautner, R.; Andreasson, U.; Öhrfelt, A.; Portelius, E.; Bjerke, M.; Hölttä, M.; Rosén, C.; Olsson, C.; Strobel, G.; et al. CSF and blood biomarkers for the diagnosis of Alzheimer’s disease: A systematic review and meta-analysis. Lancet Neurol. 2016, 15, 673–684. [Google Scholar] [CrossRef]
- GBD 2016. Parkinson’s Disease Collaborators. Global, regional, and national burden of Parkinson’s disease, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2018, 17, 939–953. [Google Scholar] [CrossRef] [Green Version]
- Alexander, G.E. Biology of Parkinson’s disease: Pathogenesis and pathophysiology of a multisystem neurodegenerative disorder. Dialogues Clin. Neurosci. 2004, 6, 259–280. [Google Scholar] [PubMed]
- Angelopoulou, E.; Paudel, Y.N.; Shaikh, M.F.; Piperi, C. Fractalkine (CX3CL1) signaling and neuroinflammation in Parkinson’s disease: Potential clinical and therapeutic implications. Pharmacol. Res. 2020, 158, 104930. [Google Scholar] [CrossRef] [PubMed]
- Calvani, R.; Picca, A.; Landi, G.; Marini, F.; Biancolillo, A.; Coelho-Junior, H.J.; Gervasoni, J.; Persichilli, S.; Primiano, A.; Arcidiacono, A.; et al. A novel multi-marker discovery approach identifies new serum biomarkers for Parkinson’s disease in older people: An EXosomes in PArkiNson Disease (EXPAND) ancillary study. GeroScience 2020. online ahead of print. [Google Scholar] [CrossRef]
- Devi, L.; Raghavendran, V.; Prabhu, B.M.; Avadhani, N.G.; Anandatheerthavarada, H.K. Mitochondrial import and accumulation of α-synuclein impair complex I in human dopaminergic neuronal cultures and Parkinson disease brain. J. Biol. Chem. 2008, 283, 9089–9100. [Google Scholar] [CrossRef] [Green Version]
- Dodson, M.W.; Guo, M. Pink1, Parkin, DJ-1 and mitochondrial dysfunction in Parkinson’s disease. Curr. Opin. Neurobiol. 2007, 17, 331–337. [Google Scholar] [CrossRef] [PubMed]
- Ostrerova-Golts, N.; Petrucelli, L.; Hardy, J.; Lee, J.M.; Farer, M.; Wolozin, B. The A53T α-synuclein mutation increases iron-dependent aggregation and toxicity. J. Neurosci. 2000, 20, 6048–6054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orth, M.; Tabrizi, S.J.; Tomlinson, C.; Messmer, K.; Korlipara, L.V.P.; Schapira, A.H.V.; Cooper, J.M. G209A mutant alpha synuclein expression specifically enhances dopamine induced oxidative damage. Neurochem. Int. 2004, 45, 669–676. [Google Scholar] [CrossRef] [PubMed]
- Junn, E.; Mouradian, M.M. Human α-synuclein over-expression increases intracellular reactive oxygen species levels and susceptibility to dopamine. Neurosci. Lett. 2002, 320, 146–150. [Google Scholar] [CrossRef]
- Paxinou, E.; Chen, Q.; Weisse, M.; Giasson, B.I.; Norris, E.H.; Rueter, S.M.; Trojanowski, J.Q.; Lee, V.M.Y.; Ischiropoulos, H. Induction of α-synuclein aggregation by intracellular nitrative insult. J. Neurosci. 2001, 21, 8053–8061. [Google Scholar] [CrossRef]
- Ahn, T.B.; Kim, S.Y.; Kim, J.Y.; Park, S.S.; Lee, D.S.; Min, H.J.; Kim, Y.K.; Kim, S.E.; Kim, J.M.; Kim, H.J.; et al. α-Synuclein gene duplication is present in sporadic Parkinson disease. Neurology 2008, 70, 43–49. [Google Scholar] [CrossRef]
- Park, J.S.; Davis, R.L.; Sue, C.M. Mitochondrial dysfunction in Parkinson’s disease: New mechanistic insights and therapeutic perspectives. Curr. Neurol. Neurosci. Rep. 2018, 18, 21. [Google Scholar] [CrossRef] [Green Version]
- Grünblatt, E.; Mandel, S.; Youdim, M.B.H. Neuroprotective Strategies in Parkinson’s disease using the models of 6-hydroxydopamine and MPTPa. Ann. N.Y. Acad. Sci. 2006, 899, 262–273. [Google Scholar] [CrossRef]
