Cardiovascular Manifestations of COVID-19 Infection



Abstract
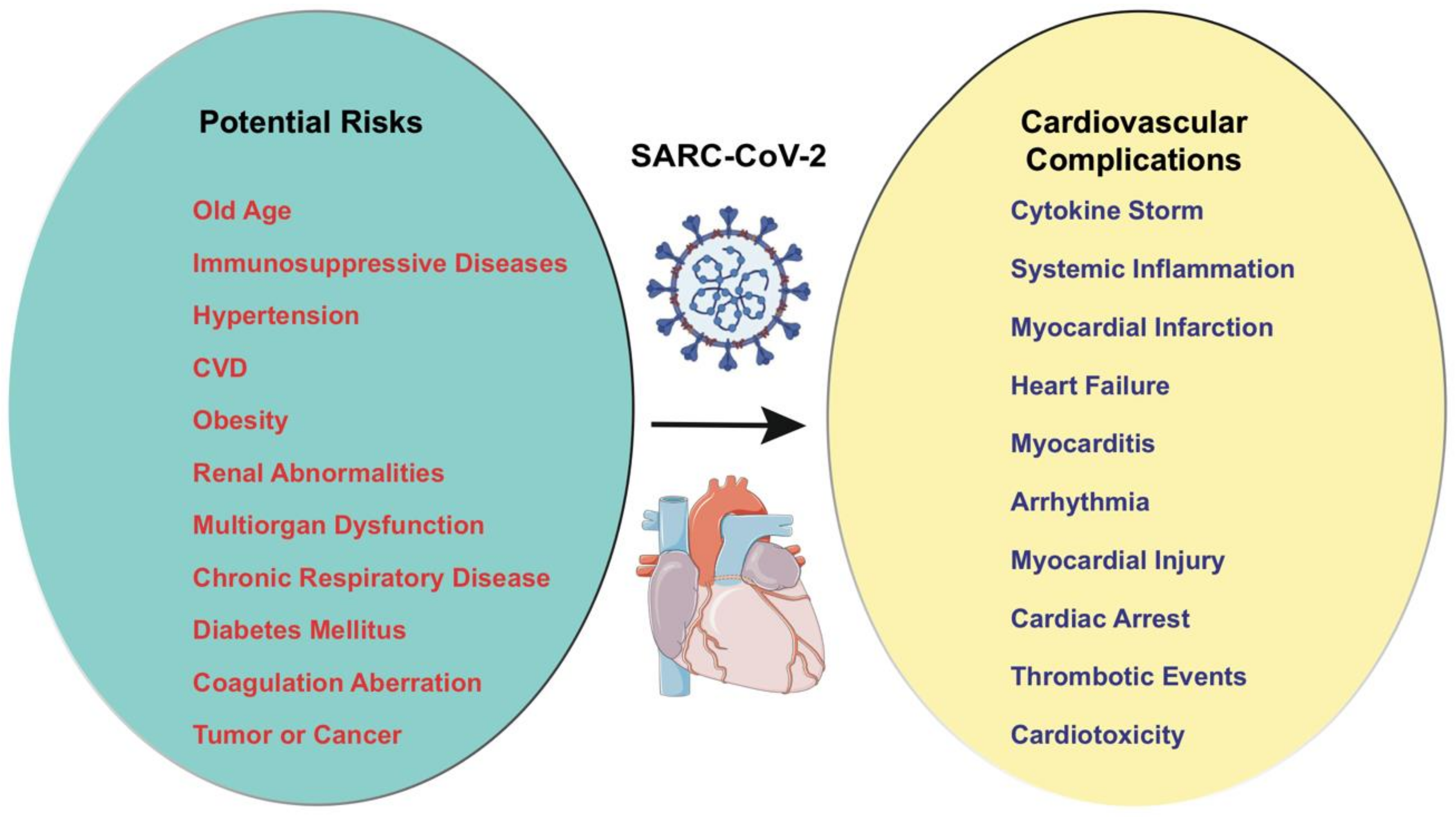
:1. Introduction
2. SARS-CoV-2 Infection
3. COVID-19 in Heart Disease
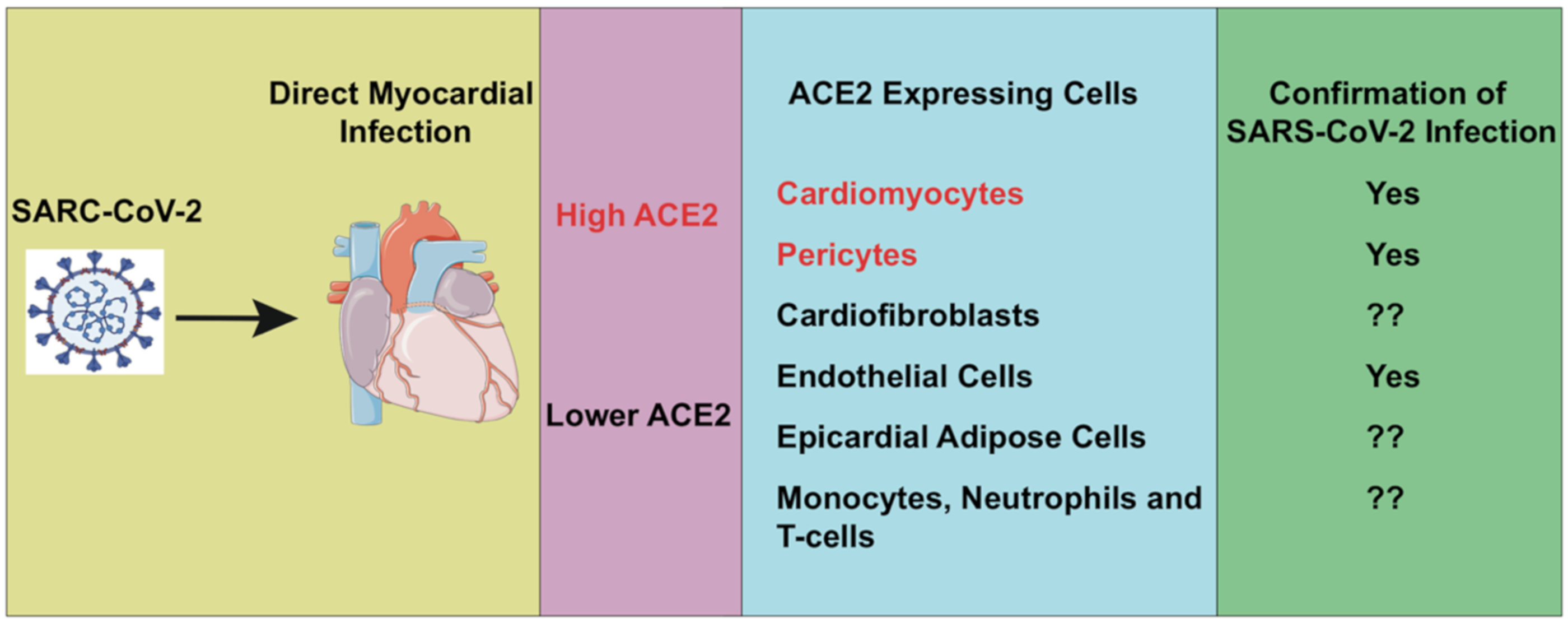
4. ACE2, CVD and COVID-19
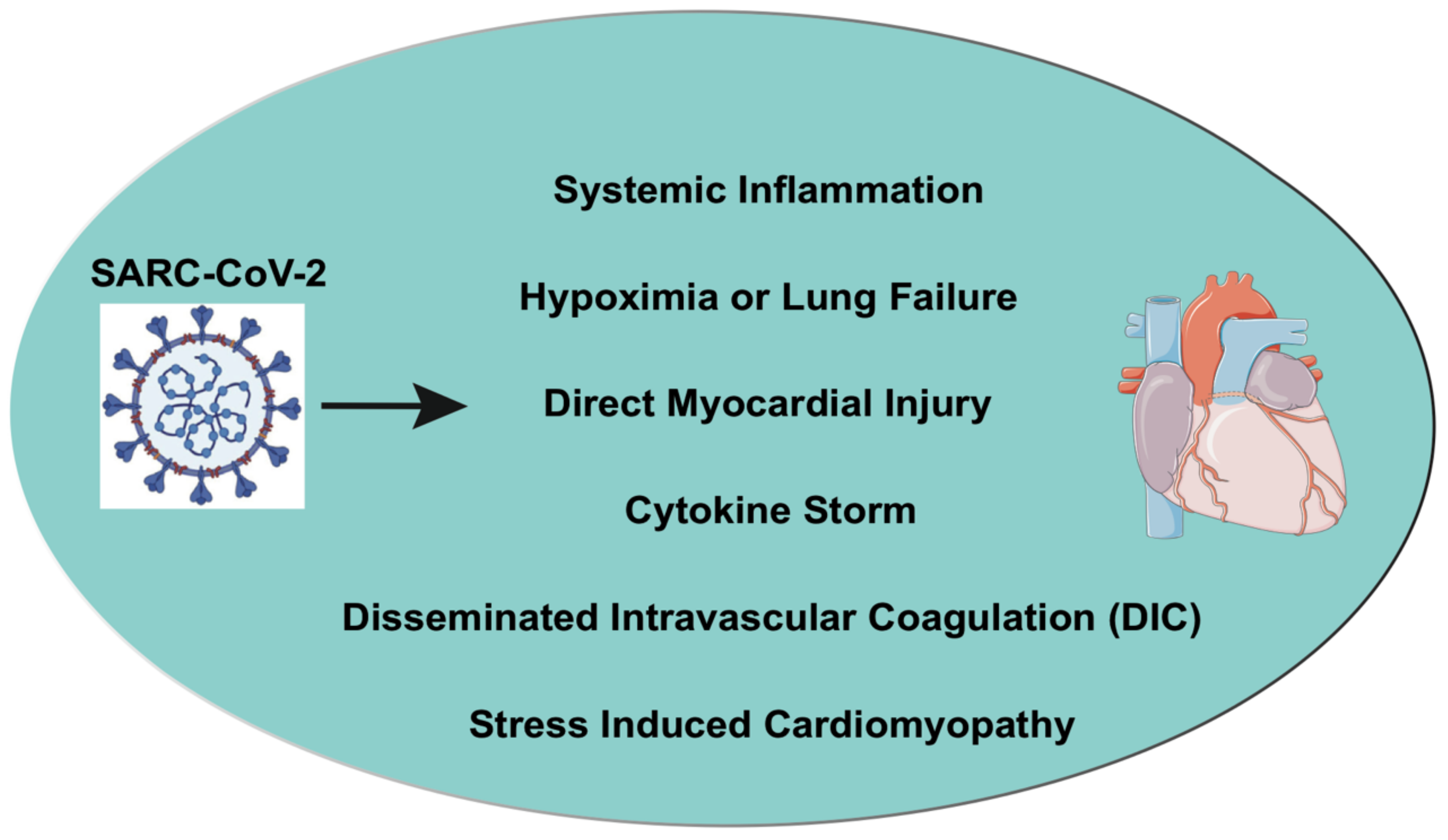
5. Cardiac Injury in COVID-19
6. Endothelial Damage in COVID-19
7. Microvascular Coagulopathy and Platelets in COVID-19
8. Oxidative Stress and Apoptosis in COVID-19
9. Inflammation and Cytokine Storm Induced by COVID-19
10. Limitations of Animal Models
11. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Graham, R.L.; Donaldson, E.F.; Baric, R.S. A decade after SARS: Strategies for controlling emerging coronaviruses. Nat. Rev. Microbiol. 2013, 11, 836–848. [Google Scholar] [CrossRef] [Green Version]
- Su, S.; Wong, G.; Shi, W.; Liu, J.; Lai, A.C.K.; Zhou, J.; Liu, W.; Bi, Y.; Gao, G.F. Epidemiology, Genetic Recombination, and Pathogenesis of Coronaviruses. Trends Microbiol. 2016, 24, 490–502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Hu, B.; Hu, C.; Zhu, F.; Liu, X.; Zhang, J.; Wang, B.; Xiang, H.; Cheng, Z.; Xiong, Y.; et al. Clinical Characteristics of 138 Hospitalized Patients with 2019 Novel Coronavirus-Infected Pneumonia in Wuhan, China. JAMA 2020, 323, 1061. [Google Scholar] [CrossRef]
- Zeng, J.H.; Liu, Y.X.; Yuan, J.; Wang, F.X.; Wu, W.B.; Li, J.X.; Wang, L.F.; Gao, H.; Wang, Y.; Dong, C.F.; et al. First case of COVID-19 complicated with fulminant myocarditis: A case report and insights. Infection 2020, 48, 773–777. [Google Scholar] [CrossRef] [Green Version]
- Zhou, F.; Yu, T.; Du, R.; Fan, G.; Liu, Y.; Liu, Z.; Xiang, J.; Wang, Y.; Song, B.; Gu, X.; et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: A retrospective cohort study. Lancet 2020, 395, 1054–1062. [Google Scholar] [CrossRef]
- Xu, Z.; Shi, L.; Wang, Y.; Zhang, J.; Huang, L.; Zhang, C.; Liu, S.; Zhao, P.; Liu, H.; Zhu, L.; et al. Pathological findings of COVID-19 associated with acute respiratory distress syndrome. Lancet Respir. Med. 2020, 8, 420–422. [Google Scholar] [CrossRef]
- Fang, L.; Karakiulakis, G.; Roth, M. Are patients with hypertension and diabetes mellitus at increased risk for COVID-19 infection? Lancet Respir. Med. 2020, 8, e21. [Google Scholar] [CrossRef]
- Chen, N.; Zhou, M.; Dong, X.; Qu, J.; Gong, F.; Han, Y.; Qiu, Y.; Wang, J.; Liu, Y.; Wei, Y.; et al. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: A descriptive study. Lancet 2020, 395, 507–513. [Google Scholar] [CrossRef] [Green Version]
- Murthy, S.; Gomersall, C.D.; Fowler, R.A. Care for Critically Ill Patients with COVID-19. JAMA 2020, 323, 1499. [Google Scholar] [CrossRef] [Green Version]
- Weiss, P.; Murdoch, D.R. Clinical course and mortality risk of severe COVID-19. Lancet 2020, 395, 1014–1015. [Google Scholar] [CrossRef]
- Donoghue, M.; Hsieh, F.; Baronas, E.; Godbout, K.; Gosselin, M.; Stagliano, N.; Donovan, M.; Woolf, B.; Robison, K.; Jeyaseelan, R.; et al. A novel angiotensin-converting enzyme-related carboxypeptidase (ACE2) converts angiotensin I to angiotensin 1-9. Circ. Res. 2000, 87, E1–E9. [Google Scholar] [CrossRef] [PubMed]
- Nicin, L.; Abplanalp, W.T.; Mellentin, H.; Kattih, B.; Tombor, L.; John, D.; Schmitto, J.D.; Heineke, J.; Emrich, F.; Arsalan, M.; et al. Cell type-specific expression of the putative SARS-CoV-2 receptor ACE2 in human hearts. Eur. Heart J. 2020, 41, 1804–1806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, A.; Peng, Y.; Huang, B.; Ding, X.; Wang, X.; Niu, P.; Meng, J.; Zhu, Z.; Zhang, Z.; Wang, J.; et al. Genome Composition and Divergence of the Novel Coronavirus (2019-nCoV) Originating in China. Cell Host Microbe 2020, 27, 325–328. [Google Scholar] [CrossRef] [Green Version]
- Yan, R.; Zhang, Y.; Li, Y.; Xia, L.; Guo, Y.; Zhou, Q. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science 2020, 367, 1444–1448. [Google Scholar] [CrossRef] [Green Version]
- Shang, J.; Ye, G.; Shi, K.; Wan, Y.; Luo, C.; Aihara, H.; Geng, Q.; Auerbach, A.; Li, F. Structural basis of receptor recognition by SARS-CoV-2. Nature 2020, 581, 221–224. [Google Scholar] [CrossRef] [Green Version]
- Millet, J.K.; Whittaker, G.R. Host cell proteases: Critical determinants of coronavirus tropism and pathogenesis. Virus Res. 2015, 202, 120–134. [Google Scholar] [CrossRef]
- Li, W.; Moore, M.J.; Vasilieva, N.; Sui, J.; Wong, S.K.; Berne, M.A.; Somasundaran, M.; Sullivan, J.L.; Luzuriaga, K.; Greenough, T.C.; et al. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature 2003, 426, 450–454. [Google Scholar] [CrossRef] [Green Version]
- Walls, A.C.; Park, Y.J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell 2020, 181, 281–292.e286. [Google Scholar] [CrossRef]
- Burrell, L.M.; Risvanis, J.; Kubota, E.; Dean, R.G.; MacDonald, P.S.; Lu, S.; Tikellis, C.; Grant, S.L.; Lew, R.A.; Smith, A.I.; et al. Myocardial infarction increases ACE2 expression in rat and humans. Eur. Heart J. 2005, 26, 369–375, discussion 322–364. [Google Scholar] [CrossRef] [Green Version]
- Kassiri, Z.; Zhong, J.; Guo, D.; Basu, R.; Wang, X.; Liu, P.P.; Scholey, J.W.; Penninger, J.M.; Oudit, G.Y. Loss of angiotensin-converting enzyme 2 accelerates maladaptive left ventricular remodeling in response to myocardial infarction. Circ. Heart Fail. 2009, 2, 446–455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertram, S.; Heurich, A.; Lavender, H.; Gierer, S.; Danisch, S.; Perin, P.; Lucas, J.M.; Nelson, P.S.; Pohlmann, S.; Soilleux, E.J. Influenza and SARS-coronavirus activating proteases TMPRSS2 and HAT are expressed at multiple sites in human respiratory and gastrointestinal tracts. PLoS ONE 2012, 7, e35876. [Google Scholar] [CrossRef] [PubMed]
- Iwata-Yoshikawa, N.; Okamura, T.; Shimizu, Y.; Hasegawa, H.; Takeda, M.; Nagata, N. TMPRSS2 Contributes to Virus Spread and Immunopathology in the Airways of Murine Models after Coronavirus Infection. J. Virol. 2019, 93, 93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Y.; Vedantham, P.; Lu, K.; Agudelo, J.; Carrion, R., Jr.; Nunneley, J.W.; Barnard, D.; Pohlmann, S.; McKerrow, J.H.; Renslo, A.R.; et al. Protease inhibitors targeting coronavirus and filovirus entry. Antivir. Res. 2015, 116, 76–84. [Google Scholar] [CrossRef]
- Driggin, E.; Madhavan, M.V.; Bikdeli, B.; Chuich, T.; Laracy, J.; Biondi-Zoccai, G.; Brown, T.S.; Der Nigoghossian, C.; Zidar, D.A.; Haythe, J.; et al. Cardiovascular Considerations for Patients, Health Care Workers, and Health Systems during the COVID-19 Pandemic. J. Am. Coll. Cardiol. 2020, 75, 2352–2371. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; McGoogan, J.M. Characteristics of and Important Lessons from the Coronavirus Disease 2019 (COVID-19) Outbreak in China: Summary of a Report of 72314 Cases from the Chinese Center for Disease Control and Prevention. JAMA 2020, 323, 1239–1242. [Google Scholar] [CrossRef]
- Kucharski, A.J.; Russell, T.W.; Diamond, C.; Liu, Y.; Edmunds, J.; Funk, S.; Eggo, R.M. Centre for Mathematical Modelling of Infectious Diseases COVID-19 Working Group. Early dynamics of transmission and control of COVID-19: A mathematical modelling study. Lancet Infect. Dis 2020, 20, 553–558. [Google Scholar] [CrossRef] [Green Version]
- Guan, W.J.; Ni, Z.Y.; Hu, Y.; Liang, W.H.; Ou, C.Q.; He, J.X.; Liu, L.; Shan, H.; Lei, C.L.; Hui, D.S.C.; et al. Clinical Characteristics of Coronavirus Disease 2019 in China. N. Engl. J. Med. 2020, 382, 1708–1720. [Google Scholar] [CrossRef]
- Pung, R.; Chiew, C.J.; Young, B.E.; Chin, S.; Chen, M.I.; Clapham, H.E.; Cook, A.R.; Maurer-Stroh, S.; Toh, M.; Poh, C.; et al. Investigation of three clusters of COVID-19 in Singapore: Implications for surveillance and response measures. Lancet 2020, 395, 1039–1046. [Google Scholar] [CrossRef]
- Lauer, S.A.; Grantz, K.H.; Bi, Q.; Jones, F.K.; Zheng, Q.; Meredith, H.R.; Azman, A.S.; Reich, N.G.; Lessler, J. The Incubation Period of Coronavirus Disease 2019 (COVID-19) from Publicly Reported Confirmed Cases: Estimation and Application. Ann. Intern. Med. 2020, 172, 577–582. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Guan, X.; Wu, P.; Wang, X.; Zhou, L.; Tong, Y.; Ren, R.; Leung, K.S.M.; Lau, E.H.Y.; Wong, J.Y.; et al. Early Transmission Dynamics in Wuhan, China, of Novel Coronavirus-Infected Pneumonia. N. Engl. J. Med. 2020, 382, 1199–1207. [Google Scholar] [CrossRef]
- Chan, J.F.; Yuan, S.; Kok, K.H.; To, K.K.; Chu, H.; Yang, J.; Xing, F.; Liu, J.; Yip, C.C.; Poon, R.W.; et al. A familial cluster of pneumonia associated with the 2019 novel coronavirus indicating person-to-person transmission: A study of a family cluster. Lancet 2020, 395, 514–523. [Google Scholar] [CrossRef] [Green Version]
- Chen, G.; Wu, D.; Guo, W.; Cao, Y.; Huang, D.; Wang, H.; Wang, T.; Zhang, X.; Chen, H.; Yu, H.; et al. Clinical and immunological features of severe and moderate coronavirus disease 2019. J. Clin. Investig. 2020, 130, 2620–2629. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Yang, Y.; Zhang, C.; Huang, F.; Wang, F.; Yuan, J.; Wang, Z.; Li, J.; Li, J.; Feng, C.; et al. Clinical and biochemical indexes from 2019-nCoV infected patients linked to viral loads and lung injury. Sci. China Life Sci. 2020, 63, 364–374. [Google Scholar] [CrossRef] [Green Version]
- Phan, L.T.; Nguyen, T.V.; Luong, Q.C.; Nguyen, T.V.; Nguyen, H.T.; Le, H.Q.; Nguyen, T.T.; Cao, T.M.; Pham, Q.D. Importation and Human-to-Human Transmission of a Novel Coronavirus in Vietnam. N. Engl. J. Med. 2020, 382, 872–874. [Google Scholar] [CrossRef] [Green Version]
- Pan, Y.; Zhang, D.; Yang, P.; Poon, L.L.M.; Wang, Q. Viral load of SARS-CoV-2 in clinical samples. Lancet Infect. Dis. 2020, 20, 411–412. [Google Scholar] [CrossRef]
- Kim, J.Y.; Ko, J.H.; Kim, Y.; Kim, Y.J.; Kim, J.M.; Chung, Y.S.; Kim, H.M.; Han, M.G.; Kim, S.Y.; Chin, B.S. Viral Load Kinetics of SARS-CoV-2 Infection in First Two Patients in Korea. J. Korean Med. Sci. 2020, 35, e86. [Google Scholar] [CrossRef]
- Zou, L.; Ruan, F.; Huang, M.; Liang, L.; Huang, H.; Hong, Z.; Yu, J.; Kang, M.; Song, Y.; Xia, J.; et al. SARS-CoV-2 Viral Load in Upper Respiratory Specimens of Infected Patients. N. Engl. J. Med. 2020, 382, 1177–1179. [Google Scholar] [CrossRef]
- Li, B.; Yang, J.; Zhao, F.; Zhi, L.; Wang, X.; Liu, L.; Bi, Z.; Zhao, Y. Prevalence and impact of cardiovascular metabolic diseases on COVID-19 in China. Clin. Res. Cardiol. Off. J. Ger. Card. Soc. 2020, 109, 531–538. [Google Scholar] [CrossRef]
- Guo, T.; Fan, Y.; Chen, M.; Wu, X.; Zhang, L.; He, T.; Wang, H.; Wan, J.; Wang, X.; Lu, Z. Cardiovascular Implications of Fatal Outcomes of Patients with Coronavirus Disease 2019 (COVID-19). JAMA Cardiol. 2020, 5, 811–818. [Google Scholar] [CrossRef] [Green Version]
- Shi, S.; Qin, M.; Shen, B.; Cai, Y.; Liu, T.; Yang, F.; Gong, W.; Liu, X.; Liang, J.; Zhao, Q.; et al. Association of Cardiac Injury with Mortality in Hospitalized Patients with COVID-19 in Wuhan, China. JAMA Cardiol. 2020, 5, 802. [Google Scholar] [CrossRef] [Green Version]
- Yao, X.H.; Li, T.Y.; He, Z.C.; Ping, Y.F.; Liu, H.W.; Yu, S.C.; Mou, H.M.; Wang, L.H.; Zhang, H.R.; Fu, W.J.; et al. A pathological report of three COVID-19 cases by minimal invasive autopsies. Zhonghua Bing Li Xue Za Zhi 2020, 49, 411–417. [Google Scholar] [CrossRef]
- Prabhu, S.D. Cytokine-induced modulation of cardiac function. Circ. Res. 2004, 95, 1140–1153. [Google Scholar] [CrossRef]
- Levi, M.; van der Poll, T.; Buller, H.R. Bidirectional relation between inflammation and coagulation. Circulation 2004, 109, 2698–2704. [Google Scholar] [CrossRef] [Green Version]
- Ding, Y.; Wang, H.; Shen, H.; Li, Z.; Geng, J.; Han, H.; Cai, J.; Li, X.; Kang, W.; Weng, D.; et al. The clinical pathology of severe acute respiratory syndrome (SARS): A report from China. J. Pathol. 2003, 200, 282–289. [Google Scholar] [CrossRef]
- Alsaad, K.O.; Hajeer, A.H.; Al Balwi, M.; Al Moaiqel, M.; Al Oudah, N.; Al Ajlan, A.; AlJohani, S.; Alsolamy, S.; Gmati, G.E.; Balkhy, H.; et al. Histopathology of Middle East respiratory syndrome coronovirus (MERS-CoV) infection-clinicopathological and ultrastructural study. Histopathology 2018, 72, 516–524. [Google Scholar] [CrossRef]
- Patel, V.B.; Zhong, J.C.; Grant, M.B.; Oudit, G.Y. Role of the ACE2/Angiotensin 1–7 Axis of the Renin-Angiotensin System in Heart Failure. Circ. Res. 2016, 118, 1313–1326. [Google Scholar] [CrossRef] [Green Version]
- Tipnis, S.R.; Hooper, N.M.; Hyde, R.; Karran, E.; Christie, G.; Turner, A.J. A human homolog of angiotensin-converting enzyme. Cloning and functional expression as a captopril-insensitive carboxypeptidase. J. Biol. Chem. 2000, 275, 33238–33243. [Google Scholar] [CrossRef] [Green Version]
- Zou, X.; Chen, K.; Zou, J.; Han, P.; Hao, J.; Han, Z. Single-cell RNA-seq data analysis on the receptor ACE2 expression reveals the potential risk of different human organs vulnerable to 2019-nCoV infection. Front. Med. 2020, 14, 185–192. [Google Scholar] [CrossRef] [Green Version]
- Crackower, M.A.; Sarao, R.; Oudit, G.Y.; Yagil, C.; Kozieradzki, I.; Scanga, S.E.; Oliveira-dos-Santos, A.J.; da Costa, J.; Zhang, L.; Pei, Y.; et al. Angiotensin-converting enzyme 2 is an essential regulator of heart function. Nature 2002, 417, 822–828. [Google Scholar] [CrossRef]
- Oudit, G.Y.; Kassiri, Z.; Jiang, C.; Liu, P.P.; Poutanen, S.M.; Penninger, J.M.; Butany, J. SARS-coronavirus modulation of myocardial ACE2 expression and inflammation in patients with SARS. Eur. J. Clin. Investig. 2009, 39, 618–625. [Google Scholar] [CrossRef]
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.R.; Zhu, Y.; Li, B.; Huang, C.L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.; Gheblawi, M.; Oudit, G.Y. Angiotensin Converting Enzyme 2: A Double-Edged Sword. Circulation 2020, 142, 426–428. [Google Scholar] [CrossRef]
- Patel, S.K.; Velkoska, E.; Freeman, M.; Wai, B.; Lancefield, T.F.; Burrell, L.M. From gene to protein-experimental and clinical studies of ACE2 in blood pressure control and arterial hypertension. Front. Physiol. 2014, 5, 227. [Google Scholar] [CrossRef]
- Qi, Y.F.; Zhang, J.; Wang, L.; Shenoy, V.; Krause, E.; Oh, S.P.; Pepine, C.J.; Katovich, M.J.; Raizada, M.K. Angiotensin-converting enzyme 2 inhibits high-mobility group box 1 and attenuates cardiac dysfunction post-myocardial ischemia. J. Mol. Med. 2016, 94, 37–49. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Qian, C.; Roks, A.J.; Westermann, D.; Schumacher, S.M.; Escher, F.; Schoemaker, R.G.; Reudelhuber, T.L.; van Gilst, W.H.; Schultheiss, H.P.; et al. Circulating rather than cardiac angiotensin-(1-7) stimulates cardioprotection after myocardial infarction. Circ. Heart Fail. 2010, 3, 286–293. [Google Scholar] [CrossRef] [Green Version]
- Patel, V.B.; Zhong, J.C.; Fan, D.; Basu, R.; Morton, J.S.; Parajuli, N.; McMurtry, M.S.; Davidge, S.T.; Kassiri, Z.; Oudit, G.Y. Angiotensin-converting enzyme 2 is a critical determinant of angiotensin II-induced loss of vascular smooth muscle cells and adverse vascular remodeling. Hypertension 2014, 64, 157–164. [Google Scholar] [CrossRef]
- Patel, V.B.; Mori, J.; McLean, B.A.; Basu, R.; Das, S.K.; Ramprasath, T.; Parajuli, N.; Penninger, J.M.; Grant, M.B.; Lopaschuk, G.D.; et al. ACE2 Deficiency Worsens Epicardial Adipose Tissue Inflammation and Cardiac Dysfunction in Response to Diet-Induced Obesity. Diabetes 2016, 65, 85–95. [Google Scholar] [CrossRef] [Green Version]
- Babuin, L.; Vasile, V.C.; Rio Perez, J.A.; Alegria, J.R.; Chai, H.S.; Afessa, B.; Jaffe, A.S. Elevated cardiac troponin is an independent risk factor for short- and long-term mortality in medical intensive care unit patients. Crit. Care Med. 2008, 36, 759–765. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Kruger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e278. [Google Scholar] [CrossRef]
- Sharma, A.; Garcia, G.; Arumugaswami, V.; Svendsen, C.N. Human iPSC-Derived Cardiomyocytes Are Susceptible to SARS-CoV-2 Infection. bioRxiv Prepr. Serv. Biol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Cooper, L.T., Jr. Myocarditis. N. Engl. J. Med. 2009, 360, 1526–1538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sugiura, M.; Hiraoka, K.; Ohkawa, S.; Ueda, K.; Matsuda, T. A clinicopathological study on cardiac lesions in 64 cases of disseminated intravascular coagulation. Jpn. Heart J. 1977, 18, 57–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, N.; Li, D.; Wang, X.; Sun, Z. Abnormal coagulation parameters are associated with poor prognosis in patients with novel coronavirus pneumonia. J. Thromb. Haemost. 2020, 18, 844–847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.; Zhou, Y.; Wang, D.W. SARS-CoV-2: A potential novel etiology of fulminant myocarditis. Herz 2020, 45, 230–232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Li, X.; Chen, M.; Feng, Y.; Xiong, C. The ACE2 expression in human heart indicates new potential mechanism of heart injury among patients infected with SARS-CoV-2. Cardiovasc. Res. 2020, 116, 1097–1100. [Google Scholar] [CrossRef] [Green Version]
- Zhong, J.; Basu, R.; Guo, D.; Chow, F.L.; Byrns, S.; Schuster, M.; Loibner, H.; Wang, X.H.; Penninger, J.M.; Kassiri, Z.; et al. Angiotensin-converting enzyme 2 suppresses pathological hypertrophy, myocardial fibrosis, and cardiac dysfunction. Circulation 2010, 122, 717–728. [Google Scholar] [CrossRef]
- Zhong, J.; Guo, D.; Chen, C.B.; Wang, W.; Schuster, M.; Loibner, H.; Penninger, J.M.; Scholey, J.W.; Kassiri, Z.; Oudit, G.Y. Prevention of angiotensin II-mediated renal oxidative stress, inflammation, and fibrosis by angiotensin-converting enzyme 2. Hypertension 2011, 57, 314–322. [Google Scholar] [CrossRef]
- Lupi, L.; Adamo, M.; Inciardi, R.M.; Metra, M. ACE2 down-regulation may contribute to the increased thrombotic risk in COVID-19. Eur. Heart J. 2020, 41, 3200. [Google Scholar] [CrossRef]
- Zhao, X.; Nicholls, J.M.; Chen, Y.G. Severe acute respiratory syndrome-associated coronavirus nucleocapsid protein interacts with Smad3 and modulates transforming growth factor-beta signaling. J. Biol. Chem. 2008, 283, 3272–3280. [Google Scholar] [CrossRef]
- Flammer, A.J.; Anderson, T.; Celermajer, D.S.; Creager, M.A.; Deanfield, J.; Ganz, P.; Hamburg, N.M.; Luscher, T.F.; Shechter, M.; Taddei, S.; et al. The assessment of endothelial function: From research into clinical practice. Circulation 2012, 126, 753–767. [Google Scholar] [CrossRef] [PubMed]
- Bonetti, P.O.; Lerman, L.O.; Lerman, A. Endothelial dysfunction: A marker of atherosclerotic risk. Arter. Thromb. Vasc. Biol. 2003, 23, 168–175. [Google Scholar] [CrossRef] [PubMed]
- Danzi, G.B.; Loffi, M.; Galeazzi, G.; Gherbesi, E. Acute pulmonary embolism and COVID-19 pneumonia: A random association? Eur. Heart J. 2020, 41, 1858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evans, P.C.; Ed Rainger, G.; Mason, J.C.; Guzik, T.J.; Osto, E.; Stamataki, Z.; Neil, D.; Hoefer, I.E.; Fragiadaki, M.; Waltenberger, J.; et al. Endothelial Dysfunction in COVID-19: A Position Paper of the ESC Working Group for Atherosclerosis and Vascular Biology, and the ESC Council of Basic Cardiovascular Science. Cardiovasc. Res. 2020, in press. [Google Scholar] [CrossRef] [PubMed]
- Fauvel, C.; Weizman, O.; Trimaille, A.; Mika, D.; Pommier, T.; Pace, N.; Douair, A.; Barbin, E.; Fraix, A.; Bouchot, O.; et al. Pulmonary embolism in COVID-19 patients: A French multicentre cohort study. Eur. Heart J. 2020, 41, 3058–3068. [Google Scholar] [CrossRef]
- Ramlall, V.; Thangaraj, P.M.; Meydan, C.; Foox, J.; Butler, D.; Kim, J.; May, B.; De Freitas, J.K.; Glicksberg, B.S.; Mason, C.E.; et al. Immune complement and coagulation dysfunction in adverse outcomes of SARS-CoV-2 infection. Nat. Med. 2020, 26, 1609–1615. [Google Scholar] [CrossRef] [PubMed]
- Boisrame-Helms, J.; Kremer, H.; Schini-Kerth, V.; Meziani, F. Endothelial dysfunction in sepsis. Curr. Vasc. Pharmacol. 2013, 11, 150–160. [Google Scholar]
- Varga, Z.; Flammer, A.J.; Steiger, P.; Haberecker, M.; Andermatt, R.; Zinkernagel, A.S.; Mehra, M.R.; Schuepbach, R.A.; Ruschitzka, F.; Moch, H. Endothelial cell infection and endotheliitis in COVID-19. Lancet 2020, 395, 1417–1418. [Google Scholar] [CrossRef]
- Klok, F.A.; Kruip, M.; van der Meer, N.J.M.; Arbous, M.S.; Gommers, D.; Kant, K.M.; Kaptein, F.H.J.; van Paassen, J.; Stals, M.A.M.; Huisman, M.V.; et al. Incidence of thrombotic complications in critically ill ICU patients with COVID-19. Thromb. Res. 2020, 191, 145–147. [Google Scholar] [CrossRef]
- Grillet, F.; Behr, J.; Calame, P.; Aubry, S.; Delabrousse, E. Acute Pulmonary Embolism Associated with COVID-19 Pneumonia Detected by Pulmonary CT Angiography. Radiology 2020, 296, 201544. [Google Scholar] [CrossRef] [Green Version]
- Uthman, I.W.; Gharavi, A.E. Viral infections and antiphospholipid antibodies. Semin. Arthritis Rheum. 2002, 31, 256–263. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Xiao, M.; Zhang, S.; Xia, P.; Cao, W.; Jiang, W.; Chen, H.; Ding, X.; Zhao, H.; Zhang, H.; et al. Coagulopathy and Antiphospholipid Antibodies in Patients with Covid-19. N. Engl. J. Med. 2020, 382, e38. [Google Scholar] [CrossRef] [PubMed]
- O’Sullivan, J.M.; Gonagle, D.M.; Ward, S.E.; Preston, R.J.S.; O’Donnell, J.S. Endothelial cells orchestrate COVID-19 coagulopathy. Lancet Haematol. 2020, 7, e553–e555. [Google Scholar] [CrossRef]
- Goshua, G.; Pine, A.B.; Meizlish, M.L.; Chang, C.H.; Zhang, H.; Bahel, P.; Baluha, A.; Bar, N.; Bona, R.D.; Burns, A.J.; et al. Endotheliopathy in COVID-19-associated coagulopathy: Evidence from a single-centre, cross-sectional study. Lancet Haematol. 2020, 7, e575–e582. [Google Scholar] [CrossRef]
- Llitjos, J.F.; Leclerc, M.; Chochois, C.; Monsallier, J.M.; Ramakers, M.; Auvray, M.; Merouani, K. High incidence of venous thromboembolic events in anticoagulated severe COVID-19 patients. J. Thromb. Haemost. 2020, 18, 1743–1746. [Google Scholar] [CrossRef] [PubMed]
- Klok, F.A.; Kruip, M.; van der Meer, N.J.M.; Arbous, M.S.; Gommers, D.; Kant, K.M.; Kaptein, F.H.J.; van Paassen, J.; Stals, M.A.M.; Huisman, M.V.; et al. Confirmation of the high cumulative incidence of thrombotic complications in critically ill ICU patients with COVID-19: An updated analysis. Thromb. Res. 2020, 191, 148–150. [Google Scholar] [CrossRef]
- Kayal, S.; Jais, J.P.; Aguini, N.; Chaudiere, J.; Labrousse, J. Elevated circulating E-selectin, intercellular adhesion molecule 1, and von Willebrand factor in patients with severe infection. Am. J. Respir. Crit. Care Med. 1998, 157, 776–784. [Google Scholar] [CrossRef]
- Aird, W.C. The role of the endothelium in severe sepsis and multiple organ dysfunction syndrome. Blood 2003, 101, 3765–3777. [Google Scholar] [CrossRef]
- Massberg, S.; Enders, G.; Leiderer, R.; Eisenmenger, S.; Vestweber, D.; Krombach, F.; Messmer, K. Platelet-endothelial cell interactions during ischemia/reperfusion: The role of P-selectin. Blood 1998, 92, 507–515. [Google Scholar] [CrossRef]
- Springer, T.A. Traffic signals for lymphocyte recirculation and leukocyte emigration: The multistep paradigm. Cell 1994, 76, 301–314. [Google Scholar] [CrossRef]
- Gupta, N.; Zhao, Y.Y.; Evans, C.E. The stimulation of thrombosis by hypoxia. Thromb. Res. 2019, 181, 77–83. [Google Scholar] [CrossRef] [PubMed]
- Amraie, R.; Napoleon, M.A.; Yin, W.; Berrigan, J.; Suder, E.; Zhao, G.; Olejnik, J.; Gummuluru, S.; Muhlberger, E.; Chitalia, V.; et al. CD209L/L-SIGN and CD209/DC-SIGN act as receptors for SARS-CoV-2 and are differentially expressed in lung and kidney epithelial and endothelial cells. bioRxiv 2020. [Google Scholar] [CrossRef]
- Daly, J.L.; Simonetti, B.; Antón-Plágaro, C.; Williamson, M.K.; Shoemark, D.K.; Simón-Gracia, L.; Klein, K.; Bauer, M.; Hollandi, R.; Greber, U.F.; et al. Neuropilin-1 is a host factor for SARS-CoV-2 infection. bioRxiv 2020. [Google Scholar] [CrossRef] [PubMed]
- Cantuti-Castelvetri, L.; Ojha, R.; Pedro, L.D.; Djannatian, M.; Franz, J.; Kuivanen, S.; Kallio, K.; Kaya, T.; Anastasina, M.; Smura, T.; et al. Neuropilin-1 facilitates SARS-CoV-2 cell entry and provides a possible pathway into the central nervous system. bioRxiv 2020. [Google Scholar] [CrossRef]
- Wang, K.; Chen, W.; Zhou, Y.-S.; Lian, J.-Q.; Zhang, Z.; Du, P.; Gong, L.; Zhang, Y.; Cui, H.-Y.; Geng, J.-J.; et al. SARS-CoV-2 invades host cells via a novel route: CD147-spike protein. bioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Kerr, J.R. Cell adhesion molecules in the pathogenesis of and host defence against microbial infection. Mol. Pathol. 1999, 52, 220–230. [Google Scholar] [CrossRef] [Green Version]
- Lippi, G.; Plebani, M.; Henry, B.M. Thrombocytopenia is associated with severe coronavirus disease 2019 (COVID-19) infections: A meta-analysis. Clin. Chim. Acta 2020, 506, 145–148. [Google Scholar] [CrossRef]
- Semple, J.W.; Italiano, J.E., Jr.; Freedman, J. Platelets and the immune continuum. Nat. Rev. Immunol. 2011, 11, 264–274. [Google Scholar] [CrossRef]
- McFadyen, J.D.; Kaplan, Z.S. Platelets are not just for clots. Transfus. Med. Rev. 2015, 29, 110–119. [Google Scholar] [CrossRef]
- Engelmann, B.; Massberg, S. Thrombosis as an intravascular effector of innate immunity. Nat. Rev. Immunol. 2013, 13, 34–45. [Google Scholar] [CrossRef]
- Zuo, Y.; Yalavarthi, S.; Shi, H.; Gockman, K.; Zuo, M.; Madison, J.A.; Blair, C.; Weber, A.; Barnes, B.J.; Egeblad, M.; et al. Neutrophil extracellular traps in COVID-19. JCI Insight 2020, 5, e138999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foley, J.H.; Conway, E.M. Cross Talk Pathways between Coagulation and Inflammation. Circ. Res. 2016, 118, 1392–1408. [Google Scholar] [CrossRef] [PubMed]
- Hori, M.; Nishida, K. Oxidative stress and left ventricular remodelling after myocardial infarction. Cardiovasc. Res. 2008, 81, 457–464. [Google Scholar] [CrossRef] [Green Version]
- Kuhl, U.; Pauschinger, M.; Noutsias, M.; Seeberg, B.; Bock, T.; Lassner, D.; Poller, W.; Kandolf, R.; Schultheiss, H.P. High prevalence of viral genomes and multiple viral infections in the myocardium of adults with “idiopathic” left ventricular dysfunction. Circulation 2005, 111, 887–893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loffredo, L.; Martino, F.; Zicari, A.M.; Carnevale, R.; Battaglia, S.; Martino, E.; