Channel-Forming Bacterial Toxins in Biosensing and Macromolecule Delivery

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction: Channel-Forming Bacterial Toxins
2. Biosensing and Polymer Translocation with Nanopores
3. GrA, αHL, and PA63 as Nanopores of Choice in Biotechnology
3.1. Gramicidin A of Bacillus Brevis
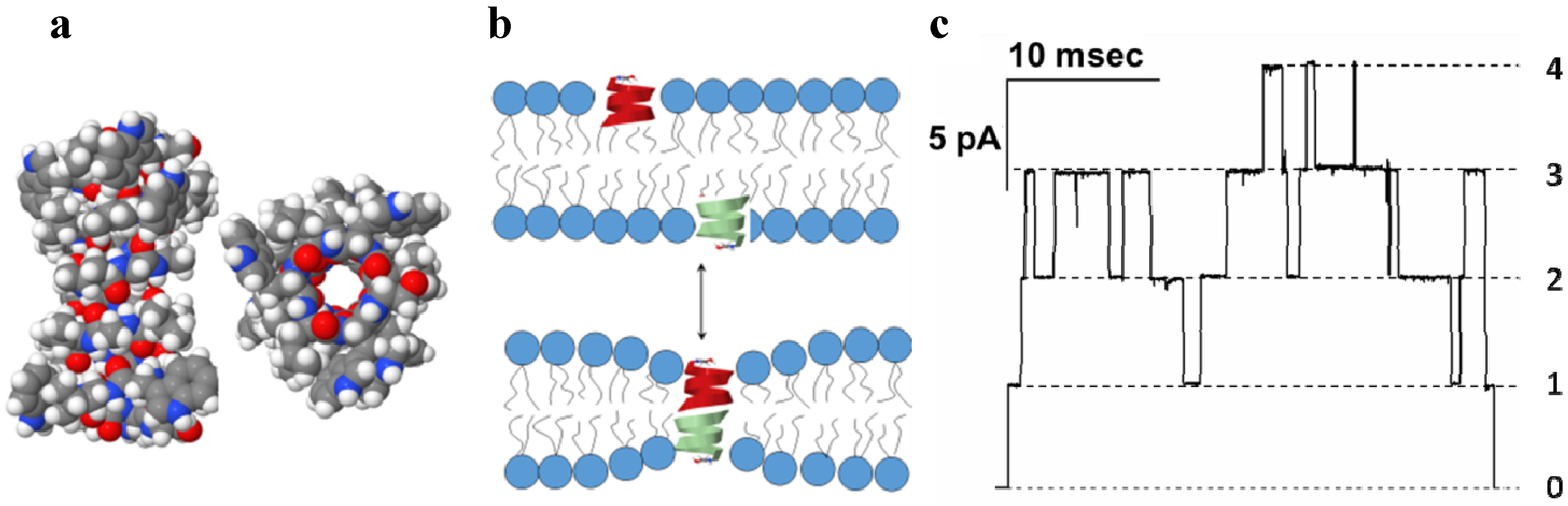
3.2. α-Hemolysin of Staphylococcus Aureus

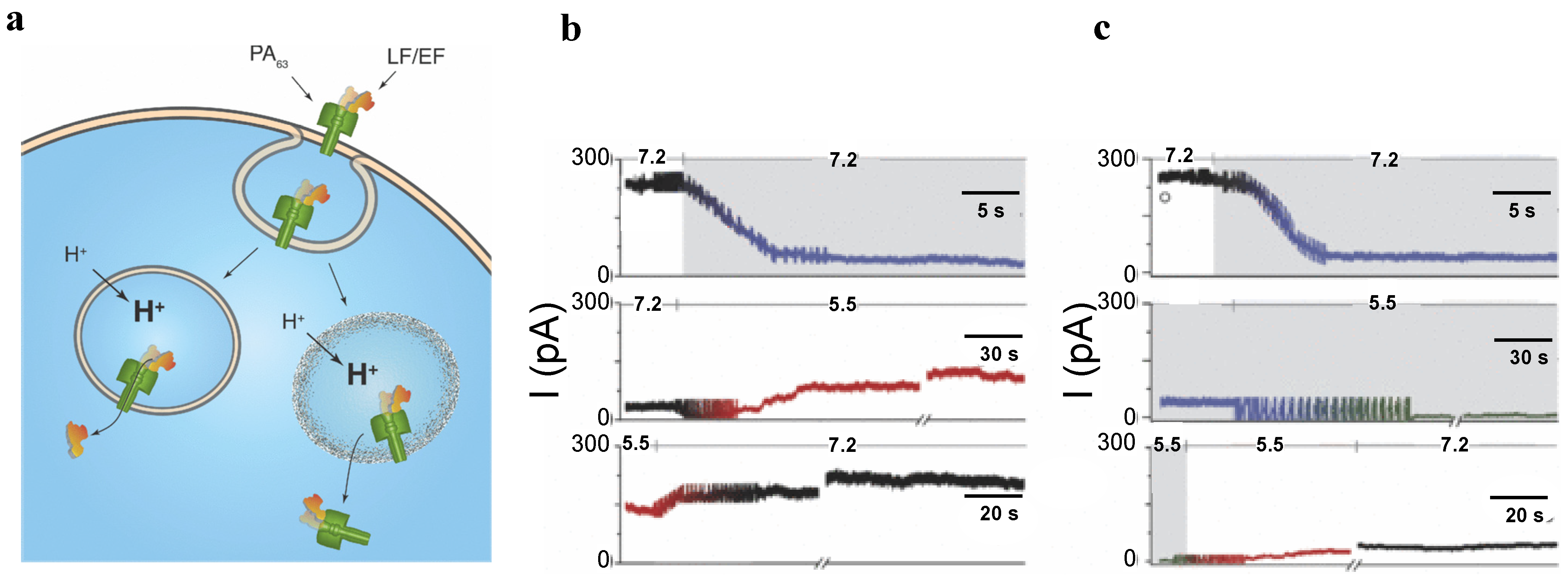
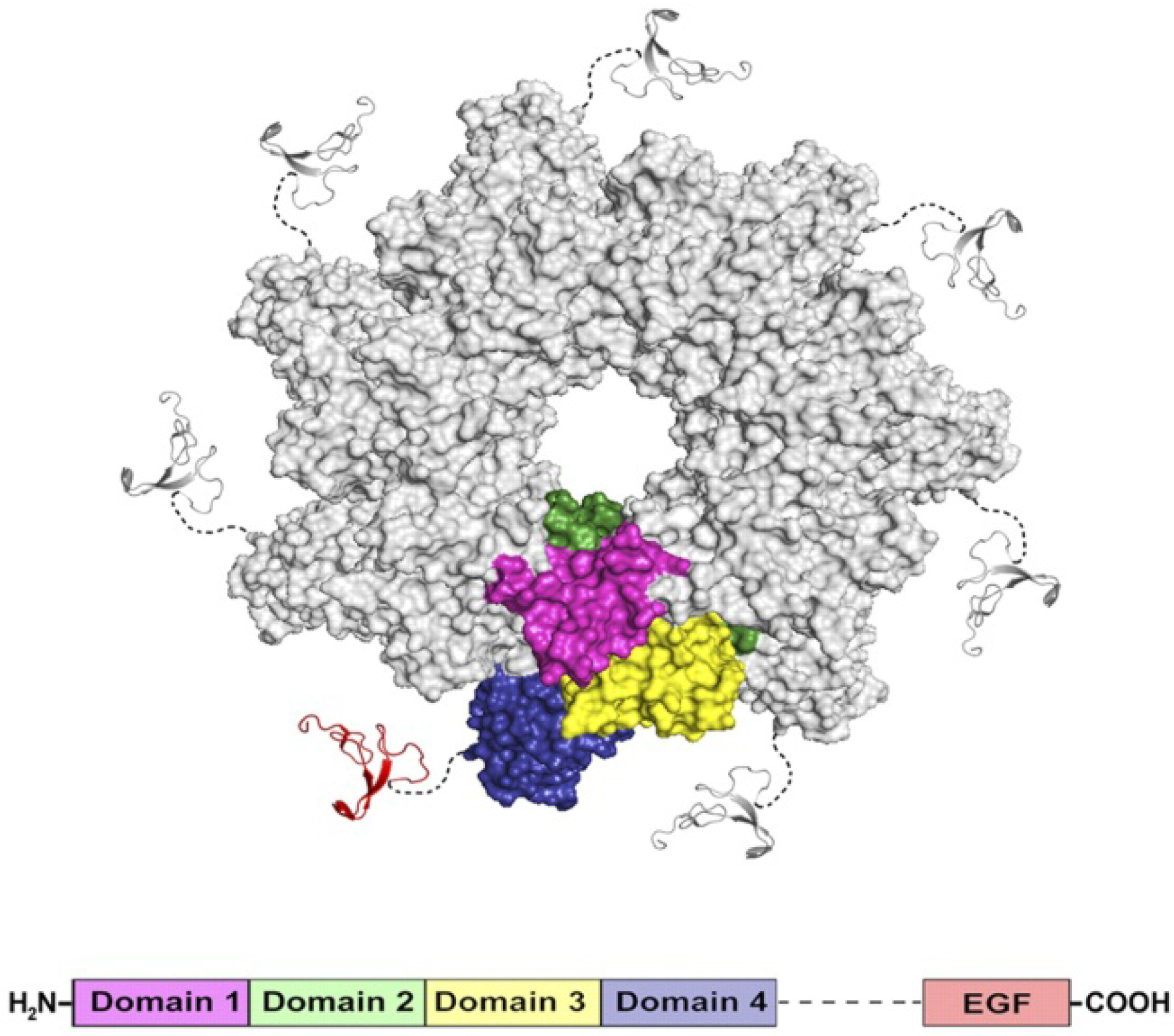
3.3. PA63 Component of Anthrax Toxin of Bacillus Anthracis

4. Channel Forming Bacterial Toxins for Molecular Sensing
4.1. Probing Structure, Charge and Physical State of the Membranes with GrA


4.2. Molecular Sensing with αHL, PA63, and GrA

4.3. Sequencing of Polynucleotides with αHL


5. Channel-Forming Bacterial Toxins to Investigate Protein and Other Macromolecule Transport
5.1. Studying Polymer and Protein Transport with aHL

5.2. Studying Protein Transport with PA63


6. Channel-Forming Bacterial Toxins for Cancer Therapy


7. Concluding Remarks
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Parker, M.W.; Feil, S.C. Pore-forming protein toxins: From structure to function. Prog. Biophys. Mol. Biol. 2005, 88, 91–142. [Google Scholar] [PubMed]
- Geny, B.; Popoff, M.R. Bacterial protein toxins and lipids: Pore formation or toxin entry into cells. Biol. Cell 2006, 98, 667–678. [Google Scholar] [CrossRef] [PubMed]
- Van der Goot, G. (Ed.) Pore-Forming Toxins; Springer-Verlag: New York, NY, USA, 2001; p. 168.
- Los, F.C.; Randis, T.M.; Aroian, R.V.; Ratner, A.J. Role of pore-forming toxins in bacterial infectious diseases. Microbiol. Mol. Biol. Rev. 2013, 77, 173–207. [Google Scholar] [CrossRef] [PubMed]
- Alouf, J.E. Pore-forming bacterial protein toxins: An overview. In Pore-Forming Toxins; Gisou van der Goot, F., Ed.; Springer-Verlag Berlin Heidelberg: Berlin, Germany, 2001; pp. 1–14. [Google Scholar]
- Gilbert, R.J. Pore-forming toxins. Cell Mol. Life Sci. 2002, 59, 832–844. [Google Scholar] [CrossRef] [PubMed]
- Gouaux, E. Channel-forming toxins: Tales of transformation. Curr. Opin. Struct. Biol. 1997, 7, 566–573. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, M.R.; Bischofberger, M.; Pernot, L.; van der Goot, F.G.; Freche, B. Bacterial pore-forming toxins: The (W)Hole story? Cell Mol. Life Sci. 2008, 65, 493–507. [Google Scholar]
- Barth, H.; Aktories, K.; Popoff, M.R.; Stiles, B.G. Binary bacterial toxins: Biochemistry, biology, and applications of common clostridium and bacillus proteins. Microbiol. Mol. Biol. Rev. 2004, 68, 373–402. [Google Scholar] [CrossRef] [PubMed]
- Donovan, J.J.; Simon, M.I.; Draper, R.K.; Montal, M. Diphtheria toxin forms transmembrane channels in planar lipid bilayers. Proc. Natl. Acad. Sci. USA 1981, 78, 172–176. [Google Scholar] [CrossRef] [PubMed]
- Kagan, B.L.; Finkelstein, A.; Colombini, M. Diphtheria toxin fragment forms large pores in phospholipid bilayer membranes. Proc. Natl. Acad. Sci. USA 1981, 78, 4950–4954. [Google Scholar] [CrossRef] [PubMed]
- Deleers, M.; Beugnier, N.; Falmagne, P.; Cabiaux, V.; Ruysschaert, J.M. Localization in diphtheria toxin fragment b of a region that induces pore formation in planar lipid bilayers at low pH. FEBS Lett. 1983, 160, 82–86. [Google Scholar] [PubMed]
- Oblatt-Montal, M.; Yamazaki, M.; Nelson, R.; Montal, M. Formation of ion channels in lipid bilayers by a peptide with the predicted transmembrane sequence of botulinum neurotoxin A. Protein Sci. 1995, 4, 1490–1497. [Google Scholar]
- Donovan, J.J.; Middlebrook, J.L. Ion-Conducting channels produced by botulinum toxin in planar lipid membranes. Biochemistry 1986, 25, 2872–2876. [Google Scholar] [PubMed]
- Blaustein, R.O.; Koehler, T.M.; Collier, R.J.; Finkelstein, A. Anthrax toxin: Channel-Forming activity of protective antigen in planar phospholipid bilayers. Proc. Natl. Acad. Sci. USA 1989, 86, 2209–2213. [Google Scholar] [CrossRef] [PubMed]
- Schmid, A.; Benz, R.; Just, I.; Aktories, K. Interaction of clostridium botulinum C2 toxin with lipid bilayer membranes. Formation of cation-selective channels and inhibition of channel function by chloroquine. J. Biol. Chem 1994, 269, 16706–16711. [Google Scholar]
- Knapp, O.; Benz, R.; Gibert, M.; Marvaud, J.C.; Popoff, M.R. Interaction of clostridium perfringens iota-toxin with lipid bilayer membranes. Demonstration of channel formation by the activated binding component Ib and channel block by the enzyme component Ia. J. Biol. Chem. 2002, 277, 6143–6152. [Google Scholar]
- Nablo, B.J.; Panchal, R.G.; Bavari, S.; Nguyen, T.L.; Gussio, R.; Ribot, W.; Friedlander, A.; Chabot, D.; Reiner, J.E.; Robertson, J.W.; et al. Anthrax toxin-induced rupture of artificial lipid bilayer membranes. J. Chem. Phys. 2013, 139. [Google Scholar] [CrossRef]
- Majd, S.; Yusko, E.C.; Billeh, Y.N.; Macrae, M.X.; Yang, J.; Mayer, M. Applications of biological pores in nanomedicine, sensing, and nanoelectronics. Curr. Opin. Biotechnol. 2010, 21, 439–476. [Google Scholar] [CrossRef] [PubMed]
- Panchal, R.G.; Smart, M.L.; Bowser, D.N.; Williams, D.A.; Petrou, S. Pore-forming proteins and their application in biotechnology. Curr. Pharm. Biotechnol. 2002, 3, 99–115. [Google Scholar] [CrossRef] [PubMed]
- Reiner, J.E.; Balijepalli, A.; Robertson, J.W.; Campbell, J.; Suehle, J.; Kasianowicz, J.J. Disease detection and management via single nanopore-based sensors. Chem. Rev. 2012, 112, 6431–6451. [Google Scholar] [CrossRef] [PubMed]
- Delcour, A.H. Electrophysiology of bacteria. Annu. Rev. Microbiol. 2013, 67, 179–197. [Google Scholar] [CrossRef] [PubMed]
- Steller, L.; Kreir, M.; Salzer, R. Natural and artificial ion channels for biosensing platforms. Anal. Bioanal Chem. 2012, 402, 209–230. [Google Scholar] [CrossRef] [PubMed]
- Bezrukov, S.M.; Kasianowicz, J.J. Current noise reveals protonation kinetics and number of ionizable sites in an open protein ion channel. Phys. Rev. Lett. 1993, 70, 2352–2355. [Google Scholar] [CrossRef] [PubMed]
- Kasianowicz, J.J.; Bezrukov, S.M. Protonation dynamics of the alpha-toxin ion channel from spectral analysis of pH-dependent current fluctuations. Biophys. J. 1995, 69, 94–105. [Google Scholar] [CrossRef] [PubMed]
- Bayley, H. Pore-forming proteins with built-in triggers and switches. Bioorg. Chem. 1995, 23, 340–354. [Google Scholar] [CrossRef]
- Coulter, H.W. Means for counting particles suspended in a fluid. U.S. Patent US2656508, 20 October 1953. [Google Scholar]
- Hotchkiss, R.D. Gramicidin, tyrocidine, and tyrothricin. Adv. Enzymol. Relat. Areas Mol. Biol. 1944, 4, 153–199. [Google Scholar]
- Ishii, S.I.; Witkop, B. Gramicidin A. I. Determination of composition and amino acid configuration by enzymatic and gas chromatographic methods. J. Am. Chem. Soc. 1963, 85, 1832–1834. [Google Scholar]
- Ishii, S.I.; Witkop, B. Gramicidin A. II. Preparation and properties of “seco-Gramicidin A”. J. Am. Chem. Soc. 1964, 86, 1848–1853. [Google Scholar]
- Sarges, R.; Witkop, B. Gramicidin A. IV. Primary sequence of valine and isoleucine gramicidin A. J. Am. Chem. Soc. 1964, 86, 1862–1863. [Google Scholar]
- Harold, F.M.; Baarda, J.R. Gramicidin, valinomycin, and cation permeability of streptococcus faecalis. J. Bacteriol. 1967, 94, 53–60. [Google Scholar] [PubMed]
- Hladky, S.B.; Haydon, D.A. Discreteness of conductance change in bimolecular lipid membranes in the presence of certain antibiotics. Nature 1970, 225, 451–453. [Google Scholar] [PubMed]
- Gurnev, P.A.; Bezrukov, S.M. Inversion of membrane surface charge by trivalent cations probed with a cation-selective channel. Langmuir 2012, 28, 15824–15830. [Google Scholar] [CrossRef] [PubMed]
- Urry, D.W. The gramicidin A transmembrane channel: A proposed Pi(L,D) helix. Proc. Natl. Acad. Sci. USA 1971, 68, 672–676. [Google Scholar] [CrossRef] [PubMed]
- Ketchem, R.R.; Hu, W.; Cross, T.A. High-resolution conformation of gramicidin A in a lipid bilayer by solid-state NMR. Science 1993, 261, 1457–1460. [Google Scholar] [CrossRef] [PubMed]
- Hladky, S.B.; Haydon, D.A. Ion transfer across lipid membranes in the presence of gramicidin A.I. Studies of the unit conductance channel. Biochim. Biophys. Acta 1972, 274, 294–312. [Google Scholar]
- Myers, V.B.; Haydon, D.A. Ion transfer across lipid membranes in the presence of gramicidin A. II. the ion selectivity. Biochim. Biophys. Acta 1972, 274, 313–322. [Google Scholar] [CrossRef]
- Mazet, J.L.; Andersen, O.S.; Koeppe, R.E., 2nd. Single-Channel studies on linear gramicidins with altered amino acid sequences. A comparison of phenylalanine, tryptophane, and tyrosine substitutions at positions 1 and 11. Biophys. J. 1984, 45, 263–276. [Google Scholar]
- Koeppe, R.E., 2nd; Mazet, J.L.; Andersen, O.S. Distinction between dipolar and inductive effects in modulating the conductance of gramicidin channels. Biochemistry 1990, 29, 512–520. [Google Scholar]
- Schagina, L.V.; Grinfeldt, A.E.; Lev, A.A. Interaction of cation fluxes in gramicidin A channels in lipid bilayer membranes. Nature 1978, 273, 243–245. [Google Scholar] [CrossRef] [PubMed]
- Elliott, J.R.; Needham, D.; Dilger, J.P.; Haydon, D.A. The effects of bilayer thickness and tension on gramicidin single-channel lifetime. Biochim. Biophys. Acta 1983, 735, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Lundbaek, J.A.; Birn, P.; Girshman, J.; Hansen, A.J.; Andersen, O.S. Membrane stiffness and channel function. Biochemistry 1996, 35, 3825–3830. [Google Scholar] [CrossRef] [PubMed]
- Helfrich, P.; Jakobsson, E. Calculation of deformation energies and conformations in lipid membranes containing gramicidin channels. Biophys. J. 1990, 57, 1075–1084. [Google Scholar] [CrossRef] [PubMed]
- Lundbaek, J.A.; Andersen, O.S. Spring constants for channel-induced lipid bilayer deformations. estimates using gramicidin channels. Biophys. J. 1999, 76, 889–895. [Google Scholar]
- Killian, J.A. Gramicidin and gramicidin-lipid interactions. Biochim. Biophys. Acta 1992, 1113, 391–425. [Google Scholar] [CrossRef]
- Lewis, J.C.; Dimick, K.P.; Feustel, I.C.; Fevold, H.L.; Olcott, H.S.; Fraenkel-Conrat, H. Modification of gramicidin through reaction with formaldehyde. Science 1945, 102, 274–275. [Google Scholar] [CrossRef] [PubMed]
- Lundbaek, J.A.; Collingwood, S.A.; Ingolfsson, H.I.; Kapoor, R.; Andersen, O.S. Lipid bilayer regulation of membrane protein function: Gramicidin channels as molecular force probes. J. R. Soc. Interface 2010, 7, 373–395. [Google Scholar] [CrossRef] [PubMed]
- Rostovtseva, T.K.; Aguilella, V.M.; Vodyanoy, I.; Bezrukov, S.M.; Parsegian, V.A. Membrane surface-charge titration probed by gramicidin a channel conductance. Biophys. J. 1998, 75, 1783–1792. [Google Scholar] [PubMed]
- Borisenko, V.; Zhang, Z.; Woolley, G.A. Gramicidin derivatives as membrane-based pH sensors. Biochim. Biophys. Acta 2002, 1558, 26–33. [Google Scholar] [CrossRef]
- Gurnev, P.A.; Yang, S.T.; Melikov, K.C.; Chernomordik, L.V.; Bezrukov, S.M. Cationic cell-penetrating peptide binds to planar lipid bilayers containing negatively charged lipids but does not induce conductive pores. Biophys. J. 2013, 104, 1933–1939. [Google Scholar] [CrossRef] [PubMed]
- Majd, S.; Yusko, E.C.; MacBriar, A.D.; Yang, J.; Mayer, M. Gramicidin pores report the activity of membrane-active enzymes. J. Am. Chem. Soc. 2009, 131, 16119–16126. [Google Scholar] [CrossRef] [PubMed]
- Macrae, M.X.; Blake, S.; Mayer, M.; Yang, J. Nanoscale ionic diodes with tunable and switchable rectifying behavior. J. Am. Chem. Soc. 2010, 132, 1766–1767. [Google Scholar] [CrossRef] [PubMed]
- Legendre, J.Y.; Szoka, F.C., Jr. Cyclic amphipathic peptide-DNA complexes mediate high-efficiency transfection of adherent mammalian cells. Proc. Natl. Acad. Sci. USA 1993, 90, 893–897. [Google Scholar] [CrossRef] [PubMed]
- Arbuthnott, J.P.; Freer, J.H.; Bernheimer, A.W. Physical states of staphylococcal alpha-toxin. J. Bacteriol. 1967, 94, 1170–1177. [Google Scholar] [PubMed]
- Bhakdi, S.; Fussle, R.; Tranum-Jensen, J. Staphylococcal alpha-toxin: Oligomerization of hydrophilic monomers to form amphiphilic hexamers induced through contact with deoxycholate detergent micelles. Proc. Natl. Acad. Sci. USA 1981, 78, 5475–5479. [Google Scholar] [CrossRef] [PubMed]
- Cassidy, P.; Harshman, S. Studies on the binding of staphylococcal 125I-Labeled alpha-toxin to rabbit erythrocytes. Biochemistry 1976, 15, 2348–2355. [Google Scholar] [CrossRef] [PubMed]
- Wilke, G.A.; Bubeck Wardenburg, J. Role of a disintegrin and metalloprotease 10 in staphylococcus aureus alpha-hemolysin-mediated cellular injury. Proc. Natl. Acad. Sci. USA 2010, 107, 13473–13478. [Google Scholar] [CrossRef] [PubMed]
- Valeva, A.; Hellmann, N.; Walev, I.; Strand, D.; Plate, M.; Boukhallouk, F.; Brack, A.; Hanada, K.; Decker, H.; Bhakdi, S. Evidence that clustered phosphocholine head groups serve as sites for binding and assembly of an oligomeric protein pore. J. Biol. Chem. 2006, 281, 26014–26021. [Google Scholar] [CrossRef] [PubMed]
- Schwiering, M.; Brack, A.; Stork, R.; Hellmann, N. Lipid and phase specificity of alpha-toxin from S. Aureus. Aureus. Biochim. Biophys. Acta 2013, 1828, 1962–1972. [Google Scholar]
- Fussle, R.; Bhakdi, S.; Sziegoleit, A.; Tranum-Jensen, J.; Kranz, T.; Wellensiek, H.J. On the mechanism of membrane damage by staphylococcus aureus alpha-toxin. J. Cell Biol. 1981, 91, 83–94. [Google Scholar] [CrossRef] [PubMed]
- Bhakdi, S.; Tranum-Jensen, J. Alpha-Toxin of staphylococcus aureus. Microbiol. Rev. 1991, 55, 733–751. [Google Scholar] [PubMed]
- Berube, B.J.; Bubeck Wardenburg, J. Staphylococcus aureus alpha-toxin: Nearly a century of intrigue. Toxins 2013, 5, 1140–1166. [Google Scholar] [CrossRef] [PubMed]
- Inoshima, I.; Inoshima, N.; Wilke, G.A.; Powers, M.E.; Frank, K.M.; Wang, Y.; Bubeck Wardenburg, J. A staphylococcus aureus pore-forming toxin subverts the activity of ADAM10 to cause lethal infection in mice. Nat. Med. 2011, 17, 1310–1314. [Google Scholar] [CrossRef] [PubMed]
- Krasilnikov, O.; Ternovsky, V.I.; Musaev, Y.M.; Tashmukhamedov, B. Influence of staphylotoxin on conductance of bilayer phospholipid membranes. Dokl. Akad. Nauk UzSSR 1980, 7, 66–68. [Google Scholar]
- Menestrina, G. Ionic channels formed by staphylococcus aureus alpha-toxin: Voltage-Dependent inhibition by divalent and trivalent cations. J. Membr. Biol. 1986, 90, 177–190. [Google Scholar] [CrossRef]
- Belmonte, G.; Cescatti, L.; Ferrari, B.; Nicolussi, T.; Ropele, M.; Menestrina, G. Pore formation by staphylococcus aureus alpha-toxin in lipid bilayers. Dependence upon temperature and toxin concentration. Eur. Biophys. J. 1987, 14, 349–358. [Google Scholar]
- Krasilnikov, O.V.; Merzliak, P.G.; Sabirov, R.Z.; Tashmuk-Hamedov, B.A. Memory is a property of an ion channels pool: Ion channels formed by staphylococcus aureus alpha-toxin. Gen. Physiol. Biophys. 1990, 9, 569–575. [Google Scholar] [PubMed]
- Korchev, Y.E.; Alder, G.M.; Bakhramov, A.; Bashford, C.L.; Joomun, B.S.; Sviderskaya, E.V.; Usherwood, P.N.; Pasternak, C.A. Staphylococcus aureus alpha-toxin-induced pores: Channel-Like Behavior in lipid bilayers and patch clamped cells. J. Membr. Biol. 1995, 143, 143–151. [Google Scholar] [CrossRef] [PubMed]
- Bezrukov, S.; Vodyanoy, I.; Kasianowicz, J. Dynamics and free energy of polymers partitioning into a nanoscale pore. Macromolecules 1996, 29, 8517–8522. [Google Scholar] [CrossRef]
- Merzlyak, P.G.; Yuldasheva, L.N.; Rodrigues, C.G.; Carneiro, C.M.; Krasilnikov, O.V.; Bezrukov, S.M. Polymeric nonelectrolytes to probe pore geometry: Application to the alpha-toxin transmembrane channel. Biophys. J. 1999, 77, 3023–3033. [Google Scholar] [CrossRef] [PubMed]
- Krasilnikov, O.V.; Merzlyak, P.G.; Yuldasheva, L.N.; Rodrigues, C.G.; Bhakdi, S.; Valeva, A. Electrophysiological Evidence for heptameric stoichiometry of ion channels formed by staphylococcus aureus alpha-toxin in planar lipid bilayers. Mol. Microbiol. 2000, 37, 1372–1378. [Google Scholar] [CrossRef] [PubMed]
- Gu, L.Q.; Bayley, H. Interaction of the noncovalent molecular adapter, beta-cyclodextrin, with the staphylococcal alpha-hemolysin pore. Biophys. J. 2000, 79, 1967–1975. [Google Scholar] [PubMed]
- Movileanu, L.; Cheley, S.; Howorka, S.; Braha, O.; Bayley, H. Location of a constriction in the lumen of a transmembrane pore by targeted covalent attachment of polymer molecules. J. Gen. Physiol. 2001, 117, 239–252. [Google Scholar] [CrossRef] [PubMed]
- Menestrina, G.; Dalla Serra, M.; Comai, M.; Coraiola, M.; Viero, G.; Werner, S.; Colin, D.A.; Monteil, H.; Prevost, G. Ion channels and bacterial infection: The Case of beta-barrel pore-forming protein toxins of staphylococcus aureus. FEBS Lett. 2003, 552, 54–60. [Google Scholar] [CrossRef] [PubMed]
- Gu, L.Q.; Cheley, S.; Bayley, H. Electroosmotic enhancement of the binding of a neutral molecule to a transmembrane pore. Proc. Natl. Acad. Sci. USA 2003, 100, 15498–15503. [Google Scholar] [CrossRef] [PubMed]
- Movileanu, L.; Cheley, S.; Bayley, H. Partitioning of individual flexible polymers into a nanoscopic protein pore. Biophys. J. 2003, 85, 897–910. [Google Scholar] [CrossRef] [PubMed]
- Song, L.; Hobaugh, M.R.; Shustak, C.; Cheley, S.; Bayley, H.; Gouaux, J.E. Structure of staphylococcal alpha-hemolysin, a heptameric transmembrane pore. Science 1996, 274, 1859–1866. [Google Scholar] [CrossRef] [PubMed]
- Aksimentiev, A.; Schulten, K. Imaging alpha-hemolysin with molecular dynamics: Ionic conductance, osmotic permeability, and the electrostatic potential map. Biophys. J. 2005, 88, 3745–3761. [Google Scholar] [CrossRef] [PubMed]
- Young, J.A.; Collier, R.J. Anthrax toxin: Receptor Binding, internalization, pore formation, and translocation. Annu. Rev. Biochem. 2007, 76, 243–265. [Google Scholar] [CrossRef] [PubMed]
- Moayeri, M.; Leppla, S.H. Cellular and systemic effects of anthrax lethal toxin and edema toxin. Mol. Aspects Med. 2009, 30, 439–455. [Google Scholar] [CrossRef] [PubMed]
- Fouet, A. The surface of bacillus anthracis. Mol. Aspects Med. 2009, 30, 374–385. [Google Scholar] [CrossRef] [PubMed]
- Bouzianas, D.G. Medical countermeasures to protect humans from anthrax bioterrorism. Trends Microbiol. 2009, 17, 522–528. [Google Scholar] [CrossRef] [PubMed]
- Brook, I.; Elliott, T.B.; Pryor, H.I., 2nd; Sautter, T.E.; Gnade, B.T.; Thakar, J.H.; Knudson, G.B. In vitro resistance of bacillus anthracis sterne to doxycycline, macrolides and quinolones. Int. J. Antimicrob. Agents 2001, 18, 559–562. [Google Scholar]
- Athamna, A.; Athamna, M.; Abu-Rashed, N.; Medlej, B.; Bast, D.J.; Rubinstein, E. Selection of bacillus anthracis isolates resistant to antibiotics. J. Antimicrob. Chemother. 2004, 54, 424–428. [Google Scholar] [CrossRef] [PubMed]
- Nestorovich, E.M.; Bezrukov, S.M. Designing inhibitors of anthrax toxin. Expert Opin. Drug Discov. 2014, 9, 299–318. [Google Scholar] [CrossRef] [PubMed]
- Bartlett, J.G.; Inglesby, T.V., Jr.; Borio, L. Management of anthrax. Clin. Infect. Dis. 2002, 35, 851–858. [Google Scholar] [CrossRef]
- Bull, J.J.; Parrish, C.R. Microbiology. A binding contract for anthrax. Science 2002, 297, 201–202. [Google Scholar]
- Mock, M.; Fouet, A. Anthrax. Annu. Rev. Microbiol. 2001, 55, 647–671. [Google Scholar] [CrossRef] [PubMed]
- Bann, J.G. Anthrax toxin protective antigen—Insights into molecular switching from prepore to pore. Protein Sci. 2012, 21, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Frankel, A.E.; Kuo, S.R.; Dostal, D.; Watson, L.; Duesbery, N.S.; Cheng, C.P.; Cheng, H.J.; Leppla, S.H. Pathophysiology of anthrax. Front. Biosci. Landmark Ed. 2009, 14, 4516–4524. [Google Scholar] [CrossRef] [PubMed]
- Guichard, A.; Nizet, V.; Bier, E. New insights into the biological effects of anthrax toxins: Linking cellular to organismal responses. Microbes Infect. 2012, 14, 97–118. [Google Scholar] [CrossRef] [PubMed]
- Hicks, C.W.; Sweeney, D.A.; Cui, X.; Li, Y.; Eichacker, P.Q. An overview of anthrax infection including the recently identified form of disease in injection drug users. Intensive Care Med. 2012, 38, 1092–1104. [Google Scholar] [CrossRef] [PubMed]
- Lowe, D.E.; Glomski, I.J. Cellular and physiological effects of anthrax exotoxin and its relevance to disease. Front. Cell. Infect. Microbiol. 2012, 2. [Google Scholar] [CrossRef]
- Moayeri, M.; Sastalla, I.; Leppla, S.H. Anthrax and the inflammasome. Microbes Infect. 2012, 14, 392–400. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, D.A.; Hicks, C.W.; Cui, X.; Li, Y.; Eichacker, P.Q. Anthrax infection. Am. J. Respir. Crit. Care Med. 2011, 184, 1333–1341. [Google Scholar] [PubMed]
- Van der Goot, G.; Young, J.A. Receptors of anthrax toxin and cell entry. Mol. Aspects Med. 2009, 30, 406–412. [Google Scholar]
- Xie, T.; Auth, R.D.; Frucht, D.M. The effects of anthrax lethal toxin on host barrier function. Toxins 2011, 3, 591–607. [Google Scholar] [CrossRef] [PubMed]
- Zakowska, D.; Bartoszcze, M.; Niemcewicz, M.; Bielawska-Drozd, A.; Kocik, J. New aspects of the infection mechanisms of bacillus anthracis. Ann. Agric. Environ. Med. 2012, 19, 613–618. [Google Scholar] [PubMed]
- Thoren, K.L.; Krantz, B.A. The unfolding story of anthrax toxin translocation. Mol. Microbiol. 2011, 80, 588–595. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.I.; Jo, S.; Rui, H.; Egwolf, B.; Roux, B.; Pastor, R.W.; Im, W. Web interface for brownian dynamics simulation of ion transport and its applications to beta-barrel pores. J. Comput. Chem. 2012, 33, 331–339. [Google Scholar] [CrossRef] [PubMed]
- Bezrukov, S.M.; Liu, X.; Karginov, V.A.; Wein, A.N.; Leppla, S.H.; Popoff, M.R.; Barth, H.; Nestorovich, E.M. Interactions of high-affinity cationic blockers with the translocation pores of B. Anthracis, C. Botulinum, and C. Perfringens binary toxins. Biophys. J. 2012, 13, 1208–1217. [Google Scholar]
- Bradley, K.A.; Mogridge, J.; Mourez, M.; Collier, R.J.; Young, J.A. Identification of the cellular receptor for anthrax toxin. Nature 2001, 414, 225–229. [Google Scholar] [CrossRef] [PubMed]
- Scobie, H.M.; Rainey, G.J.; Bradley, K.A.; Young, J.A. Human capillary morphogenesis protein 2 functions as an anthrax toxin receptor. Proc. Natl. Acad. Sci. USA 2003, 100, 5170–5174. [Google Scholar] [CrossRef] [PubMed]
- Klimpel, K.R.; Molloy, S.S.; Thomas, G.; Leppla, S.H. Anthrax toxin protective antigen is activated by a cell surface protease with the sequence specificity and catalytic properties of furin. Proc. Natl. Acad. Sci. USA 1992, 89, 10277–10281. [Google Scholar] [CrossRef] [PubMed]
- Petosa, C.; Collier, R.J.; Klimpel, K.R.; Leppla, S.H.; Liddington, R.C. Crystal structure of the anthrax toxin protective antigen. Nature 1997, 385, 833–838. [Google Scholar] [CrossRef] [PubMed]
- Kintzer, A.F.; Thoren, K.L.; Sterling, H.J.; Dong, K.C.; Feld, G.K.; Tang, I.I.; Zhang, T.T.; Williams, E.R.; Berger, J.M.; Krantz, B.A. The protective antigen component of anthrax toxin forms functional octameric complexes. J. Mol. Biol. 2009, 392, 614–629. [Google Scholar] [CrossRef] [PubMed]
- Mogridge, J.; Cunningham, K.; Collier, R.J. Stoichiometry of anthrax toxin complexes. Biochemistry 2002, 41, 1079–1082. [Google Scholar] [CrossRef] [PubMed]
- Pilpa, R.M.; Bayrhuber, M.; Marlett, J.M.; Riek, R.; Young, J.A. A receptor-based switch that regulates anthrax toxin pore formation. PLoS Pathog. 2011, 7. [Google Scholar] [CrossRef]
- Katayama, H.; Janowiak, B.E.; Brzozowski, M.; Juryck, J.; Falke, S.; Gogol, E.P.; Collier, R.J.; Fisher, M.T. GroEL as a molecular scaffold for structural analysis of the anthrax toxin pore. Nat. Struct. Mol. Biol. 2008, 15, 754–760. [Google Scholar] [CrossRef] [PubMed]
- Orlik, F.; Schiffler, B.; Benz, R. Anthrax toxin protective antigen: Inhibition of channel function by chloroquine and related compounds and study of binding kinetics using the current noise analysis. Biophys. J. 2005, 88, 1715–1724. [Google Scholar] [PubMed]
- Panchal, R.G.; Halverson, K.M.; Ribot, W.; Lane, D.; Kenny, T.; Abshire, T.G.; Ezzell, J.W.; Hoover, T.A.; Powell, B.; Little, S.; et al. Purified bacillus anthracis lethal toxin complex formed in vitro and during infection exhibits functional and biological activity. J. Biol. Chem. 2005, 280, 10834–10839. [Google Scholar] [PubMed]
- Blaustein, R.O.; Lea, E.J.; Finkelstein, A. Voltage-Dependent block of anthrax toxin channels in planar phospholipid bilayer membranes by symmetric tetraalkylammonium ions. Single-Channel analysis. J. Gen. Physiol. 1990, 96, 921–942. [Google Scholar]
- Nestorovich, E.M.; Karginov, V.A.; Berezhkovskii, A.M.; Bezrukov, S.M. Blockage of anthrax PA63 pore by a multicharged high-affinity toxin inhibitor. Biophys. J. 2010, 99, 134–143. [Google Scholar] [PubMed]
- Nestorovich, E.M.; Bezrukov, S.M. Obstructing Toxin pathways by targeted pore blockage. Chem. Rev. 2012, 112, 6388–6430. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Finkelstein, A.; Collier, R.J. Evidence that translocation of anthrax Toxin’s lethal factor is initiated by entry of its N terminus into the protective antigen channel. Proc. Natl. Acad. Sci. USA 2004, 101, 16756–16761. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Udho, E.; Wu, Z.; Collier, R.J.; Finkelstein, A. Protein translocation through anthrax toxin channels formed in planar lipid bilayers. Biophys. J. 2004, 87, 3842–3849. [Google Scholar] [CrossRef] [PubMed]
- Krantz, B.A.; Melnyk, R.A.; Zhang, S.; Juris, S.J.; Lacy, D.B.; Wu, Z.; Finkelstein, A.; Collier, R.J. A phenylalanine clamp catalyzes protein translocation through the anthrax toxin pore. Science 2005, 309, 777–781. [Google Scholar] [CrossRef] [PubMed]
- Basilio, D.; Juris, S.J.; Collier, R.J.; Finkelstein, A. Evidence for a proton-protein symport mechanism in the anthrax toxin channel. J. Gen. Physiol. 2009, 133, 307–314. [Google Scholar] [CrossRef] [PubMed]
- Basilio, D.; Kienker, P.K.; Briggs, S.W.; Finkelstein, A. A kinetic analysis of protein transport through the anthrax toxin channel. J. Gen. Physiol. 2011, 137, 521–531. [Google Scholar] [CrossRef] [PubMed]
- Basilio, D.; Jennings-Antipov, L.D.; Jakes, K.S.; Finkelstein, A. Trapping a translocating protein within the anthrax toxin channel: Implications for the secondary structure of permeating proteins. J. Gen. Physiol. 2011, 137, 343–356. [Google Scholar] [CrossRef] [PubMed]
- Duesbery, N.S.; Webb, C.P.; Leppla, S.H.; Gordon, V.M.; Klimpel, K.R.; Copeland, T.D.; Ahn, N.G.; Oskarsson, M.K.; Fukasawa, K.; Paull, K.D.; et al. Proteolytic inactivation of MAP-Kinase-Kinase by anthrax lethal factor. Science 1998, 280, 734–737. [Google Scholar]
- Vitale, G.; Bernardi, L.; Napolitani, G.; Mock, M.; Montecucco, C. Susceptibility of mitogen-activated protein kinase kinase family members to proteolysis by anthrax lethal factor. Biochem. J. 2000, 352, 739–745. [Google Scholar] [CrossRef] [PubMed]
- Levinsohn, J.L.; Newman, Z.L.; Hellmich, K.A.; Fattah, R.; Getz, M.A.; Liu, S.; Sastalla, I.; Leppla, S.H.; Moayeri, M. Anthrax lethal factor cleavage of Nlrp1 is required for activation of the inflammasome. PLoS Pathog. 2012, 8. [Google Scholar] [CrossRef]
- Dumetz, F.; Jouvion, G.; Khun, H.; Glomski, I.J.; Corre, J.P.; Rougeaux, C.; Tang, W.J.; Mock, M.; Huerre, M.; Goossens, P.L. Noninvasive imaging technologies reveal edema toxin as a key virulence factor in anthrax. Am. J. Pathol. 2011, 178, 2523–2535. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Zhang, Y.; Moayeri, M.; Liu, J.; Crown, D.; Fattah, R.J.; Wein, A.N.; Yu, Z.X.; Finkel, T.; Leppla, S.H. Key tissue targets responsible for anthrax-toxin-induced lethality. Nature 2013, 501, 63–68. [Google Scholar] [CrossRef] [PubMed]
- Gruner, S.M. Intrinsic curvature hypothesis for biomembrane lipid composition: A role for nonbilayer lipids. Proc. Natl. Acad. Sci. USA 1985, 82, 3665–3669. [Google Scholar] [CrossRef] [PubMed]
- Andersen, O.S.; Koeppe, R.E., 2nd. Bilayer thickness and membrane protein function: An energetic perspective. Annu. Rev. Biophys. Biomol. Struct. 2007, 36, 107–130. [Google Scholar] [CrossRef] [PubMed]
- McLaughlin, S.G.; Szabo, G.; Eisenman, G.; Ciani, S.M. Surface charge and the conductance of phospholipid membranes. Proc. Natl. Acad. Sci. USA 1970, 67, 1268–1275. [Google Scholar] [PubMed]
- McLaughlin, S. The electrostatic properties of membranes. Annu. Rev. Biophys. Biophys. Chem. 1989, 18, 113–136. [Google Scholar] [PubMed]
- Apell, H.J.; Bamberg, E.; Lauger, P. Effects of surface charge on the conductance of the gramicidin channel. Biochim. Biophys. Acta 1979, 552, 369–378. [Google Scholar] [CrossRef] [PubMed]
- Franklin, J.C.; Cafiso, D.S.; Flewelling, R.F.; Hubbell, W.L. Probes of membrane electrostatics: Synthesis and voltage-dependent partitioning of negative hydrophobic ion spin labels in lipid vesicles. Biophys. J. 1993, 64, 642–653. [Google Scholar] [CrossRef] [PubMed]
- Rokitskaya, T.I.; Sorochkina, A.I.; Kovalchuk, S.I.; Egorova, N.S.; Kotova, E.A.; Sychev, S.V.; Antonenko, Y.N. The pH-Dependent induction of lipid membrane ionic permeability by n-terminally lysine-substituted analogs of gramicidin A. Eur. Biophys. J. 2012, 41, 129–138. [Google Scholar] [CrossRef] [PubMed]
- Macrae, M.X.; Blake, S.; Jiang, X.; Capone, R.; Estes, D.J.; Mayer, M.; Yang, J. A semi-synthetic ion channel platform for detection of phosphatase and protease activity. ACS Nano 2009, 3, 3567–3580. [Google Scholar] [CrossRef] [PubMed]
- Malev, V.V.; Schagina, L.V.; Gurnev, P.A.; Takemoto, J.Y.; Nestorovich, E.M.; Bezrukov, S.M. Syringomycin E channel: A lipidic pore stabilized by lipopeptide? Biophys. J. 2002, 82, 1985–1994. [Google Scholar]
- Bessonov, A.; Takemoto, J.Y.; Simmel, F.C. Probing DNA-Lipid membrane interactions with a lipopeptide nanopore. ACS Nano 2012, 6, 3356–3363. [Google Scholar] [CrossRef] [PubMed]
- Kasianowicz, J.J.; Brandin, E.; Branton, D.; Deamer, D.W. Characterization of individual polynucleotide molecules using a membrane channel. Proc. Natl. Acad. Sci. USA 1996, 93, 13770–13773. [Google Scholar] [CrossRef] [PubMed]
- Movileanu, L.; Schmittschmitt, J.P.; Scholtz, J.M.; Bayley, H. Interactions of peptides with a protein pore. Biophys. J. 2005, 89, 1030–1045. [Google Scholar] [CrossRef] [PubMed]
- Gu, L.Q.; Braha, O.; Conlan, S.; Cheley, S.; Bayley, H. Stochastic sensing of organic analytes by a pore-forming protein containing a molecular adapter. Nature 1999, 398, 686–690. [Google Scholar] [CrossRef] [PubMed]
- Gu, L.Q.; Dalla Serra, M.; Vincent, J.B.; Vigh, G.; Cheley, S.; Braha, O.; Bayley, H. Reversal of charge selectivity in transmembrane protein pores by using noncovalent molecular adapters. Proc. Natl. Acad. Sci. USA 2000, 97, 3959–3964. [Google Scholar] [CrossRef] [PubMed]
- Braha, O.; Walker, B.; Cheley, S.; Kasianowicz, J.J.; Song, L.; Gouaux, J.E.; Bayley, H. Designed protein pores as components for biosensors. Chem. Biol. 1997, 4, 497–505. [Google Scholar] [CrossRef] [PubMed]
- Braha, O.; Gu, L.Q.; Zhou, L.; Lu, X.; Cheley, S.; Bayley, H. Simultaneous stochastic sensing of divalent metal ions. Nat. Biotechnol. 2000, 18, 1005–1007. [Google Scholar] [CrossRef] [PubMed]
- Choi, L.S.; Mach, T.; Bayley, H. Rates and stoichiometries of metal ion probes of cysteine residues within ion channels. Biophys. J. 2013, 105, 356–364. [Google Scholar] [CrossRef] [PubMed]
- Cheley, S.; Gu, L.Q.; Bayley, H. Stochastic sensing of nanomolar inositol 1,4,5-trisphosphate with an engineered pore. Chem. Biol. 2002, 9, 829–838. [Google Scholar] [CrossRef] [PubMed]
- Guan, X.; Gu, L.Q.; Cheley, S.; Braha, O.; Bayley, H. Stochastic sensing of TNT with a genetically engineered pore. Chembiochem 2005, 6, 1875–1881. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Ma, L.; Cheley, S.; Bayley, H.; Cui, Q.; Chapman, E.R. Permeation of styryl dyes through nanometer-scale pores in membranes. Biochemistry 2011, 50, 7493–7502. [Google Scholar] [CrossRef] [PubMed]
- Asandei, A.; Mereuta, L.; Luchian, T. The kinetics of ampicillin complexation by gamma-cyclodextrins. A single molecule approach. J. Phys. Chem. B 2011, 115, 10173–10181. [Google Scholar]
- Asandei, A.; Apetrei, A.; Luchian, T. Uni-Molecular detection and quantification of selected beta-lactam antibiotics with a hybrid alpha-hemolysin protein pore. J. Mol. Recognit. 2011, 24, 199–207. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, L.R.; Merzlyak, P.G.; Valeva, A.; Krasilnikov, O.V. Interaction of heparins and dextran sulfates with a mesoscopic protein nanopore. Biophys. J. 2009, 97, 2894–2903. [Google Scholar] [PubMed]
- Wu, H.C.; Bayley, H. Single-Molecule detection of nitrogen mustards by covalent reaction within a protein nanopore. J. Am. Chem. Soc. 2008, 130, 6813–6819. [Google Scholar] [CrossRef] [PubMed]
- Bacri, L.; Oukhaled, A.; Hemon, E.; Bassafoula, F.B.; Auvray, L.; Daniel, R. Discrimination of neutral oligosaccharides through a nanopore. Biochem. Biophys. Res. Commun. 2011, 412, 561–564. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Egwolf, B.; Walters, D.E.; Roux, B. Ion selectivity of alpha-hemolysin with a beta-cyclodextrin adapter. I. Single ion potential of mean force and diffusion coefficient. J. Phys. Chem. B 2010, 114, 952–958. [Google Scholar]
- Egwolf, B.; Luo, Y.; Walters, D.E.; Roux, B. Ion selectivity of alpha-hemolysin with beta-cyclodextrin adapter. II. Multi-Ion effects studied with grand canonical monte carlo/brownian dynamics simulations. J. Phys. Chem. B 2010, 114, 2901–2909. [Google Scholar]
- Banerjee, A.; Mikhailova, E.; Cheley, S.; Gu, L.Q.; Montoya, M.; Nagaoka, Y.; Gouaux, E.; Bayley, H. Molecular bases of cyclodextrin adapter interactions with engineered protein nanopores. Proc. Natl. Acad. Sci. USA 2010, 107, 8165–8170. [Google Scholar] [CrossRef] [PubMed]
- Gu, L.Q.; Cheley, S.; Bayley, H. Prolonged residence time of a noncovalent molecular adapter, beta-cyclodextrin, within the lumen of mutant alpha-hemolysin pores. J. Gen. Physiol. 2001, 118, 481–494. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.C.; Astier, Y.; Maglia, G.; Mikhailova, E.; Bayley, H. Protein nanopores with covalently attached molecular adapters. J. Am. Chem. Soc. 2007, 129, 16142–16148. [Google Scholar] [CrossRef] [PubMed]
- Li, W.W.; Claridge, T.D.; Li, Q.; Wormald, M.R.; Davis, B.G.; Bayley, H. Tuning the cavity of cyclodextrins: Altered sugar adaptors in protein pores. J. Am. Chem. Soc. 2011, 133, 1987–2001. [Google Scholar] [CrossRef] [PubMed]
- Gu, L.Q.; Cheley, S.; Bayley, H. Capture of a single molecule in a nanocavity. Science 2001, 291, 636–640. [Google Scholar] [CrossRef] [PubMed]
- Gurnev, P.A.; Harries, D.; Parsegian, V.A.; Bezrukov, S.M. The dynamic side of the hofmeister effect: A single-molecule nanopore study of specific complex formation. Chem. Phys. Chem. 2009, 10, 1445–1449. [Google Scholar] [PubMed]
- Kang, X.F.; Cheley, S.; Guan, X.; Bayley, H. Stochastic detection of enantiomers. J. Am. Chem. Soc. 2006, 128, 10684–10685. [Google Scholar] [CrossRef] [PubMed]
- Karginov, V.A.; Nestorovich, E.M.; Schmidtmann, F.; Robinson, T.M.; Yohannes, A.; Fahmi, N.E.; Bezrukov, S.M.; Hecht, S.M. Inhibition of S. Aureus alpha-hemolysin and B. anthracis lethal toxin by beta-cyclodextrin derivatives. Bioorg. Med. Chem. 2007, 15, 5424–5431. [Google Scholar]
- Ragle, B.E.; Karginov, V.A.; Bubeck Wardenburg, J. Prevention and treatment of staphylococcus aureus pneumonia with a beta-cyclodextrin derivative. Antimicrob. Agents Chemother. 2010, 54, 298–304. [Google Scholar] [CrossRef] [PubMed]
- Karginov, V.A.; Nestorovich, E.M.; Moayeri, M.; Leppla, S.H.; Bezrukov, S.M. Blocking anthrax lethal toxin at the protective antigen channel by using structure-inspired drug design. Proc. Natl. Acad. Sci. USA 2005, 102, 15075–15080. [Google Scholar] [CrossRef] [PubMed]
- Karginov, V.A.; Yohannes, A.; Robinson, T.M.; Fahmi, N.E.; Alibek, K.; Hecht, S.M. Beta-Cyclodextrin Derivatives that inhibit anthrax lethal toxin. bioorg. Med. Chem. 2006, 14, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Karginov, V.A.; Nestorovich, E.M.; Yohannes, A.; Robinson, T.M.; Fahmi, N.E.; Schmidtmann, F.; Hecht, S.M.; Bezrukov, S.M. Search for cyclodextrin-based inhibitors of anthrax toxins: synthesis, structural features, and relative activities. Antimicrob. Agents Chemother. 2006, 50, 3740–3753. [Google Scholar] [CrossRef] [PubMed]
- Backer, M.V.; Patel, V.; Jehning, B.T.; Claffey, K.P.; Karginov, V.A.; Backer, J.M. Inhibition of Anthrax protective antigen outside and inside the cell. Antimicrob. Agents Chemother. 2007, 51, 245–251. [Google Scholar] [CrossRef] [PubMed]
- Moayeri, M.; Robinson, T.M.; Leppla, S.H.; Karginov, V.A. In vivo efficacy of beta-cyclodextrin derivatives against anthrax lethal toxin. Antimicrob. Agents Chemother. 2008, 52, 2239–2241. [Google Scholar] [CrossRef] [PubMed]
- Nestorovich, E.M.; Karginov, V.A.; Popoff, M.R.; Bezrukov, S.M.; Barth, H. Tailored Ss-Cyclodextrin blocks the translocation pores of binary exotoxins from C. Botulinum and C. Perfringens and protects cells from intoxication. PLoS One 2011, 6. [Google Scholar] [CrossRef]
- Yannakopoulou, K.; Jicsinszky, L.; Aggelidou, C.; Mourtzis, N.; Robinson, T.M.; Yohannes, A.; Nestorovich, E.M.; Bezrukov, S.M.; Karginov, V.A. Symmetry requirements for effective blocking of pore-forming toxins: Comparative study with alpha-, beta-, and gamma-cyclodextrin derivatives. Antimicrob. Agents Chemother. 2011, 55, 3594–3597. [Google Scholar] [CrossRef] [PubMed]
- Nestorovich, E.M.; Karginov, V.A.; Berezhkovskii, A.M.; Parsegian, V.A.; Bezrukov, S.M. Kinetics and thermodynamics of binding reactions as exemplified by anthrax toxin channel blockage with a cationic cyclodextrin derivative. Proc. Natl. Acad. Sci. USA 2012, 109, 18453–18458. [Google Scholar] [CrossRef] [PubMed]
- Karginov, V.A. Cyclodextrin derivatives as anti-infectives. Curr. Opin. Pharmacol. 2013, 13, 717–725. [Google Scholar] [CrossRef] [PubMed]
- Berezhkovskii, A.; Bezrukov, S. Channel-facilitated membrane transport: Constructive role of particle attraction to the channel pore. Chem. Phys. 2005, 319, 342–349. [Google Scholar]
- Berezhkovskii, A.M.; Bezrukov, S.M. Optimizing transport of metabolites through large channels: Molecular sieves with and without binding. Biophys. J. 2005, 88, L17–L19. [Google Scholar] [CrossRef] [PubMed]
- Blaustein, R.O.; Finkelstein, A. Diffusion limitation in the block by symmetric tetraalkylammonium ions of anthrax toxin channels in planar phospholipid bilayer membranes. J. Gen. Physiol. 1990, 96, 943–957. [Google Scholar] [CrossRef] [PubMed]
- Blaustein, R.O.; Finkelstein, A. Voltage-dependent block of anthrax toxin channels in planar phospholipid bilayer membranes by symmetric tetraalkylammonium ions. Effects on macroscopic conductance. J. Gen. Physiol. 1990, 96, 905–919. [Google Scholar]
- Beitzinger, C.; Bronnhuber, A.; Duscha, K.; Riedl, Z.; Huber-Lang, M.; Benz, R.; Hajos, G.; Barth, H. Designed azolopyridinium salts block protective antigen pores in vitro and protect cells from anthrax toxin. PLoS One 2013, 8. [Google Scholar] [CrossRef] [Green Version]
- Halverson, K.M.; Panchal, R.G.; Nguyen, T.L.; Gussio, R.; Little, S.F.; Misakian, M.; Bavari, S.; Kasianowicz, J.J. Anthrax Biosensor, protective antigen ion channel asymmetric blockade. J. Biol. Chem. 2005, 280, 34056–34062. [Google Scholar] [CrossRef] [PubMed]
- Cornell, B.A.; Braach-Maksvytis, V.L.; King, L.G.; Osman, P.D.; Raguse, B.; Wieczorek, L.; Pace, R.J. A biosensor that uses ion-channel switches. Nature 1997, 387, 580–583. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.Y.; Cornell, B.; Smith, D.; Higgins, G.; Burrell, C.J.; Kok, T.W. Rapid detection of influenza a virus in clinical samples using an ion channel switch biosensor. Biosens. Bioelectron. 2008, 23, 1161–1165. [Google Scholar] [CrossRef] [PubMed]
- Krishnamurthy, V.; Monfared, S.M.; Cornell, B. Ion-Channel Biosensors—Part I: Construction, operation, and clinical studies. Nanotechnol. IEEE Trans. 2010, 9, 303–312. [Google Scholar] [CrossRef]
- Krishnamurthy, V.; Monfared, S.M.; Cornell, B. Ion channel biosensors—Part II: Dynamic modeling, analysis, and statistical signal processing. Nanotechnol. IEEE Trans. 2010, 9, 313–321. [Google Scholar] [CrossRef]
- Church, G.; Deamer, D.W.; Branton, D.; Baldarelli, R.; Kasianowicz, J. Characterization of individual polymer molecules based on monomer-interface interactions. U.S. Patent US5795782, 18 August 1998. [Google Scholar]
- Akeson, M.; Branton, D.; Kasianowicz, J.J.; Brandin, E.; Deamer, D.W. Microsecond time-scale discrimination among polycytidylic acid, polyadenylic acid, and polyuridylic acid as homopolymers or as segments within single RNA molecules. Biophys. J. 1999, 77, 3227–3233. [Google Scholar] [CrossRef] [PubMed]
- Deamer, D.W.; Akeson, M. Nanopores and nucleic acids: Prospects for ultrarapid sequencing. Trends Biotechnol. 2000, 18, 147–151. [Google Scholar] [CrossRef] [PubMed]
- Bayley, H. Sequencing single molecules of DNA. Curr. Opin. Chem. Biol. 2006, 10, 628–637. [Google Scholar] [CrossRef] [PubMed]
- Branton, D.; Deamer, D.W.; Marziali, A.; Bayley, H.; Benner, S.A.; Butler, T.; di Ventra, M.; Garaj, S.; Hibbs, A.; Huang, X.; et al. The potential and challenges of nanopore sequencing. Nat. Biotechnol. 2008, 26, 1146–1153. [Google Scholar] [CrossRef] [PubMed]
- Meller, A.; Nivon, L.; Branton, D. Voltage-Driven DNA translocations through a nanopore. Phys. Rev. Lett. 2001, 86, 3435–3438. [Google Scholar] [CrossRef] [PubMed]
- Meller, A.; Branton, D. Single molecule measurements of DNA transport through a nanopore. Electrophoresis 2002, 23, 2583–2591. [Google Scholar] [CrossRef] [PubMed]
- Aksimentiev, A.; Heng, J.B.; Timp, G.; Schulten, K. Microscopic kinetics of DNA translocation through synthetic nanopores. Biophys. J. 2004, 87, 2086–2097. [Google Scholar] [CrossRef] [PubMed]
- Muthukumar, M.; Kong, C.Y. Simulation of polymer translocation through protein channels. Proc. Natl. Acad. Sci. USA 2006, 103, 5273–5278. [Google Scholar] [CrossRef] [PubMed]
- Saenger, W.; Riecke, J.; Suck, D. A structural model for the polyadenylic acid single helix. J. Mol. Biol. 1975, 93, 529–534. [Google Scholar] [CrossRef] [PubMed]
- Meller, A.; Nivon, L.; Brandin, E.; Golovchenko, J.; Branton, D. Rapid nanopore discrimination between single polynucleotide molecules. Proc. Natl. Acad. Sci. USA 2000, 97, 1079–1084. [Google Scholar] [CrossRef] [PubMed]
- Wiggin, M.; Tropini, C.; Tabard-Cossa, V.; Jetha, N.N.; Marziali, A. Nonexponential kinetics of DNA escape from alpha-hemolysin nanopores. Biophys. J. 2008, 95, 5317–5323. [Google Scholar] [CrossRef] [PubMed]
- Butler, T.Z.; Pavlenok, M.; Derrington, I.M.; Niederweis, M.; Gundlach, J.H. Single-Molecule DNA detection with an engineered mspa protein nanopore. Proc. Natl. Acad. Sci. USA 2008, 105, 20647–20652. [Google Scholar] [CrossRef] [PubMed]
- Howorka, S.; Siwy, Z. Nanopore analytics: Sensing of single molecules. Chem. Soc. Rev. 2009, 38, 2360–2384. [Google Scholar] [CrossRef] [PubMed]
- Wanunu, M. Nanopores: A journey towards DNA sequencing. Phys. Life. Rev. 2012, 9, 125–158. [Google Scholar] [CrossRef] [PubMed]
- Ashkenasy, N.; Sanchez-Quesada, J.; Bayley, H.; Ghadiri, M.R. Recognizing a single base in an individual DNA strand: A step toward DNA sequencing in nanopores. Angew. Chem. Int. Ed. Engl. 2005, 44, 1401–1404. [Google Scholar] [PubMed]
- Purnell, R.F.; Schmidt, J.J. Discrimination of single base substitutions in a DNA strand immobilized in a biological nanopore. ACS Nano 2009, 3, 2533–2538. [Google Scholar] [CrossRef] [PubMed]
- Stoddart, D.; Heron, A.J.; Mikhailova, E.; Maglia, G.; Bayley, H. Single-Nucleotide discrimination in immobilized DNA oligonucleotides with a biological nanopore. Proc. Natl. Acad. Sci. USA 2009, 106, 7702–7707. [Google Scholar] [PubMed]
- Cockroft, S.L.; Chu, J.; Amorin, M.; Ghadiri, M.R. A single-molecule nanopore device detects DNA polymerase activity with single-nucleotide resolution. J. Am. Chem. Soc. 2008, 130, 818–820. [Google Scholar] [CrossRef] [PubMed]
- Chu, J.; Gonzalez-Lopez, M.; Cockroft, S.L.; Amorin, M.; Ghadiri, M.R. Real-Time monitoring of DNA polymerase function and stepwise single-nucleotide DNA strand translocation through a protein nanopore. Angew. Chem. Int. Ed. Engl. 2010, 49, 10106–10109. [Google Scholar] [CrossRef] [PubMed]
- Lieberman, K.R.; Cherf, G.M.; Doody, M.J.; Olasagasti, F.; Kolodji, Y.; Akeson, M. Processive replication of single DNA molecules in a nanopore catalyzed by phi29 DNA polymerase. J. Am. Chem. Soc. 2010, 132, 17961–17972. [Google Scholar] [CrossRef] [PubMed]
- Cherf, G.M.; Lieberman, K.R.; Rashid, H.; Lam, C.E.; Karplus, K.; Akeson, M. Automated forward and reverse ratcheting of DNA in a nanopore at 5-A precision. Nat. Biotechnol. 2012, 30, 344–348. [Google Scholar] [CrossRef] [PubMed]
- Astier, Y.; Braha, O.; Bayley, H. Toward single molecule DNA sequencing: Direct identification of ribonucleoside and deoxyribonucleoside 5'-Monophosphates by using an engineered protein nanopore equipped with a molecular adapter. J. Am. Chem. Soc. 2006, 128, 1705–1710. [Google Scholar] [CrossRef] [PubMed]
- Ayub, M.; Hardwick, S.W.; Luisi, B.F.; Bayley, H. Nanopore-based identification of individual nucleotides for direct RNA sequencing. Nano Lett. 2013, 13, 6144–6150. [Google Scholar] [CrossRef] [PubMed]
- Muthukumar, M. Polymer Translocation; CRC Press: New York, NY, USA, 2011. [Google Scholar]
- Movileanu, L. Interrogating single proteins through nanopores: Challenges and opportunities. Trends Biotechnol. 2009, 27, 333–341. [Google Scholar] [CrossRef] [PubMed]
- Krasilnikov, O.V.; Sabirov, R.Z.; Ternovsky, V.I.; Merzliak, P.G.; Muratkhodjaev, J.N. A simple method for the determination of the pore radius of ion channels in planar lipid bilayer membranes. FEMS Microbiol. Immunol. 1992, 5, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Nablo, B.J.; Halverson, K.M.; Robertson, J.W.; Nguyen, T.L.; Panchal, R.G.; Gussio, R.; Bavari, S.; Krasilnikov, O.V.; Kasianowicz, J.J. Sizing the bacillus anthracis PA63 channel with nonelectrolyte Poly(Ethylene Glycols). Biophys. J. 2008, 95, 1157–1164. [Google Scholar] [CrossRef] [PubMed]
- Howorka, S.; Movileanu, L.; Lu, X.; Magnon, M.; Cheley, S.; Braha, O.; Bayley, H. A protein pore with a single polymer chain tethered within the lumen. J. Am. Chem. Soc. 2000, 122, 2411–2416. [Google Scholar] [CrossRef]
- Movileanu, L.; Howorka, S.; Braha, O.; Bayley, H. Detecting protein analytes that modulate transmembrane movement of a polymer chain within a single protein pore. Nat. Biotechnol. 2000, 18, 1091–1095. [Google Scholar] [CrossRef] [PubMed]
- Bezrukov, S.M.; Krasilnikov, O.V.; Yuldasheva, L.N.; Berezhkovskii, A.M.; Rodrigues, C.G. Field-Dependent effect of crown ether (18-Crown-6) on ionic conductance of alpha-hemolysin channels. Biophys. J. 2004, 87, 3162–3171. [Google Scholar] [CrossRef] [PubMed]
- Krasilnikov, O.V.; Rodrigues, C.G.; Bezrukov, S.M. Single polymer molecules in a protein nanopore in the limit of a strong polymer-pore attraction. Phys. Rev. Lett. 2006, 97. [Google Scholar] [CrossRef]
- Merzlyak, P.G.; Capistrano, M.F.; Valeva, A.; Kasianowicz, J.J.; Krasilnikov, O.V. Conductance and ion selectivity of a mesoscopic protein nanopore probed with cysteine scanning mutagenesis. Biophys. J. 2005, 89, 3059–3070. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.T.; Muthukumar, M. Polymer translocation through alpha-hemolysin pore with tunable polymer-pore electrostatic interaction. J. Chem. Phys. 2010, 133. [Google Scholar] [CrossRef]
- Jeon, B.J.; Muthukumar, M. Polymer capture by alpha-hemolysin pore upon salt concentration gradient. J. Chem. Phys. 2014, 140. [Google Scholar] [CrossRef]
- Sutherland, T.C.; Long, Y.-T.; Stefureac, R.-L.; Bediako-Amoa, I.; Kraatz, H.-B.; Lee, J.S. Structure of peptides investigated by nanopore analysis. Nano Lett. 2004, 4, 1273–1277. [Google Scholar] [CrossRef]
- Stefureac, R.; Long, Y.T.; Kraatz, H.B.; Howard, P.; Lee, J.S. Transport of alpha-helical peptides through alpha-hemolysin and aerolysin pores. Biochemistry 2006, 45, 9172–9179. [Google Scholar] [CrossRef] [PubMed]
- Oukhaled, G.; Mathe, J.; Biance, A.L.; Bacri, L.; Betton, J.M.; Lairez, D.; Pelta, J.; Auvray, L. Unfolding of proteins and long transient conformations detected by single nanopore recording. Phys. Rev. Lett. 2007, 98. [Google Scholar] [CrossRef]
- Goodrich, C.P.; Kirmizialtin, S.; Huyghues-Despointes, B.M.; Zhu, A.; Scholtz, J.M.; Makarov, D.E.; Movileanu, L. Single-Molecule electrophoresis of beta-hairpin peptides by electrical recordings and langevin dynamics simulations. J. Phys. Chem. B 2007, 111, 3332–3335. [Google Scholar] [PubMed]
- Nivala, J.; Marks, D.B.; Akeson, M. Unfoldase-mediated protein translocation through an alpha-hemolysin nanopore. Nat. Biotechnol. 2013, 31, 247–250. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Larrea, D.; Bayley, H. Multistep protein unfolding during nanopore translocation. Nat. Nanotechnol. 2013, 8, 288–295. [Google Scholar]
- Thoren, K.L.; Worden, E.J.; Yassif, J.M.; Krantz, B.A. Lethal factor unfolding is the most force-dependent step of anthrax toxin translocation. Proc. Natl. Acad. Sci. USA 2009, 106, 21555–21560. [Google Scholar] [CrossRef] [PubMed]
- Rosen, C.B.; Rodriguez-Larrea, D.; Bayley, H. Single-Molecule site-specific detection of protein phosphorylation with a nanopore. Nat. Biotechnol. 2014, 32, 179–181. [Google Scholar] [CrossRef] [PubMed]
- Wolfe, A.J.; Mohammad, M.M.; Cheley, S.; Bayley, H.; Movileanu, L. Catalyzing the translocation of polypeptides through attractive interactions. J. Am. Chem. Soc. 2007, 129, 14034–14041. [Google Scholar] [CrossRef] [PubMed]
- Mohammad, M.M.; Movileanu, L. Excursion of a single polypeptide into a protein pore: Simple physics, but complicated biology. Eur. Biophys. J. 2008, 37, 913–925. [Google Scholar] [CrossRef] [PubMed]
- Bikwemu, R.; Wolfe, A.J.; Xing, X.; Movileanu, L. Facilitated translocation of polypeptides through a single nanopore. J. Phys. Condens Matter 2010, 22. [Google Scholar] [CrossRef]
- Mohammad, M.M.; Movileanu, L. Impact of distant charge reversals within a robust beta-barrel protein pore. J. Phys. Chem. B 2010, 114, 8750–8759. [Google Scholar] [PubMed]
- Zhao, Q.; Jayawardhana, D.A.; Wang, D.; Guan, X. Study of peptide transport through engineered protein channels. J. Phys. Chem. B 2009, 113, 3572–3578. [Google Scholar] [PubMed]
- Madampage, C.; Tavassoly, O.; Christensen, C.; Kumari, M.; Lee, J.S. Nanopore analysis: An emerging technique for studying the folding and misfolding of proteins. Prion 2012, 6, 116–123. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.Y.; Ying, Y.L.; Li, Y.; Kraatz, H.B.; Long, Y.T. Nanopore analysis of beta-amyloid peptide aggregation transition induced by small molecules. Anal. Chem. 2011, 83, 1746–1752. [Google Scholar] [CrossRef] [PubMed]
- Gurnev, P.A.; Yap, T.L.; Pfefferkorn, C.M.; Rostovtseva, T.K.; Berezhkovskii, A.M.; Lee, J.C.; Parsegian, V.A.; Bezrukov, S.M. Alpha-Synuclein lipid-dependent membrane binding and translocation through the alpha-hemolysin channel. Biophys. J. 2014, 106, 556–565. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.Y.; Gu, Z.; Cao, C.; Wang, J.; Long, Y.T. Analysis of a single alpha-synuclein fibrillation by the interaction with a protein nanopore. Anal. Chem. 2013, 85, 8254–8261. [Google Scholar] [CrossRef] [PubMed]
- Rotem, D.; Jayasinghe, L.; Salichou, M.; Bayley, H. Protein detection by nanopores equipped with aptamers. J. Am. Chem. Soc. 2012, 134, 2781–2787. [Google Scholar] [CrossRef] [PubMed]
- Collier, R.J. Membrane translocation by anthrax toxin. Mol. Aspects Med. 2009, 30, 413–422. [Google Scholar] [CrossRef]
- Feld, G.K.; Brown, M.J.; Krantz, B.A. Ratcheting up protein translocation with anthrax toxin. Protein Sci. 2012, 21, 606–624. [Google Scholar] [CrossRef] [PubMed]
- Oh, K.J.; Senzel, L.; Collier, R.J.; Finkelstein, A. Translocation of the catalytic domain of diphtheria toxin across planar phospholipid bilayers by its own T domain. Proc. Natl. Acad. Sci. USA 1999, 96, 8467–8470. [Google Scholar] [CrossRef] [PubMed]
- Koriazova, L.K.; Montal, M. Translocation of botulinum neurotoxin light chain protease through the heavy chain channel. Nat. Struct. Biol. 2003, 10, 13–18. [Google Scholar] [CrossRef] [PubMed]
- Krantz, B.A.; Finkelstein, A.; Collier, R.J. Protein translocation through the anthrax toxin transmembrane pore is driven by a proton gradient. J. Mol. Biol. 2006, 355, 968–979. [Google Scholar] [CrossRef] [PubMed]
- Finkelstein, A. Proton-Coupled protein transport through the anthrax toxin channel. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2009, 364, 209–215. [Google Scholar] [CrossRef] [PubMed]
- Benson, E.L.; Huynh, P.D.; Finkelstein, A.; Collier, R.J. Identification of residues lining the anthrax protective antigen channel. Biochemistry 1998, 37, 3941–3948. [Google Scholar] [CrossRef] [PubMed]
- Sellman, B.R.; Nassi, S.; Collier, R.J. Point mutations in anthrax protective antigen that block translocation. J. Biol. Chem. 2001, 276, 8371–8376. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.L. Three-Dimensional model of the pore form of anthrax protective antigen. Structure and biological implications. J. Biomol. Struct. Dyn. 2004, 22, 253–265. [Google Scholar]
- Krantz, B.A.; Trivedi, A.D.; Cunningham, K.; Christensen, K.A.; Collier, R.J. Acid-Induced unfolding of the amino-terminal domains of the lethal and edema factors of anthrax toxin. J. Mol. Biol. 2004, 344, 739–756. [Google Scholar] [CrossRef] [PubMed]
- Pentelute, B.L.; Sharma, O.; Collier, R.J. Chemical dissection of protein translocation through the anthrax toxin pore. Angew. Chem. Int. Ed. Engl. 2011, 50, 2294–2296. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Moscoso, A.; Mendez-Ardoy, A.; Ortega-Caballero, F.; Benito, J.M.; Ortiz Mellet, C.; Defaye, J.; Robinson, T.M.; Yohannes, A.; Karginov, V.A.; Garcia Fernandez, J.M. Symmetry complementarity-guided design of anthrax toxin inhibitors based on beta-cyclodextrin: synthesis and relative activities of face-selective functionalized polycationic clusters. Chem. Med. Chem. 2011, 6, 181–192. [Google Scholar]
- Sellman, B.R.; Mourez, M.; Collier, R.J. Dominant-Negative mutants of a toxin subunit: An approach to therapy of anthrax. Science 2001, 292, 695–697. [Google Scholar] [CrossRef] [PubMed]
- Singh, Y.; Khanna, H.; Chopra, A.P.; Mehra, V. A dominant negative mutant of bacillus anthracis protective antigen inhibits anthrax toxin action in vivo. J. Biol. Chem. 2001, 276, 22090–22094. [Google Scholar] [CrossRef] [PubMed]
- Yan, M.; Collier, R.J. Characterization of dominant-negative forms of anthrax protective antigen. Mol. Med. 2003, 9, 46–51. [Google Scholar] [PubMed]
- Cao, S.; Guo, A.; Liu, Z.; Tan, Y.; Wu, G.; Zhang, C.; Zhao, Y.; Chen, H. Investigation of new dominant-negative inhibitors of anthrax protective antigen mutants for use in therapy and vaccination. Infect. Immun. 2009, 77, 4679–4687. [Google Scholar] [CrossRef] [PubMed]
- Krogsgaard-Larsen, P.; Strømgaard, K.; Madsen, U. (Eds.) Textbook of Drug Design and Discovery; CRC Press LLC, Taylor & Francis Group: New York, NY, USA, 2010.
- Panchal, R.G. Novel therapeutic strategies to selectively kill cancer cells. Biochem. Pharmacol. 1998, 55, 247–252. [Google Scholar] [CrossRef] [PubMed]
- Yamaizumi, M.; Mekada, E.; Uchida, T.; Okada, Y. One molecule of diphtheria toxin fragment A introduced into a cell can kill the cell. Cell 1978, 15, 245–250. [Google Scholar] [CrossRef] [PubMed]
- Mathew, M.; Verma, R.S. Humanized immunotoxins: A new generation of immunotoxins for targeted cancer therapy. Cancer Sci. 2009, 100, 1359–1365. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.M.; Hall, W.A. Targeted toxins in brain tumor therapy. Toxins 2010, 2, 2645–2662. [Google Scholar] [CrossRef] [PubMed]
- Shapira, A.; Benhar, I. Toxin-Based therapeutic approaches. Toxins 2010, 2, 2519–2583. [Google Scholar] [CrossRef] [PubMed]
- Iyer, U.; Kadambi, V.J. Antibody drug conjugates—Trojan horses in the war on cancer. J. Pharmacol. Toxicol. Methods 2011, 64, 207–212. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, S.; Mathew, M.; Verma, R.S. Therapeutic potential of anticancer immunotoxins. Drug Discov. Today 2011, 16, 495–503. [Google Scholar] [CrossRef] [PubMed]
- Weldon, J.E.; Pastan, I. A guide to taming a toxin—Recombinant immunotoxins constructed from pseudomonas exotoxin A for the treatment of cancer. FEBS J. 2011, 278, 4683–4700. [Google Scholar] [CrossRef] [PubMed]
- Adkins, I.; Holubova, J.; Kosova, M.; Sadilkova, L. Bacteria and their toxins tamed for immunotherapy. Curr. Pharm. Biotechnol. 2012, 13, 1446–1473. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.M.; Vallera, D.A.; Hall, W.A. Diphtheria toxin-based targeted toxin therapy for brain tumors. J. Neurooncol. 2013, 114, 155–164. [Google Scholar] [CrossRef] [PubMed]
- Antignani, A.; Fitzgerald, D. Immunotoxins: The role of the toxin. Toxins 2013, 5, 1486–1502. [Google Scholar] [CrossRef] [PubMed]
- Dosio, F.; Stella, B.; Cerioni, S.; Gastaldi, D.; Arpicco, S. Advances in anticancer antibody-drug conjugates and immunotoxins. Recent. Pat. Anticancer Drug Discov. 2014, 9, 35–65. [Google Scholar] [PubMed]
- Weidle, U.H.; Tiefenthaler, G.; Schiller, C.; Weiss, E.H.; Georges, G.; Brinkmann, U. Prospects of bacterial and plant protein-based immunotoxins for treatment of cancer. Cancer Genomics Proteomics 2014, 11, 25–38. [Google Scholar] [PubMed]
- Frankel, A.E.; Powell, B.L.; Duesbery, N.S.; Vande Woude, G.F.; Leppla, S.H. Anthrax fusion protein therapy of cancer. Curr. Protein Pept. Sci. 2002, 3, 399–407. [Google Scholar] [CrossRef] [PubMed]
- Varughese, M.; Chi, A.; Teixeira, A.V.; Nicholls, P.J.; Keith, J.M.; Leppla, S.H. Internalization of a bacillus anthracis protective Antigen-C-Myc fusion protein mediated by cell surface Anti-C-Myc antibodies. Mol. Med. 1998, 4, 87–95. [Google Scholar] [PubMed]
- Liu, S.; Schubert, R.L.; Bugge, T.H.; Leppla, S.H. Anthrax toxin: Structures, functions and tumour targeting. Expert Opin. Biol. Ther. 2003, 3, 843–853. [Google Scholar] [CrossRef] [PubMed]
- Mechaly, A.; McCluskey, A.J.; Collier, R.J. Changing the receptor specificity of anthrax toxin. MBio 2012, 3. [Google Scholar] [CrossRef]
- McCluskey, A.J.; Olive, A.J.; Starnbach, M.N.; Collier, R.J. Targeting HER2-Positive cancer cells with receptor-redirected anthrax protective antigen. Mol. Oncol. 2013, 7, 440–451. [Google Scholar] [CrossRef] [PubMed]
- McCluskey, A.J.; Collier, R.J. Receptor-Directed chimeric toxins created by sortase-mediated protein fusion. Mol. Cancer. Ther. 2013, 12, 2273–2281. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Netzel-Arnett, S.; Birkedal-Hansen, H.; Leppla, S.H. Tumor cell-selective cytotoxicity of matrix metalloproteinase-activated anthrax toxin. Cancer Res. 2000, 60, 6061–6067. [Google Scholar] [PubMed]
- Koo, H.M.; VanBrocklin, M.; McWilliams, M.J.; Leppla, S.H.; Duesbery, N.S.; Vande Woude, G.F. Apoptosis and melanogenesis in human melanoma cells induced by anthrax lethal factor inactivation of mitogen-activated protein kinase kinase. Proc. Natl. Acad. Sci. USA 2002, 99, 3052–3057. [Google Scholar] [CrossRef] [PubMed]
- Alfano, R.W.; Leppla, S.H.; Liu, S.; Bugge, T.H.; Duesbery, N.S.; Frankel, A.E. Potent inhibition of tumor angiogenesis by the matrix metalloproteinase-activated anthrax lethal toxin: Implications for broad anti-tumor efficacy. Cell Cycle 2008, 7, 745–749. [Google Scholar] [CrossRef] [PubMed]
- Alfano, R.W.; Leppla, S.H.; Liu, S.; Bugge, T.H.; Ortiz, J.M.; Lairmore, T.C.; Duesbery, N.S.; Mitchell, I.C.; Nwariaku, F.; Frankel, A.E. Inhibition of tumor angiogenesis by the matrix metalloproteinase-activated anthrax lethal toxin in an orthotopic model of anaplastic thyroid carcinoma. Mol. Cancer Ther. 2010, 9, 190–201. [Google Scholar] [CrossRef] [PubMed]
- Schafer, J.M.; Peters, D.E.; Morley, T.; Liu, S.; Molinolo, A.A.; Leppla, S.H.; Bugge, T.H. Efficient targeting of head and neck squamous cell carcinoma by systemic administration of a dual uPA and MMP-Activated engineered anthrax toxin. PLoS One 2011, 6. [Google Scholar] [CrossRef]
- Liu, S.; Bugge, T.H.; Leppla, S.H. Targeting of tumor cells by cell surface urokinase plasminogen activator-dependent anthrax toxin. J. Biol. Chem. 2001, 276, 17976–17984. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Aaronson, H.; Mitola, D.J.; Leppla, S.H.; Bugge, T.H. Potent antitumor activity of a urokinase-activated engineered anthrax toxin. Proc. Natl. Acad. Sci. USA 2003, 100, 657–662. [Google Scholar] [CrossRef] [PubMed]
- Rono, B.; Romer, J.; Liu, S.; Bugge, T.H.; Leppla, S.H.; Kristjansen, P.E. Antitumor efficacy of a urokinase activation-dependent anthrax toxin. Mol. Cancer. Ther. 2006, 5, 89–96. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.; Ortiz, J.; Liu, S.; Bugge, T.H.; Singh, R.; Leppla, S.H.; Frankel, A.E. Systematic urokinase-activated anthrax toxin therapy produces regressions of subcutaneous human non-small cell lung tumor in athymic nude mice. Cancer Res. 2007, 67, 3329–3336. [Google Scholar] [CrossRef] [PubMed]
- Wein, A.N.; Liu, S.; Zhang, Y.; McKenzie, A.T.; Leppla, S.H. Tumor therapy with a urokinase plasminogen activator-activated anthrax lethal toxin alone and in combination with paclitaxel. Invest. New Drugs 2013, 31, 206–212. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Redeye, V.; Kuremsky, J.G.; Kuhnen, M.; Molinolo, A.; Bugge, T.H.; Leppla, S.H. Intermolecular complementation achieves high-specificity tumor targeting by anthrax toxin. Nat. Biotechnol. 2005, 23, 725–730. [Google Scholar] [CrossRef] [PubMed]
- Phillips, D.D.; Fattah, R.J.; Crown, D.; Zhang, Y.; Liu, S.; Moayeri, M.; Fischer, E.R.; Hansen, B.T.; Ghirlando, R.; Nestorovich, E.M.; et al. Engineering anthrax toxin variants that exclusively form octamers and their application to targeting tumors. J. Biol. Chem. 2013, 288, 9058–9065. [Google Scholar] [CrossRef] [PubMed]
- Feld, G.K.; Kintzer, A.F.; Tang, I.I.; Thoren, K.L.; Krantz, B.A. Domain flexibility modulates the heterogeneous assembly mechanism of anthrax toxin protective antigen. J. Mol. Biol. 2012, 415, 159–174. [Google Scholar] [CrossRef] [PubMed]
- Arora, N.; Klimpel, K.R.; Singh, Y.; Leppla, S.H. Fusions of anthrax toxin lethal factor to the ADP-Ribosylation domain of pseudomonas exotoxin A are potent cytotoxins which are translocated to the cytosol of mammalian cells. J. Biol. Chem. 1992, 267, 15542–15548. [Google Scholar] [PubMed]
- Arora, N.; Leppla, S.H. Fusions of anthrax toxin lethal factor with shiga toxin and diphtheria toxin enzymatic domains are toxic to mammalian cells. Infect. Immun. 1994, 62, 4955–4961. [Google Scholar] [PubMed]
- Milne, J.C.; Blanke, S.R.; Hanna, P.C.; Collier, R.J. Protective antigen-binding domain of anthrax lethal factor mediates translocation of a heterologous protein fused to its Amino- or carboxy-terminus. Mol. Microbiol. 1995, 15, 661–666. [Google Scholar] [CrossRef] [PubMed]
- Arora, N.; Williamson, L.C.; Leppla, S.H.; Halpern, J.L. Cytotoxic effects of a chimeric protein consisting of tetanus toxin light chain and anthrax toxin lethal factor in non-neuronal cells. J. Biol. Chem. 1994, 269, 26165–26171. [Google Scholar] [PubMed]
- Spyres, L.M.; Qa’Dan, M.; Meader, A.; Tomasek, J.J.; Howard, E.W.; Ballard, J.D. Cytosolic delivery and characterization of the TcdB glucosylating domain by using a heterologous protein fusion. Infect. Immun. 2001, 69, 599–601. [Google Scholar] [CrossRef] [PubMed]
- Bachran, C.; Hasikova, R.; Leysath, C.E.; Sastalla, I.; Zhang, Y.; Fattah, R.J.; Liu, S.; Leppla, S.H. Cytolethal distending toxin B as a cell-killing component of tumor-targeted anthrax toxin fusion proteins. Cell Death Dis. 2014, 5. [Google Scholar] [CrossRef]
- Liu, X.H.; Collier, R.J.; Youle, R.J. Inhibition of axotomy-induced neuronal apoptosis by extracellular delivery of a Bcl-XL fusion protein. J. Biol. Chem. 2001, 276, 46326–46332. [Google Scholar] [CrossRef] [PubMed]
- Von Moltke, J.; Trinidad, N.J.; Moayeri, M.; Kintzer, A.F.; Wang, S.B.; van Rooijen, N.; Brown, C.R.; Krantz, B.A.; Leppla, S.H.; Gronert, K.; et al. Rapid induction of inflammatory lipid mediators by the inflammasome in vivo. Nature 2012, 490, 107–111. [Google Scholar]
- Hobson, J.P.; Liu, S.; Rono, B.; Leppla, S.H.; Bugge, T.H. Imaging specific cell-surface proteolytic activity in single living cells. Nat. Methods 2006, 3, 259–261. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Leppla, S.H. Anthrax toxin uptake by primary immune cells as determined with a lethal factor-beta-lactamase fusion protein. PLoS One 2009, 4. [Google Scholar] [CrossRef]
- Wesche, J.; Elliott, J.L.; Falnes, P.O.; Olsnes, S.; Collier, R.J. Characterization of membrane translocation by anthrax protective antigen. Biochemistry 1998, 37, 15737–15746. [Google Scholar] [CrossRef] [PubMed]
- Bachran, C.; Morley, T.; Abdelazim, S.; Fattah, R.J.; Liu, S.; Leppla, S.H. Anthrax toxin-mediated delivery of the pseudomonas exotoxin A enzymatic domain to the cytosol of tumor cells via cleavable ubiquitin fusions. MBio 2013, 4. [Google Scholar] [CrossRef]
- London, E.; Luongo, C.L. Domain-Specific bias in arginine/lysine usage by protein toxins. Biochem. Biophys. Res. Commun. 1989, 160, 333–339. [Google Scholar] [CrossRef] [PubMed]
- Falnes, P.O.; Olsnes, S. Modulation of the intracellular stability and toxicity of diphtheria toxin through degradation by the N-End rule pathway. EMBO J. 1998, 17, 615–625. [Google Scholar] [CrossRef] [PubMed]
- Gupta, P.K.; Moayeri, M.; Crown, D.; Fattah, R.J.; Leppla, S.H. Role of N-Terminal amino acids in the potency of anthrax lethal factor. PLoS One 2008, 3. [Google Scholar] [CrossRef]
- Bachmair, A.; Finley, D.; Varshavsky, A. In vivo half-life of a protein is a function of its amino-terminal residue. Science 1986, 234, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Bachran, C.; Gupta, P.K.; Bachran, S.; Leysath, C.E.; Hoover, B.; Fattah, R.J.; Leppla, S.H. Reductive methylation and mutation of an anthrax toxin fusion protein modulates its stability and cytotoxicity. Sci. Rep. 2014, 4. [Google Scholar] [CrossRef]
- Walker, B.; Bayley, H. A pore-forming protein with a protease-activated trigger. Protein Eng. 1994, 7, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Bayley, H. Triggers and switches in a self-assembling pore-forming protein. J. Cell. Biochem. 1994, 56, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Panchal, R.G.; Cusack, E.; Cheley, S.; Bayley, H. Tumor protease-activated, pore-forming toxins from a combinatorial library. Nat. Biotechnol. 1996, 14, 852–856. [Google Scholar] [CrossRef] [PubMed]
- Mizuki, E.; Park, Y.S.; Saitoh, H.; Yamashita, S.; Akao, T.; Higuchi, K.; Ohba, M. Parasporin, a human leukemic cell-recognizing parasporal protein of bacillus thuringiensis. Clin. Diagn. Lab. Immunol. 2000, 7, 625–634. [Google Scholar] [PubMed]
- Chan, K.K.; Wong, R.S.; Mohamed, S.M.; Ibrahim, T.A.; Abdullah, M.; Nadarajah, V.D. Bacillus thuringiensis parasporal proteins induce cell-cycle arrest and caspase-dependant apoptotic cell death in leukemic cells. J. Environ. Pathol. Toxicol. Oncol. 2012, 31, 75–86. [Google Scholar] [CrossRef] [PubMed]
- Ohba, M.; Mizuki, E.; Uemori, A. Parasporin, a new anticancer protein group from bacillus thuringiensis. Anticancer Res. 2009, 29, 427–433. [Google Scholar] [PubMed]
- Uemori, A.; Ohgushi, A.; Yasutake, K.; Maeda, M.; Mizuki, E.; Ohba, M. Parasporin-1Ab, a novel bacillus thuringiensis cytotoxin preferentially active on human cancer cells in vitro. Anticancer Res. 2008, 28, 91–95. [Google Scholar] [PubMed]
- Bergelt, S.; Frost, S.; Lilie, H. Listeriolysin O as cytotoxic component of an immunotoxin. Protein Sci. 2009, 18, 1210–1220. [Google Scholar] [CrossRef] [PubMed]
- Williams, S.A.; Merchant, R.F.; Garrett-Mayer, E.; Isaacs, J.T.; Buckley, J.T.; Denmeade, S.R. A prostate-specific antigen-activated channel-forming toxin as therapy for prostatic disease. J. Natl. Cancer Inst. 2007, 99, 376–385. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Browning, J.L.; Abi-Habib, R.; Wong, K.; Williams, S.A.; Merchant, R.; Denmeade, S.R.; Buckley, T.J.; Frankel, A.E. Recombinant prostate-specific antigen proaerolysin shows selective protease sensitivity and cell cytotoxicity. Anticancer Drugs 2007, 18, 809–816. [Google Scholar] [CrossRef] [PubMed]
- Bu, S.; Xie, Q.; Chang, W.; Huo, X.; Chen, F.; Ma, X. LukS-PV induces mitochondrial-mediated apoptosis and G0/G1 cell cycle arrest in human acute myeloid leukemia THP-1 cells. Int. J. Biochem. Cell Biol. 2013, 45, 1531–1537. [Google Scholar] [PubMed]
- Shekarsaraei, A.G.; Hasannia, S.; Pirooznia, N.; Ataiee, F. The investigation of epsilon toxin effects on different cancerous cell lines and its synergism effect with methotrexate. J. Cancer Res. Ther. 2014, 10, 15–20. [Google Scholar] [CrossRef] [PubMed]
- Ward, R.J.; Palmer, M.; Leonard, K.; Bhakdi, S. Identification of a putative membrane-inserted segment in the alpha-toxin of staphylococcus aureus. Biochemistry 1994, 33, 7477–7484. [Google Scholar] [CrossRef] [PubMed]
- Walker, B.; Kasianowicz, J.; Krishnasastry, M.; Bayley, H. A pore-forming protein with a metal-actuated switch. Protein Eng. 1994, 7, 655–662. [Google Scholar] [CrossRef] [PubMed]
- Valeva, A.; Weisser, A.; Walker, B.; Kehoe, M.; Bayley, H.; Bhakdi, S.; Palmer, M. Molecular architecture of a toxin pore: A 15-Residue sequence lines the transmembrane channel of staphylococcal alpha-toxin. EMBO J. 1996, 15, 1857–1864. [Google Scholar] [PubMed]
- Gondi, C.S.; Rao, J.S. Cathepsin B as a cancer target. Expert Opin. Ther. Targets 2013, 17, 281–291. [Google Scholar] [CrossRef] [PubMed]
- Provoda, C.J.; Lee, K.D. Bacterial pore-forming hemolysins and their use in the cytosolic delivery of macromolecules. Adv. Drug Deliv. Rev. 2000, 41, 209–221. [Google Scholar] [CrossRef] [PubMed]
- Barth, H.; Stiles, B.G. Binary Actin-ADP-Ribosylating toxins and their use as molecular trojan horses for drug delivery into eukaryotic cells. Curr. Med. Chem. 2008, 15, 459–469. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.D.; Oh, Y.K.; Portnoy, D.A.; Swanson, J.A. Delivery of macromolecules into cytosol using liposomes containing hemolysin from listeria monocytogenes. J. Biol. Chem. 1996, 271, 7249–7252. [Google Scholar] [CrossRef] [PubMed]
- Mandal, M.; Lee, K.D. Listeriolysin O-Liposome-Mediated cytosolic delivery of macromolecule antigen in vivo: enhancement of antigen-specific cytotoxic T lymphocyte frequency, activity, and tumor protection. Biochim. Biophys. Acta 2002, 1563, 7–17. [Google Scholar] [CrossRef] [PubMed]
- Saito, G.; Amidon, G.L.; Lee, K.D. Enhanced cytosolic delivery of plasmid DNA by a sulfhydryl-activatable listeriolysin O/Protamine conjugate utilizing cellular reducing potential. Gene Ther. 2003, 10, 72–83. [Google Scholar] [CrossRef] [PubMed]
- Mathew, E.; Hardee, G.E.; Bennett, C.F.; Lee, K.D. Cytosolic delivery of antisense oligonucleotides by listeriolysin O-Containing liposomes. Gene Ther. 2003, 10, 1105–1115. [Google Scholar] [CrossRef] [PubMed]
- Provoda, C.J.; Stier, E.M.; Lee, K.D. Tumor cell killing enabled by listeriolysin O-Liposome-Mediated delivery of the protein toxin gelonin. J. Biol. Chem. 2003, 278, 35102–35108. [Google Scholar] [CrossRef] [PubMed]
- Mandal, M.; Kawamura, K.S.; Wherry, E.J.; Ahmed, R.; Lee, K.D. Cytosolic delivery of viral nucleoprotein by listeriolysin O-Liposome induces enhanced specific cytotoxic T lymphocyte response and protective immunity. Mol. Pharm. 2004, 1, 2–8. [Google Scholar] [CrossRef] [PubMed]
- Walls, Z.F.; Goodell, S.; Andrews, C.D.; Mathis, J.; Lee, K.D. Mutants of listeriolysin O for enhanced liposomal delivery of macromolecules. J. Biotechnol. 2013, 164, 500–502. [Google Scholar] [CrossRef] [PubMed]
- Goletz, T.J.; Klimpel, K.R.; Leppla, S.H.; Keith, J.M.; Berzofsky, J.A. Delivery of antigens to the MHC class i pathway using bacterial toxins. Hum. Immunol. 1997, 54, 129–136. [Google Scholar] [CrossRef] [PubMed]
- Goletz, T.J.; Klimpel, K.R.; Arora, N.; Leppla, S.H.; Keith, J.M.; Berzofsky, J.A. Targeting HIV proteins to the major histocompatibility complex class I processing pathway with a novel gp120-Anthrax toxin fusion protein. Proc. Natl. Acad. Sci. USA 1997, 94, 12059–12064. [Google Scholar] [CrossRef] [PubMed]
- Leppla, S.H.; Arora, N.; Varughese, M. Anthrax toxin fusion proteins for intracellular delivery of macromolecules. J. Appl. Microbiol. 1999, 87. [Google Scholar] [CrossRef]
- Kelkar, D.A.; Chattopadhyay, A. The gramicidin ion channel: A model membrane protein. Biochim. Biophys. Acta 2007, 1768, 2011–2025. [Google Scholar] [CrossRef]
- Bourinbaiar, A.S.; Krasinski, K.; Borkowsky, W. Anti-HIV effect of gramicidin in vitro: Potential for spermicide use. Life Sci. 1994, 54, PL5–PL9. [Google Scholar] [CrossRef] [PubMed]
- Bourinbaiar, A.S.; Lee-Huang, S. Comparative in vitro study of contraceptive agents with Anti-HIV activity: Gramicidin, Nonoxynol-9, and gossypol. Contraception 1994, 49, 131–137. [Google Scholar] [CrossRef] [PubMed]
- Bourinbaiar, A.S.; Lee, C.H. Synergistic effect of gramicidin and EDTA in inhibiting sperm motility and cervical mucus penetration in vitro. Contraception 1996, 54, 367–372. [Google Scholar] [CrossRef] [PubMed]
- Bourinbaiar, A.S.; Coleman, C.F. The effect of gramicidin, a topical contraceptive and antimicrobial agent with Anti-HIV activity, against herpes simplex viruses Type 1 and 2 in vitro. Arch. Virol. 1997, 142, 2225–2235. [Google Scholar] [CrossRef] [PubMed]
- Stoloff, D.H.; Wanunu, M. Recent trends in nanopores for biotechnology. Curr. Opin. Biotechnol. 2013, 24, 699–704. [Google Scholar] [CrossRef] [PubMed]
- Mantri, S.; Sapra, K.T.; Cheley, S.; Sharp, T.H.; Bayley, H. An engineered dimeric protein pore that spans adjacent lipid bilayers. Nat. Commun. 2013, 4. [Google Scholar] [CrossRef]
- Stoddart, D.; Ayub, M.; Hofler, L.; Raychaudhuri, P.; Klingelhoefer, J.W.; Maglia, G.; Heron, A.; Bayley, H. Functional truncated membrane pores. Proc. Natl. Acad. Sci. USA 2014, 111, 2425–2430. [Google Scholar] [CrossRef] [PubMed]
- Bainbridge, G.; Gokce, I.; Lakey, J.H. Voltage gating is a fundamental feature of porin and toxin beta-barrel membrane channels. FEBS Lett. 1998, 431, 305–308. [Google Scholar] [CrossRef] [PubMed]
- Delcour, A.H. Structure and function of pore-forming beta-barrels from bacteria. J. Mol. Microbiol. Biotechnol. 2002, 4, 1–10. [Google Scholar] [PubMed]
- Radjainia, M.; Hyun, J.K.; Leysath, C.E.; Leppla, S.H.; Mitra, A.K. Anthrax toxin-neutralizing antibody reconfigures the protective antigen heptamer into a supercomplex. Proc. Natl. Acad. Sci. USA 2010, 107, 14070–14074. [Google Scholar] [CrossRef] [PubMed]
- Ostroumova, O.S.; Kaulin, Y.A.; Gurnev, P.A.; Schagina, L.V. Effect of agents modifying the membrane dipole potential on properties of syringomycin E channels. Langmuir 2007, 23, 6889–6892. [Google Scholar] [CrossRef] [PubMed]
- Degiacomi, M.T.; Iacovache, I.; Pernot, L.; Chami, M.; Kudryashev, M.; Stahlberg, H.; van der Goot, F.G.; dal Peraro, M. Molecular assembly of the aerolysin pore reveals a swirling membrane-insertion mechanism. Nat. Chem. Biol. 2013, 9, 623–629. [Google Scholar] [PubMed]
- Fennouri, A.; Przybylski, C.; Pastoriza-Gallego, M.; Bacri, L.; Auvray, L.; Daniel, R. Single molecule detection of glycosaminoglycan hyaluronic acid oligosaccharides and depolymerization enzyme activity using a protein nanopore. ACS Nano 2012, 6, 9672–9678. [Google Scholar] [CrossRef] [PubMed]
- Payet, L.; Martinho, M.; Pastoriza-Gallego, M.; Betton, J.M.; Auvray, L.; Pelta, J.; Mathe, J. Thermal unfolding of proteins probed at the single molecule level using nanopores. Anal. Chem. 2012, 84, 4071–4076. [Google Scholar] [CrossRef] [PubMed]
- Merstorf, C.; Cressiot, B.; Pastoriza-Gallego, M.; Oukhaled, A.; Betton, J.M.; Auvray, L.; Pelta, J. Wild type, mutant protein unfolding and phase transition detected by single-nanopore recording. ACS Chem. Biol. 2012, 7, 652–658. [Google Scholar] [CrossRef] [PubMed]
- Pastoriza-Gallego, M.; Rabah, L.; Gibrat, G.; Thiebot, B.; van der Goot, F.G.; Auvray, L.; Betton, J.M.; Pelta, J. Dynamics of unfolded protein transport through an aerolysin pore. J. Am. Chem. Soc. 2011, 133, 2923–2931. [Google Scholar]
- Yuldasheva, L.N.; Merzlyak, P.G.; Zitzer, A.O.; Rodrigues, C.G.; Bhakdi, S.; Krasilnikov, O.V. Lumen geometry of ion channels formed by vibrio cholerae EL Tor cytolysin elucidated by nonelectrolyte exclusion. Biochim. Biophys. Acta 2001, 1512, 53–63. [Google Scholar] [CrossRef] [PubMed]
- Krasilnikov, O.V.; Merzlyak, P.G.; Lima, V.L.; Zitzer, A.O.; Valeva, A.; Yuldasheva, L.N. Pore formation by vibrio cholerae cytolysin requires cholesterol in both monolayers of the target membrane. Biochimie 2007, 89, 271–277. [Google Scholar] [CrossRef] [PubMed]
- Krasilnikov, O.V.; Yuldasheva, L.N. Transmembrane cholesterol migration in planar lipid membranes measured with vibrio cholerae cytolysin as molecular tool. Biochimie 2009, 91, 620–623. [Google Scholar] [CrossRef] [PubMed]
- De, S.; Olson, R. Crystal structure of the vibrio cholerae cytolysin heptamer reveals common features among disparate pore-forming toxins. Proc. Natl. Acad. Sci. USA 2011, 108, 7385–7390. [Google Scholar] [CrossRef] [PubMed]
- Mathe, J.; Aksimentiev, A.; Nelson, D.R.; Schulten, K.; Meller, A. Orientation discrimination of single-stranded DNA inside the alpha-hemolysin membrane channel. Proc. Natl. Acad. Sci. USA 2005, 102, 12377–12382. [Google Scholar] [CrossRef] [PubMed]
- Mereuta, L.; Roy, M.; Asandei, A.; Lee, J.K.; Park, Y.; Andricioaei, I.; Luchian, T. Slowing down single-molecule trafficking through a protein nanopore reveals intermediates for peptide translocation. Sci. Rep. 2014, 4. [Google Scholar] [CrossRef]
- Mahendran, K.R.; Singh, P.R.; Arning, J.; Stolte, S.; Kleinekathofer, U.; Winterhalter, M. Permeation through nanochannels: Revealing fast kinetics. J. Phys. Condens Matter 2010, 22. [Google Scholar] [CrossRef]
- Popoff, M.R. Epsilon toxin: A fascinating pore-forming toxin. FEBS J. 2011, 278, 4602–4615. [Google Scholar] [CrossRef] [PubMed]
- Bokori-Brown, M.; Savva, C.G.; Fernandes da Costa, S.P.; Naylor, C.E.; Basak, A.K.; Titball, R.W. Molecular basis of toxicity of clostridium perfringens epsilon toxin. FEBS J. 2011, 278, 4589–4601. [Google Scholar]
- Stiles, B.G.; Barth, G.; Barth, H.; Popoff, M.R. Clostridium Perfringens epsilon toxin: A malevolent molecule for animals and man? Toxins 2013, 5, 2138–2160. [Google Scholar] [CrossRef] [PubMed]
- Petit, L.; Gibert, M.; Gourch, A.; Bens, M.; Vandewalle, A.; Popoff, M.R. Clostridium perfringens epsilon toxin rapidly decreases membrane barrier permeability of polarized MDCK cells. Cell. Microbiol. 2003, 5, 155–164. [Google Scholar] [CrossRef] [PubMed]
- Knapp, O.; Maier, E.; Benz, R.; Geny, B.; Popoff, M.R. Identification of the channel-forming domain of clostridium perfringens epsilon-toxin (ETX). Biochim. Biophys. Acta 2009, 1788, 2584–2593. [Google Scholar] [CrossRef]
- Nestorovich, E.M.; Karginov, V.A.; Bezrukov, S.M. Polymer partitioning and ion selectivity suggest asymmetrical shape for the membrane pore formed by epsilon toxin. Biophys. J. 2010, 99, 782–789. [Google Scholar] [CrossRef] [PubMed]
- Kronhardt, A.; Rolando, M.; Beitzinger, C.; Stefani, C.; Leuber, M.; Flatau, G.; Popoff, M.R.; Benz, R.; Lemichez, E. Cross-Reactivity of anthrax and C2 toxin: Protective antigen promotes the uptake of botulinum C2I toxin into human endothelial cells. PLoS One 2011, 6. [Google Scholar] [CrossRef]
- Beitzinger, C.; Stefani, C.; Kronhardt, A.; Rolando, M.; Flatau, G.; Lemichez, E.; Benz, R. Role of N-Terminal His6-Tags in binding and efficient translocation of polypeptides into cells using anthrax Protective Antigen (PA). PLoS One 2012, 7. [Google Scholar] [CrossRef]
- Fahrer, J.; Plunien, R.; Binder, U.; Langer, T.; Seliger, H.; Barth, H. Genetically engineered clostridial C2 toxin as a novel delivery system for living mammalian cells. Bioconjug. Chem. 2010, 21, 130–139. [Google Scholar] [CrossRef] [PubMed]
- Fahrer, J.; Rieger, J.; van Zandbergen, G.; Barth, H. The C2-Streptavidin delivery system promotes the uptake of biotinylated molecules in macrophages and T-Leukemia cells. Biol. Chem. 2010, 391, 1315–1325. [Google Scholar] [CrossRef] [PubMed]
- Fahrer, J.; Funk, J.; Lillich, M.; Barth, H. Internalization of biotinylated compounds into cancer cells is promoted by a molecular trojan horse based upon core streptavidin and clostridial C2 toxin. Naunyn Schmiedebergs Arch. Pharmacol. 2011, 383, 263–273. [Google Scholar] [CrossRef] [PubMed]
- Fahrer, J.; Schweitzer, B.; Fiedler, K.; Langer, T.; Gierschik, P.; Barth, H. C2-Streptavidin mediates the delivery of biotin-conjugated tumor suppressor protein p53 into tumor cells. Bioconjug. Chem. 2013, 24, 595–603. [Google Scholar] [CrossRef] [PubMed]
- Fahrer, J.; Rausch, J.; Barth, H. A cell-permeable fusion protein based on clostridium botulinum C2 toxin for delivery of p53 tumorsuppressor into cancer cells. PLoS One 2013, 8. [Google Scholar] [CrossRef]
- Kuan, S.L.; Ng, D.Y.; Wu, Y.; Fortsch, C.; Barth, H.; Doroshenko, M.; Koynov, K.; Meier, C.; Weil, T. pH responsive janus-like supramolecular fusion proteins for functional protein delivery. J. Am. Chem. Soc. 2013, 135, 17254–17257. [Google Scholar] [CrossRef] [PubMed]
- Lang, A.E.; Neumeyer, T.; Sun, J.; Collier, R.J.; Benz, R.; Aktories, K. Amino acid residues involved in membrane insertion and pore formation of clostridium botulinum C2 toxin. Biochemistry 2008, 47, 8406–8413. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, E.; Pust, S.; Kroll, C.; Barth, H. Cyclophilin A facilitates translocation of the clostridium botulinum C2 toxin across membranes of acidified endosomes into the cytosol of mammalian cells. Cell. Microbiol. 2009, 11, 780–795. [Google Scholar] [CrossRef] [PubMed]
- Dmochewitz, L.; Lillich, M.; Kaiser, E.; Jennings, L.D.; Lang, A.E.; Buchner, J.; Fischer, G.; Aktories, K.; Collier, R.J.; Barth, H. Role of CypA and Hsp90 in membrane translocation mediated by anthrax protective antigen. Cell. Microbiol. 2011, 13, 359–373. [Google Scholar] [CrossRef] [PubMed]
- Barth, H. Exploring the role of host cell chaperones/PPIases during cellular Up-Take of bacterial ADP-Ribosylating toxins as basis for novel pharmacological strategies to protect mammalian cells against these virulence factors. Naunyn Schmiedebergs Arch. Pharmacol. 2011, 383, 237–245. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Gurnev, P.A.; Nestorovich, E.M. Channel-Forming Bacterial Toxins in Biosensing and Macromolecule Delivery. Toxins 2014, 6, 2483-2540. https://doi.org/10.3390/toxins6082483
Gurnev PA, Nestorovich EM. Channel-Forming Bacterial Toxins in Biosensing and Macromolecule Delivery. Toxins. 2014; 6(8):2483-2540. https://doi.org/10.3390/toxins6082483
Chicago/Turabian StyleGurnev, Philip A., and Ekaterina M. Nestorovich. 2014. "Channel-Forming Bacterial Toxins in Biosensing and Macromolecule Delivery" Toxins 6, no. 8: 2483-2540. https://doi.org/10.3390/toxins6082483
APA StyleGurnev, P. A., & Nestorovich, E. M. (2014). Channel-Forming Bacterial Toxins in Biosensing and Macromolecule Delivery. Toxins, 6(8), 2483-2540. https://doi.org/10.3390/toxins6082483