- Bender, A.; Krishnan, K.J.; Morris, C.M.; Taylor, G.A.; Reeve, A.K.; Perry, R.H.; Jaros, E.; Hersheson, J.S.; Betts, J.; Klopstock, T.; et al. High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and Parkinson disease. Nat. Genet. 2006, 38, 515–517. [Google Scholar] [CrossRef]
- Kraytsberg, Y.; Kudryavtseva, E.; McKee, A.C.; Geula, C.; Kowall, N.W.; Khrapko, K. Mitochondrial DNA deletions are abundant and cause functional impairment in aged human substantia nigra neurons. Nat. Genet. 2006, 38, 518–520. [Google Scholar] [CrossRef]
- Dölle, C.; Flønes, I.; Nido, G.S.; Miletic, H.; Osuagwu, N.; Kristoffersen, S.; Lilleng, P.K.; Larsen, J.P.; Tysnes, O.-B.; Haugarvoll, K.; et al. Defective mitochondrial DNA homeostasis in the substantia nigra in Parkinson disease. Nat. Commun. 2016, 7, 13548. [Google Scholar] [CrossRef]
- Dias, V.; Junn, E.; Mouradian, M.M. The role of oxidative stress in parkinson’s disease. J. Parkinsons Dis. 2013, 3, 461–491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hastings, T.G. The role of dopamine oxidation in mitochondrial dysfunction: Implications for Parkinson’s disease. J. Bioenerg. Biomembr. 2009, 41, 469–472. [Google Scholar] [CrossRef] [PubMed]
- Lotharius, J.; Brundin, P. Pathogenesis of parkinson’s disease: Dopamine, vesicles and α-synuclein. Nat. Rev. Neurosci. 2002, 3, 932–942. [Google Scholar] [CrossRef] [PubMed]
- Chinta, S.J.; Kumar, M.J.; Hsu, M.; Rajagopalan, S.; Kaur, D.; Rane, A.; Nicholls, D.G.; Choi, J.; Andersen, J.K. Inducible alterations of glutathione levels in adult dopaminergic midbrain neurons result in nigrostriatal degeneration. J. Neurosci. 2007, 27, 13997–14006. [Google Scholar] [CrossRef] [Green Version]
- Cho, B.; Kim, T.; Huh, Y.J.; Lee, J.; Lee, Y. Amelioration of mitochondrial quality control and proteostasis by natural compounds in Parkinson’s disease models. Int. J. Mol. Sci. 2019, 20, 5208. [Google Scholar] [CrossRef] [Green Version]
- White, A.J.; Wijeyekoon, R.S.; Scott, K.M.; Gunawardana, N.P.; Hayat, S.; Solim, I.H.; McMahon, H.T.; Barker, R.A.; Williams-Gray, C.H. The peripheral inflammatory response to alpha-synuclein and endotoxin in Parkinson’s disease. Front. Neurol. 2018, 9, 946. [Google Scholar] [CrossRef]
- Matheoud, D.; Sugiura, A.; Bellemare-Pelletier, A.; Laplante, A.; Rondeau, C.; Chemali, M.; Fazel, A.; Bergeron, J.J.; Trudeau, L.E.; Burelle, Y.; et al. Parkinson’s disease-related proteins PINK1 and Parkin repress mitochondrial antigen presentation. Cell 2016, 166, 314–327. [Google Scholar] [CrossRef] [Green Version]
- Picca, A.; Calvani, R.; Landi, G.; Marini, F.; Biancolillo, A.; Gervasoni, J.; Persichilli, S.; Primiano, A.; Urbani, A.; Bossola, M.; et al. Circulating amino acid signature in older people with Parkinson’s disease: A metabolic complement to the EXosomes in PArkiNson Disease (EXPAND) study. Exp. Gerontol. 2019, 128, 110766. [Google Scholar] [CrossRef]
- Todkar, K.; Chikhi, L.; Germain, M. Mitochondrial interaction with the endosomal compartment in endocytosis and mitochondrial transfer. Mitochondrion 2019, 49, 284–288. [Google Scholar] [CrossRef]
- Restelli, L.M.; Oettinghaus, B.; Halliday, M.; Agca, C.; Licci, M.; Sironi, L.; Savoia, C.; Hench, J.; Tolnay, M.; Neutzner, A.; et al. Neuronal mitochondrial dysfunction activates the integrated stress response to induce fibroblast growth factor 21. Cell Rep. 2018, 24, 1407–1414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carfì, A.; Antocicco, M.; Brandi, V.; Cipriani, C.; Fiore, F.; Mascia, D.; Settanni, S.; Vetrano, D.L.; Bernabei, R.; Onder, G. Characteristics of adults with Down syndrome: Prevalence of age-related conditions. Front. Med. 2014, 1, 51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coppus, A.M.W.; Evenhuis, H.M.; Verberne, G.J.; Visser, F.E.; Oostra, B.A.; Eikelenboom, P.; Van Gool, W.A.; Janssens, A.C.J.W.; Van Duijn, C.M. Survival in elderly persons with down syndrome. J. Am. Geriatr. Soc. 2008, 56, 2311–2316. [Google Scholar] [CrossRef] [PubMed]
