Truncated PPM1D Prevents Apoptosis in the Murine Thymus and Promotes Ionizing Radiation-Induced Lymphoma



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Western Blotting
2.3. Flow Cytometry
2.4. Immunohistochemistry
2.5. Real-Time Quantitative Reverse Transcriptase PCR (RT-qPCR)
2.6. Statistical Analysis
3. Results
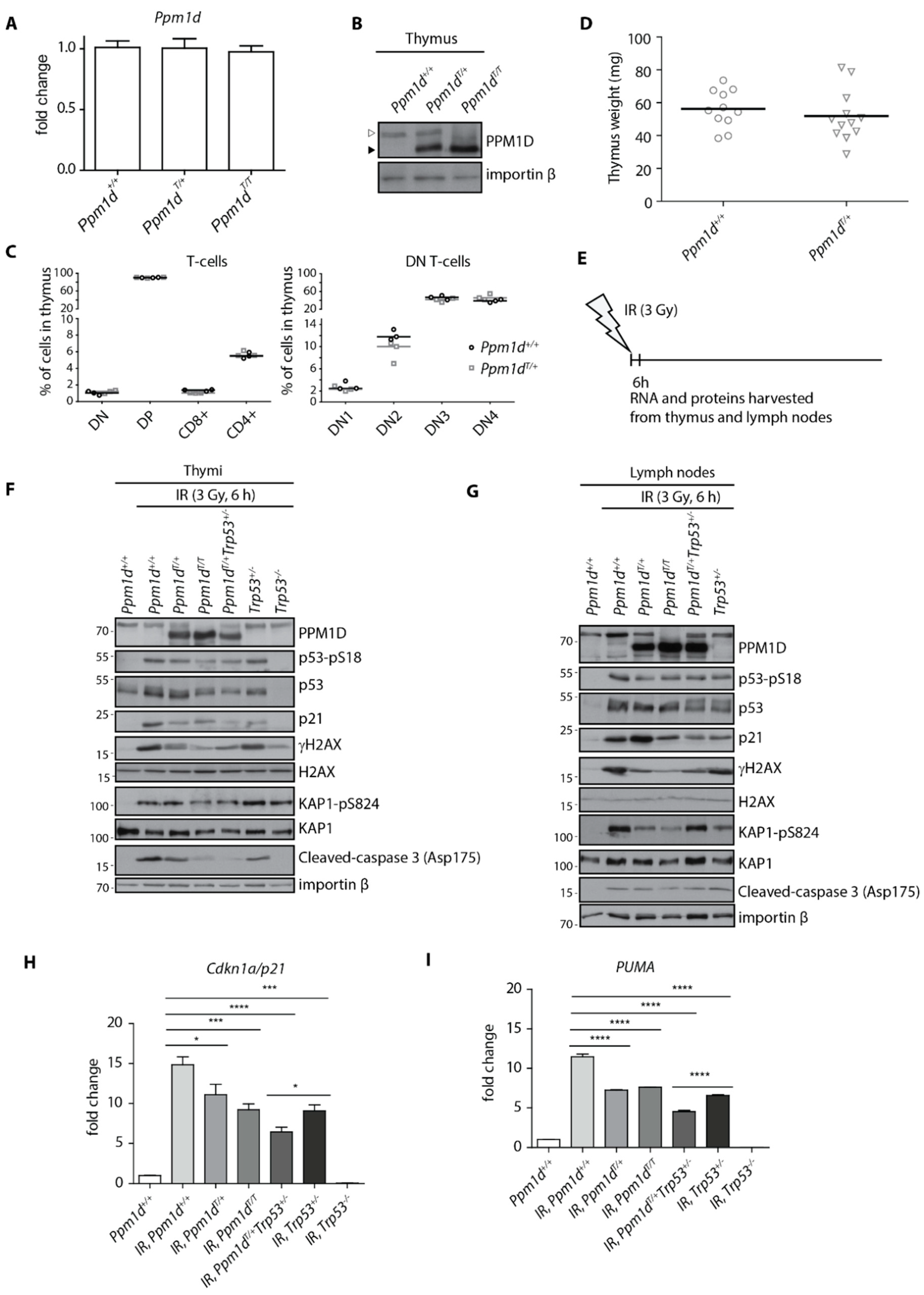
3.1. Mice with Truncated PPM1D Show Impaired Acute DNA-Damage Response (DDR) in the Thymus upon γ-Irradiation
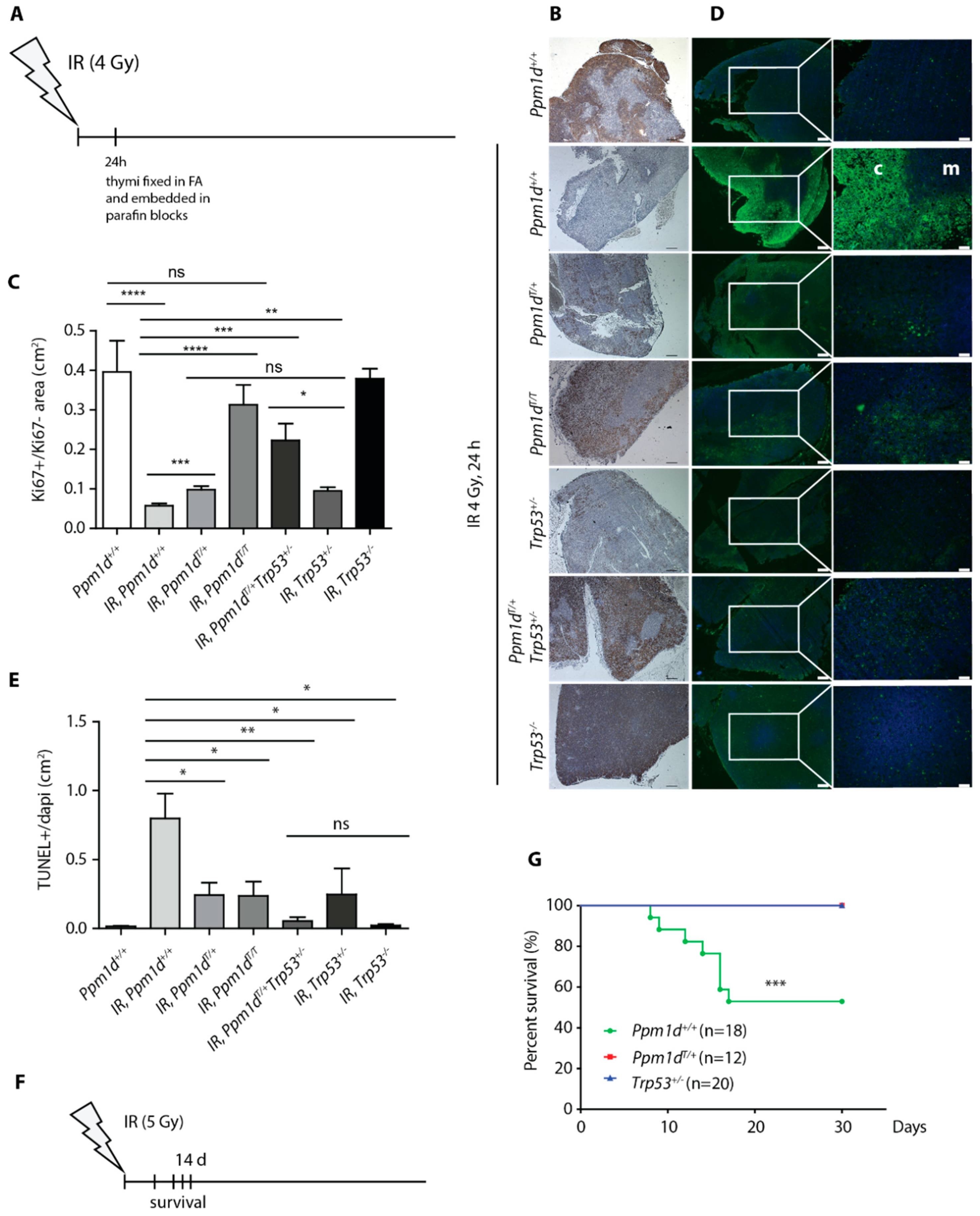
3.2. Truncated PPM1D Prevents Apoptosis and Provides a Proliferation Advantage after Genotoxic Stress
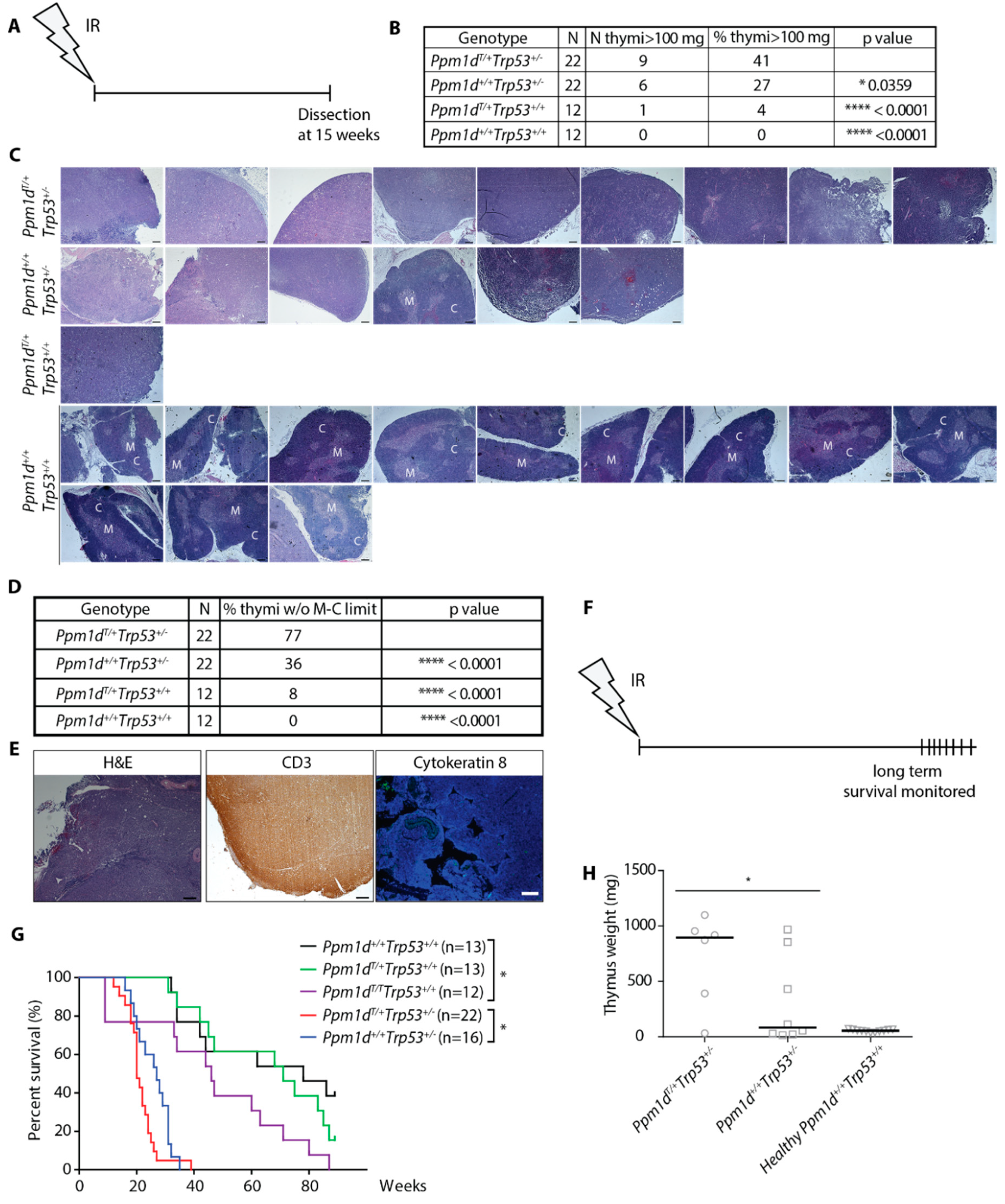
3.3. Truncated PPM1D Promotes Formation of the Ionizing Radiation-Induced Lymphoma
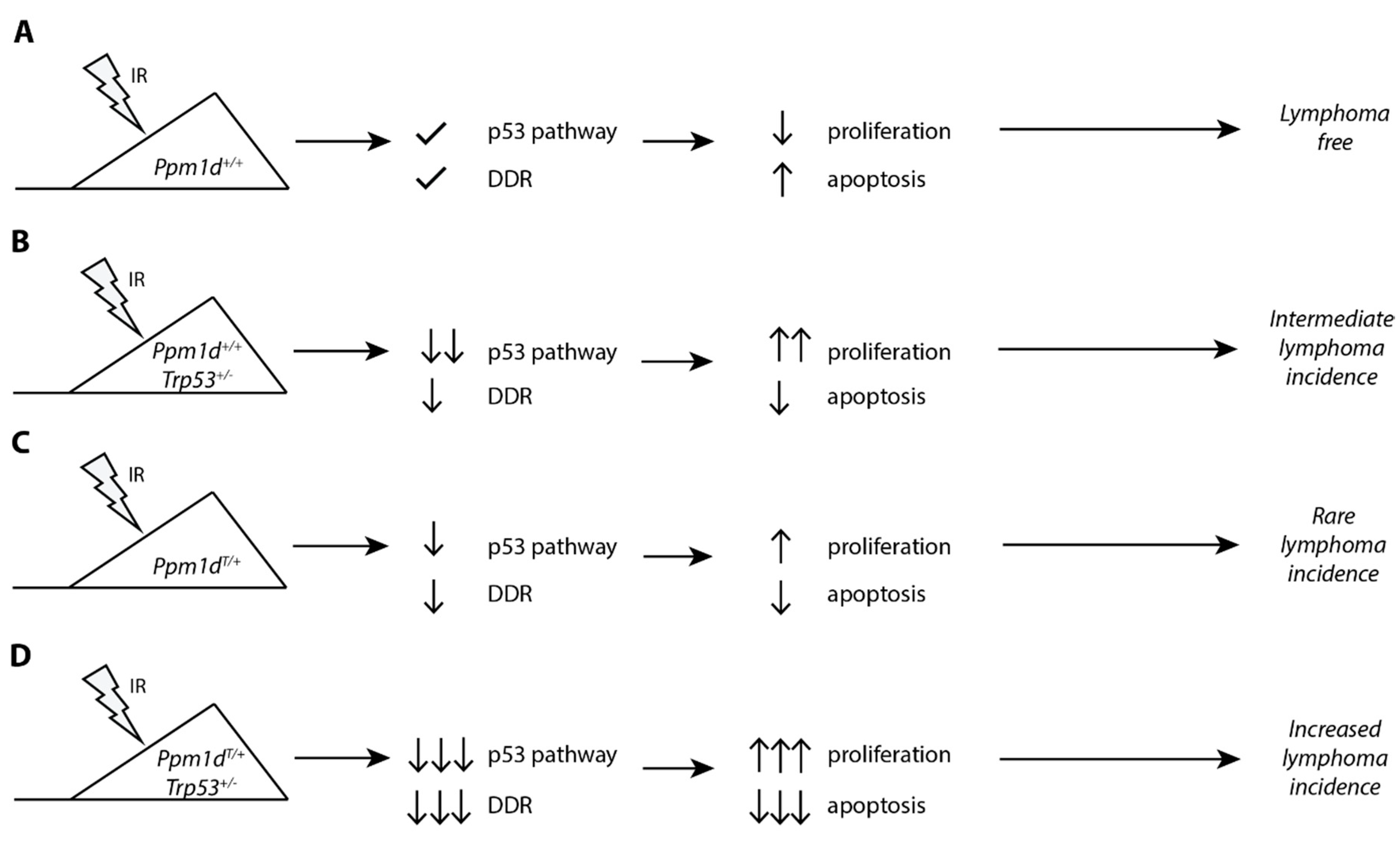
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bartek, J.; Lukas, J. DNA damage checkpoints: From initiation to recovery or adaptation. Curr. Opin. Cell Biol. 2007, 19, 238–245. [Google Scholar] [CrossRef] [PubMed]
- Bartek, J.; Lukas, J. Chk1 and Chk2 kinases in checkpoint control and cancer. Cancer Cell 2003, 3, 421–429. [Google Scholar] [CrossRef] [Green Version]
- Medema, R.H.; Macůrek, L. Checkpoint control and cancer. Oncogene 2012, 31, 2601–2613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soto, M.; Raaijmakers, J.A.; Bakker, B.; Spierings, D.C.J.; Lansdorp, P.M.; Foijer, F.; Medema, R.H. p53 Prohibits Propagation of Chromosome Segregation Errors that Produce Structural Aneuploidies. Cell Rep. 2017, 19, 2423–2431. [Google Scholar] [CrossRef] [Green Version]
- Shieh, S.-Y.; Ikeda, M.; Taya, Y.; Prives, C. DNA Damage-Induced Phosphorylation of p53 Alleviates Inhibition by MDM2. Cell 1997, 91, 325–334. [Google Scholar] [CrossRef] [Green Version]
- Shieh, S.; Ahn, J.; Tamai, K.; Taya, Y.; Prives, C. The human homologs of checkpoint kinases Chk1 and Cds1 (Chk2) phosphorylate p53 at multiple DNA damage-inducible sites. Genes Dev. 2000, 14, 289–300. [Google Scholar]
- el-Deiry, W.S.; Tokino, T.; Velculescu, V.E.; Levy, D.B.; Parsons, R.; Trent, J.M.; Lin, D.; Mercer, W.E.; Kinzler, K.W.; Vogelstein, B. WAF1, a potential mediator of p53 tumor suppression. Cell 1993, 75, 817–825. [Google Scholar] [CrossRef]
- Sørensen, C.S.; Syljuåsen, R.G.; Falck, J.; Schroeder, T.; Rönnstrand, L.; Khanna, K.K.; Zhou, B.B.; Bartek, J.; Lukas, J. Chk1 regulates the S phase checkpoint by coupling the physiological turnover and ionizing radiation-induced accelerated proteolysis of Cdc25A. Cancer Cell 2003, 3, 247–258. [Google Scholar] [CrossRef] [Green Version]
- Bunz, F.; Dutriaux, A.; Lengauer, C.; Waldman, T.; Zhou, S.; Brown, J.P.; Sedivy, J.M.; Kinzler, K.W.; Vogelstein, B. Requirement for p53 and p21 to Sustain G2 Arrest after DNA Damage. Science 1998, 282, 1497–1501. [Google Scholar] [CrossRef]
- Lindqvist, A.; de Bruijn, M.; Macurek, L.; Bras, A.; Mensinga, A.; Bruinsma, W. Wip1 confers G2 checkpoint recovery competence by counteracting p53-dependent transcriptional repression. EMBO J. 2009, 28, 3196–3206. [Google Scholar] [CrossRef] [Green Version]
- Jaiswal, H.; Benada, J.; Müllers, E.; Akopyan, K.; Burdova, K.; Koolmeister, T.; Helleday, T.; Medema, R.H.; Macurek, L.; Lindqvist, A. ATM/Wip1 activities at chromatin control Plk1 re-activation to determine G2 checkpoint duration. EMBO J. 2017, 36, 2161–2176. [Google Scholar] [CrossRef] [PubMed]
- Shreeram, S.; Demidov, O.N.; Hee, W.K.; Yamaguchi, H.; Onishi, N.; Kek, C.; Timofeev, O.N.; Dudgeon, C.; Fornace, A.J.; Anderson, C.W.; et al. Wip1 Phosphatase Modulates ATM-Dependent Signaling Pathways. Mol. Cell 2006, 23, 757–764. [Google Scholar] [CrossRef] [PubMed]
- Fiscella, M.; Zhang, H.; Fan, S.; Sakaguchi, K.; Shen, S.; Mercer, W.E.; Vande Woude, G.F.; O’Connor, P.M.; Appella, E. Wip1, a novel human protein phosphatase that is induced in response to ionizing radiation in a p53-dependent manner. Proc. Natl. Acad. Sci. USA 1997, 94, 6048–6053. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krenning, L.; Feringa, F.M.; Shaltiel, I.A.; van den Berg, J.; Medema, R.H. Transient Activation of p53 in G2 Phase Is Sufficient to Induce Senescence. Mol. Cell 2014, 55, 59–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Müllers, E.; Cascales, H.S.; Jaiswal, H.; Saurin, A.T.; Lindqvist, A. Nuclear translocation of Cyclin B1 marks the restriction point for terminal cell cycle exit in G2 phase. Cell Cycle 2014, 13, 2733–2743. [Google Scholar] [CrossRef] [Green Version]
- Feringa, F.M.; Raaijmakers, J.A.; Hadders, M.A.; Vaarting, C.; Macurek, L.; Heitink, L.; Krenning, L.; Medema, R.H. Persistent repair intermediates induce senescence. Nat. Commun. 2018, 9, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Burdova, K.; Storchova, R.; Palek, M.; Macurek, L. WIP1 Promotes Homologous Recombination and Modulates Sensitivity to PARP Inhibitors. Cells 2019, 8, 1258. [Google Scholar] [CrossRef] [Green Version]
- Purvis, J.E.; Karhohs, K.W.; Mock, C.; Batchelor, E.; Loewer, A.; Lahav, G. p53 dynamics control cell fate. Science 2012, 336, 1440–1444. [Google Scholar] [CrossRef] [Green Version]
- Barr, A.R.; Cooper, S.; Heldt, F.S.; Butera, F.; Stoy, H.; Mansfeld, J.; Novák, B.; Bakal, C. DNA damage during S-phase mediates the proliferation-quiescence decision in the subsequent G1 via p21 expression. Nat. Commun. 2017, 8, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Shaltiel, I.A.; Aprelia, M.; Saurin, A.T.; Chowdhury, D.; Kops, G.J.P.L.; Voest, E.E.; Medema, R.H. Distinct phosphatases antagonize the p53 response in different phases of the cell cycle. Proc. Natl. Acad. Sci. USA 2014, 111, 7313–7318. [Google Scholar] [CrossRef] [Green Version]
- Bartkova, J.; Horejsi, Z.; Koed, K.; Kramer, A.; Tort, F.; Zieger, K.; Guldberg, P.; Sehested, M.; Nesland, J.M.; Lukas, C.; et al. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature 2005, 434, 864–870. [Google Scholar] [CrossRef] [PubMed]
- Bartkova, J.; Rezaei, N.; Liontos, M.; Karakaidos, P.; Kletsas, D.; Issaeva, N.; Vassiliou, L.-V.F.; Kolettas, E.; Niforou, K.; Zoumpourlis, V.C.; et al. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature 2006, 444, 633–637. [Google Scholar] [CrossRef] [PubMed]
- Di Micco, R.; Fumagalli, M.; Cicalese, A.; Piccinin, S.; Gasparini, P.; Luise, C.; Schurra, C.; Garre, M.; Nuciforo, P.G.; Bensimon, A.; et al. Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature 2006, 444, 638–642. [Google Scholar] [CrossRef] [PubMed]
- Bulavin, D.V.; Demidov, O.N.; Saito, S.I.; Kauraniemi, P.; Phillips, C.; Amundson, S.A.; Ambrosino, C.; Sauter, G.; Nebreda, A.R.; Anderson, C.W.; et al. Amplification of PPM1D in human tumors abrogates p53 tumor-suppressor activity. Nat. Genet. 2002, 31, 210–215. [Google Scholar] [CrossRef]
- Emelyanov, A.; Bulavin, D.V. Wip1 phosphatase in breast cancer. Oncogene 2015, 34, 4429–4438. [Google Scholar] [CrossRef] [Green Version]
- Yu, E.; Ahn, Y.S.; Jang, S.J.; Kim, M.J.; Yoon, H.S.; Gong, G.; Choi, J. Overexpression of the wip1 gene abrogates the p38 MAPK/p53/Wip1 pathway and silences p16 expression in human breast cancers. Breast Cancer Res. Treat. 2007, 101, 269–278. [Google Scholar] [CrossRef]
- Bulavin, D.V.; Phillips, C.; Nannenga, B.; Timofeev, O.; Donehower, L.A.; Anderson, C.W.; Appella, E.; Fornace, A.J. Inactivation of the Wip1 phosphatase inhibits mammary tumorigenesis through p38 MAPK-mediated activation of the p16Ink4a-p19Arf pathway. Nat. Genet. 2004, 36, 343–350. [Google Scholar] [CrossRef]
- Demidov, O.N.; Kek, C.; Shreeram, S.; Timofeev, O.; Fornace, A.J.; Appella, E.; Bulavin, D.V. The role of the MKK6//p38 MAPK pathway in Wip1-dependent regulation of ErbB2-driven mammary gland tumorigenesis. Oncogene 2006, 26, 2502–2506. [Google Scholar] [CrossRef] [Green Version]
- Pechackova, S.; Burdova, K.; Benada, J.; Kleiblova, P.; Jenikova, G.; Macurek, L. Inhibition of WIP1 phosphatase sensitizes breast cancer cells to genotoxic stress and to MDM2 antagonist nutlin-3. Oncotarget 2016, 7, 14458–14475. [Google Scholar] [CrossRef] [Green Version]
- Pecháčková, S.; Burdová, K.; Macurek, L. WIP1 phosphatase as pharmacological target in cancer therapy. J. Mol. Med. 2017, 95, 589–599. [Google Scholar] [CrossRef] [Green Version]
- Kleiblova, P.; Shaltiel, I.A.; Benada, J.; Sevčík, J.; Pecháčková, S.; Pohlreich, P.; Voest, E.E.; Dundr, P.; Bartek, J.; Kleibl, Z.; et al. Gain-of-function mutations of PPM1D/Wip1 impair the p53-dependent G1 checkpoint. J. Cell Biol. 2013, 201, 511–521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Chen, L.H.; Wan, H.; Yang, R.; Wang, Z.; Feng, J.; Yang, S.; Jones, S.; Wang, S.; Zhou, W.; et al. Exome sequencing identifies somatic gain-of-function PPM1D mutations in brainstem gliomas. Nat. Genet. 2014, 46, 726–730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruark, E.; Snape, K.; Humburg, P.; Loveday, C.; Bajrami, I.; Brough, R.; Rodrigues, D.N.; Renwick, A.; Seal, S.; Ramsay, E.; et al. Mosaic PPM1D mutations are associated with predisposition to breast and ovarian cancer. Nature 2013, 493, 406–410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kahn, J.D.; Miller, P.G.; Silver, A.J.; Sellar, R.S.; Bhatt, S.; Gibson, C.; McConkey, M.; Adams, D.; Mar, B.; Mertins, P.; et al. PPM1D truncating mutations confer resistance to chemotherapy and sensitivity to PPM1D inhibition in hematopoietic cells. Blood 2018, 132, 1095–1105. [Google Scholar] [CrossRef] [PubMed]
- Burocziova, M.; Burdova, K.; Martinikova, A.S.; Kasparek, P.; Kleiblova, P.; Danielsen, S.A.; Borecka, M.; Jenikova, G.; Janečková, L.; Pavel, J.; et al. Truncated PPM1D impairs stem cell response to genotoxic stress and promotes growth of APC-deficient tumors in the mouse colon. Cell Death Dis. 2019, 10, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Germain, R.N. T-cell development and the CD4–CD8 lineage decision. Nat. Rev. Immunol. 2002, 2, 309–322. [Google Scholar] [CrossRef]
- Ceredig, R.; Rolink, T. A positive look at double-negative thymocytes. Nat. Rev. Immunol. 2002, 2, 888–897. [Google Scholar] [CrossRef]
- Bogue, M.A.; Zhu, C.; Aguilar-Cordova, E.; Donehower, L.A.; Roth, D.B. p53 is required for both radiation-induced differentiation and rescue of V(D)J rearrangement in scid mouse thymocytes. Genes Dev. 1996, 10, 553–565. [Google Scholar] [CrossRef] [Green Version]
- Guidos, C.J.; Williams, C.J.; Grandal, I.; Knowles, G.; Huang, M.T.; Danska, J.S. V(D)J recombination activates a p53-dependent DNA damage checkpoint in scid lymphocyte precursors. Genes Dev. 1996, 10, 2038–2054. [Google Scholar] [CrossRef] [Green Version]
- Jiang, D.; Lenardo, M.J.; Zúñiga-Pflücker, J.C. p53 prevents maturation to the CD4+CD8+ stage of thymocyte differentiation in the absence of T cell receptor rearrangement. J. Exp. Med. 1996, 183, 1923–1928. [Google Scholar] [CrossRef] [Green Version]
- Schito, M.; Demidov, O.; Saito, S.; Ashwell, J.; Appella, E. Wip1 phosphatase-deficient mice exhibit defective T cell maturation due to sustained p53 activation. J. Immunol. 2006, 176, 4818–4825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, L.; Li, H.; Luo, H.; Zhang, L.; Hu, X.; Yang, T.; Sun, C.; Chen, H.; Zhang, L.; Zhao, Y. Phosphatase Wip1 is essential for the maturation and homeostasis of medullary thymic epithelial cells in mice. J. Immunol. 2013, 191, 3210–3220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yi, W.; Hu, X.; Chen, Z.; Liu, L.; Tian, Y.; Chen, H.; Cong, Y.S.; Yang, F.; Zhang, L.; Rudolph, K.L.; et al. Phosphatase Wip1 controls antigen-independent B-cell development in a p53-dependent manner. Blood 2015, 126, 620–628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uyanik, B.; Grigorash, B.B.; Goloudina, A.R.; Demidov, O.N. DNA damage-induced phosphatase Wip1 in regulation of hematopoiesis, immune system and inflammation. Cell Death Discov. 2017, 3, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Jacks, T.; Remington, L.; Williams, B.O.; Schmitt, E.M.; Halachmi, S.; Bronson, R.T.; Weinberg, R.A. Tumor spectrum analysis in p53-mutant mice. Curr. Biol. 1994, 4, 1–7. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [Green Version]
- Enari, M.; Sakahira, H.; Yokoyama, H.; Okawa, K.; Iwamatsu, A.; Nagata, S. A caspase-activated DNase that degrades DNA during apoptosis, and its inhibitor ICAD. Nature 1998, 391, 43–50. [Google Scholar] [CrossRef]
- Gorczyca, W.; Bruno, S.; Darzynkiewicz, R.; Gong, J.; Darzynkiewicz, Z. DNA strand breaks occurring during apoptosis—Their early insitu detection by the terminal deoxynucleotidyl transferase and nick translation assays and prevention by serine protease inhibitors. Int. J. Oncol. 1992, 1, 639–648. [Google Scholar] [CrossRef]
- Zhao, R.; Kaakati, R.; Lee, A.K.; Liu, X.; Li, F.; Li, C.Y. Novel roles of apoptotic caspases in tumor repopulation, epigenetic reprogramming, carcinogenesis, and beyond. Cancer Metastasis Rev. 2018, 37, 227–236. [Google Scholar] [CrossRef]
- Tang, H.L.; Tang, H.M.; Mak, K.H.; Hu, S.; Wang, S.S.; Wong, K.M.; Wong, C.S.; Wu, H.Y.; Law, H.T.; Liu, K.; et al. Cell survival, DNA damage, and oncogenic transformation after a transient and reversible apoptotic response. Mol. Biol. Cell 2012, 23, 2240–2252. [Google Scholar] [CrossRef]
- Kemp, C.J.; Wheldon, T.; Balmain, A. p53-deficient mice are extremely susceptible to radiation-induced tumorigenesis. Nat. Genet. 1994, 8, 66–69. [Google Scholar] [CrossRef] [PubMed]
- Donehower, L.A.; Harvey, M.; Slagle, B.L.; McArthur, M.J.; Montgomery, C.A.; Butel, J.S.; Bradley, A. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature 1992, 356, 215–221. [Google Scholar] [CrossRef]
- Ströbel, P.; Hartmann, E.; Rosenwald, A.; Kalla, J.; Ott, G.; Friedel, G.; Schalke, B.; Kasahara, M.; Tomaru, U.; Marx, A. Corticomedullary differentiation and maturational arrest in thymomas. Histopathology 2014, 64, 557–566. [Google Scholar] [CrossRef] [PubMed]
- Odaka, C.; Loranger, A.; Takizawa, K.; Ouellet, M.; Tremblay, M.J.; Murata, S.; Inoko, A.; Inagaki, M.; Marceau, N. Keratin 8 is required for the maintenance of architectural structure in thymus epithelium. PLoS ONE 2013, 8, e75101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lauder, A.J.; Jolin, H.E.; Smith, P.; van den Berg, J.G.; Jones, A.; Wisden, W.; Smith, K.G.; Dasvarma, A.; Fallon, P.G.; McKenzie, A.N. Lymphomagenesis, hydronephrosis, and autoantibodies result from dysregulation of IL-9 and are differentially dependent on Th2 cytokines. J. Immunol. 2004, 173, 113–122. [Google Scholar] [CrossRef] [Green Version]
- Hsu, J.I.; Dayaram, T.; Tovy, A.; De Braekeleer, E.; Jeong, M.; Wang, F.; Zhang, J.; Heffernan, T.P.; Gera, S.; Kovacs, J.J.; et al. PPM1D Mutations Drive Clonal Hematopoiesis in Response to Cytotoxic Chemotherapy. Cell Stem Cell 2018, 23, 700–713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rauta, J.; Alarmo, E.-L.; Kauraniemi, P.; Karhu, R.; Kuukasjärvi, T.; Kallioniemi, A. The serine-threonine protein phosphatase PPM1D is frequently activated through amplification in aggressive primary breast tumours. Breast Cancer Res. Treat. 2006, 95, 257–263. [Google Scholar] [CrossRef] [PubMed]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martinikova, A.S.; Burocziova, M.; Stoyanov, M.; Macurek, L. Truncated PPM1D Prevents Apoptosis in the Murine Thymus and Promotes Ionizing Radiation-Induced Lymphoma. Cells 2020, 9, 2068. https://doi.org/10.3390/cells9092068
Martinikova AS, Burocziova M, Stoyanov M, Macurek L. Truncated PPM1D Prevents Apoptosis in the Murine Thymus and Promotes Ionizing Radiation-Induced Lymphoma. Cells. 2020; 9(9):2068. https://doi.org/10.3390/cells9092068
Chicago/Turabian StyleMartinikova, Andra S., Monika Burocziova, Miroslav Stoyanov, and Libor Macurek. 2020. "Truncated PPM1D Prevents Apoptosis in the Murine Thymus and Promotes Ionizing Radiation-Induced Lymphoma" Cells 9, no. 9: 2068. https://doi.org/10.3390/cells9092068
APA StyleMartinikova, A. S., Burocziova, M., Stoyanov, M., & Macurek, L. (2020). Truncated PPM1D Prevents Apoptosis in the Murine Thymus and Promotes Ionizing Radiation-Induced Lymphoma. Cells, 9(9), 2068. https://doi.org/10.3390/cells9092068