- Schoufour, J.D.; Mitnitski, A.; Rockwood, K.; Evenhuis, H.M.; Echteld, M.A. Predicting 3-year survival in older people with intellectual disabilities using a frailty index. J. Am. Geriatr. Soc. 2015, 63, 531–536. [Google Scholar] [CrossRef]
- Esbensen, A.J. Health conditions associated with aging and end of life of adults with Down syndrome. Int. Rev. Res. Ment. Retard. 2010, 39, 107–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carfì, A.; Romano, A.; Zaccaria, G.; Villani, E.R.; Manes Gravina, E.; Vetrano, D.L.; Bernabei, R.; Onder, G. The burden of chronic disease, multimorbidity, and polypharmacy in adults with Down syndrome. Am. J. Med. Genet. A 2020, 182, 1735–1743. [Google Scholar] [CrossRef]
- Martin, G.M. Genetic syndromes in man with potential relevance to the pathobiology of aging. Birth Defects Orig. Artic. Ser. 1978, 14, 5–39. [Google Scholar]
- Franceschi, C.; Garagnani, P.; Gensous, N.; Bacalini, M.G.; Conte, M.; Salvioli, S. Accelerated bio-cognitive aging in Down syndrome: State of the art and possible deceleration strategies. Aging Cell 2019, 18, e12903. [Google Scholar] [CrossRef]
- Horvath, S.; Garagnani, P.; Bacalini, M.G.; Pirazzini, C.; Salvioli, S.; Gentilini, D.; Di Blasio, A.M.; Giuliani, C.; Tung, S.; Vinters, H.V.; et al. Accelerated epigenetic aging in Down syndrome. Aging Cell 2015, 14, 491–495. [Google Scholar] [CrossRef]
- Marzetti, E.; Calvani, R.; Cesari, M.; Tosato, M.; Cherubini, A.; Di Bari, M.; Pahor, M.; Savera, G.; Collamati, A.; D’Angelo, E.; et al. Operationalization of the physical frailty & sarcopenia syndrome: Rationale and clinical implementation. Transl. Med. UniSa 2015, 13, 29–32. [Google Scholar]
- Lott, I.T.; Dierssen, M. Cognitive deficits and associated neurological complications in individuals with Down’s syndrome. Lancet Neurol. 2010, 9, 623–633. [Google Scholar] [CrossRef]
- Bayen, E.; Possin, K.L.; Chen, Y.; Cleret De Langavant, L.; Yaffe, K. Prevalence of aging, dementia, and multimorbidity in older adults with Down syndrome. JAMA Neurol. 2018, 75, 1399–1406. [Google Scholar] [CrossRef] [PubMed]
- Coelho-Junior, H.J.; Villani, E.R.; Calvani, R.; Carfì, A.; Picca, A.; Landi, F.; Bernabei, R.; Onder, G.; Marzetti, E. Sarcopenia-related parameters in adults with Down syndrome: A cross-sectional exploratory study. Exp. Gerontol. 2019, 119, 93–99. [Google Scholar] [CrossRef] [PubMed]
- Foley, C.; Killeen, O.G. Musculoskeletal anomalies in children with Down syndrome: An observational study. Arch. Dis. Child. 2019, 104, 482–487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valenti, D.; de Bari, L.; de Rasmo, D.; Signorile, A.; Henrion-Caude, A.; Contestabile, A.; Vacca, R.A. The polyphenols resveratrol and epigallocatechin-3-gallate restore the severe impairment of mitochondria in hippocampal progenitor cells from a Down syndrome mouse model. Biochim. Biophys. Acta 2016, 1862, 1093–1104. [Google Scholar] [CrossRef]
- Helguera, P.; Seiglie, J.; Rodriguez, J.; Hanna, M.; Helguera, G.; Busciglio, J. Adaptive downregulation of mitochondrial function in down syndrome. Cell Metab. 2013, 17, 132–140. [Google Scholar] [CrossRef] [Green Version]
- Valenti, D.; Tullo, A.; Caratozzolo, M.F.; Merafina, R.S.; Scartezzini, P.; Marra, E.; Vacca, R.A. Impairment of F1F0-ATPase, adenine nucleotide translocator and adenylate kinase causes mitochondrial energy deficit in human skin fibroblasts with chromosome 21 trisomy. Biochem. J. 2010, 431, 299–310. [Google Scholar] [CrossRef]
- Valenti, D.; Manente, G.A.; Moro, L.; Marra, E.; Vacca, R.A. Deficit of complex I activity in human skin fibroblasts with chromosome 21 trisomy and overproduction of reactive oxygen species by mitochondria: Involvement of the cAMP/PKA signalling pathway. Biochem. J. 2011, 435, 679–688. [Google Scholar] [CrossRef] [Green Version]
- Druzhyna, N.; Nair, R.G.; Ledoux, S.P.; Wilson, G.L. Defective repair of oxidative damage in mitochondrial DNA in Down’s syndrome. Mutat. Res. 1998, 409, 81–89. [Google Scholar] [CrossRef]
- Coskun, P.E.; Busciglio, J. Oxidative stress and mitochondrial dysfunction in Down’s syndrome: Relevance to aging and dementia. Curr. Gerontol. Geriatr. Res. 2012, 2012, 383170. [Google Scholar] [CrossRef]
- Weick, J.P.; Held, D.L.; Bonadurer, G.F.; Doers, M.E.; Liu, Y.; Maguire, C.; Clark, A.; Knackert, J.A.; Molinarolo, K.; Musser, M.; et al. Deficits in human trisomy 21 iPSCs and neurons. Proc. Natl. Acad. Sci. USA 2013, 110, 9962–9967. [Google Scholar] [CrossRef] [Green Version]
- Briggs, J.A.; Sun, J.; Shepherd, J.; Ovchinnikov, D.A.; Chung, T.L.; Nayler, S.P.; Kao, L.P.; Morrow, C.A.; Thakar, N.Y.; Soo, S.Y.; et al. Integration-free induced pluripotent stem cells model genetic and neural developmental features of down syndrome etiology. Stem Cells 2013, 31, 467–478. [Google Scholar] [CrossRef] [PubMed]
- Mollo, N.; Cicatiello, R.; Aurilia, M.; Scognamiglio, R.; Genesio, R.; Charalambous, M.; Paladino, S.; Conti, A.; Nitsch, L.; Izzo, A. Targeting mitochondrial network architecture in Down syndrome and aging. Int. J. Mol. Sci. 2020, 21, 3134. [Google Scholar] [CrossRef]
- Trotta, M.B.F.; Serro Azul, J.B.; Wajngarten, M.; Fonseca, S.G.; Goldberg, A.C.; Kalil, J.E. Inflammatory and immunological parameters in adults with Down syndrome. Immun. Ageing 2011, 8, 4. [Google Scholar] [CrossRef] [Green Version]
- Huggard, D.; Kelly, L.; Ryan, E.; McGrane, F.; Lagan, N.; Roche, E.; Balfe, J.; Leahy, T.R.; Franklin, O.; Doherty, D.G.; et al. Increased systemic inflammation in children with Down syndrome. Cytokine 2020, 127, 154938. [Google Scholar] [CrossRef] [PubMed]
- Udan, M.L.D.; Ajit, D.; Crouse, N.R.; Nichols, M.R. Toll-like receptors 2 and 4 mediate Aβ(1-42) activation of the innate immune response in a human monocytic cell line. J. Neurochem. 2008, 104, 524–533. [Google Scholar] [CrossRef] [PubMed]
- Coll, R.C.; Robertson, A.A.B.; Chae, J.J.; Higgins, S.C.; Muñoz-Planillo, R.; Inserra, M.C.; Vetter, I.; Dungan, L.S.; Monks, B.G.; Stutz, A.; et al. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat. Med. 2015, 21, 248–257. [Google Scholar] [CrossRef] [Green Version]
- Marchi, N.; Granata, T.; Freri, E.; Ciusani, E.; Ragona, F.; Puvenna, V.; Teng, Q.; Alexopolous, A.; Janigro, D. Efficacy of anti-inflammatory therapy in a model of acute seizures and in a population of pediatric drug resistant epileptics. PLoS One 2011, 6, e18200. [Google Scholar] [CrossRef]
- Gordon, R.; Albornoz, E.A.; Christie, D.C.; Langley, M.R.; Kumar, V.; Mantovani, S.; Robertson, A.A.B.; Butler, M.S.; Rowe, D.B.; O’Neill, L.A.; et al. Inflammasome inhibition prevents α-synuclein pathology and dopaminergic neurodegeneration in mice. Sci. Transl. Med. 2018, 10, eaah4066. [Google Scholar] [CrossRef] [Green Version]
- Savitt, D.; Jankovic, J. Targeting α-synuclein in Parkinson’s Disease: Progress towards the development of disease-modifying therapeutics. Drugs 2019, 79, 797–810. [Google Scholar] [CrossRef]
- Prots, I.; Winner, B. Th17 cells: A promising therapeutic target for Parkinson’s disease? Expert Opin. Ther. Targets 2019, 23, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Capiralla, H.; Vingtdeux, V.; Zhao, H.; Sankowski, R.; Al-Abed, Y.; Davies, P.; Marambaud, P. Resveratrol mitigates lipopolysaccharide- and Aβ-mediated microglial inflammation by inhibiting the TLR4/NF-κB/STAT signaling cascade. J. Neurochem. 2012, 120, 461–472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schroeder, E.K.; Kelsey, N.A.; Doyle, J.; Breed, E.; Bouchard, R.J.; Loucks, F.A.; Harbison, R.A.; Linseman, D.A. Green tea epigallocatechin 3-gallate accumulates in mitochondria and displays a selective antiapoptotic effect against inducers of mitochondrial oxidative stress in neurons. Antioxidants Redox Signal. 2009, 11, 469–480. [Google Scholar] [CrossRef]
- Sutherland, B.A.; Shaw, O.M.; Clarkson, A.N.; Jackson, D.M.; Sammut, I.A.; Appleton, I. Neuroprotective effects of (−)-epigallocatechin gallate after hypoxia-ischemia-induced brain damage: Novel mechanisms of action. FASEB J. 2005, 19, 1–22. [Google Scholar] [CrossRef]
- Valenti, D.; De Rasmo, D.; Signorile, A.; Rossi, L.; de Bari, L.; Scala, I.; Granese, B.; Papa, S.; Vacca, R.A. Epigallocatechin-3-gallate prevents oxidative phosphorylation deficit and promotes mitochondrial biogenesis in human cells from subjects with Down’s syndrome. Biochim. Biophys. Acta Mol. Basis Dis. 2013, 1832, 542–552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.X.; Li, Y.B.; Zhao, R.P. Epigallocatechin gallate attenuates β-amyloid generation and oxidative stress involvement of PPARγ in N2a/APP695 cells. Neurochem. Res. 2017, 42, 468–480. [Google Scholar] [CrossRef] [PubMed]
- Izzo, A.; Nitti, M.; Mollo, N.; Paladino, S.; Procaccini, C.; Faicchia, D.; Calì, G.; Genesio, R.; Bonfiglio, F.; Cicatiello, R.; et al. Metformin restores the mitochondrial network and reverses mitochondrial dysfunction in Down syndrome cells. Hum. Mol. Genet. 2017, 26, 1056–1069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reiter, R.; Tan, D.; Rosales-Corral, S.; Galano, A.; Zhou, X.; Xu, B. Mitochondria: Central organelles for melatonin′s antioxidant and anti-aging actions. Molecules 2018, 23, 509. [Google Scholar] [CrossRef] [Green Version]
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Picca, A.; Calvani, R.; Coelho-Junior, H.J.; Landi, F.; Bernabei, R.; Marzetti, E. Mitochondrial Dysfunction, Oxidative Stress, and Neuroinflammation: Intertwined Roads to Neurodegeneration. Antioxidants 2020, 9, 647. https://doi.org/10.3390/antiox9080647
Picca A, Calvani R, Coelho-Junior HJ, Landi F, Bernabei R, Marzetti E. Mitochondrial Dysfunction, Oxidative Stress, and Neuroinflammation: Intertwined Roads to Neurodegeneration. Antioxidants. 2020; 9(8):647. https://doi.org/10.3390/antiox9080647
Chicago/Turabian StylePicca, Anna, Riccardo Calvani, Hélio José Coelho-Junior, Francesco Landi, Roberto Bernabei, and Emanuele Marzetti. 2020. "Mitochondrial Dysfunction, Oxidative Stress, and Neuroinflammation: Intertwined Roads to Neurodegeneration" Antioxidants 9, no. 8: 647. https://doi.org/10.3390/antiox9080647
APA StylePicca, A., Calvani, R., Coelho-Junior, H. J., Landi, F., Bernabei, R., & Marzetti, E. (2020). Mitochondrial Dysfunction, Oxidative Stress, and Neuroinflammation: Intertwined Roads to Neurodegeneration. Antioxidants, 9(8), 647. https://doi.org/10.3390/antiox9080647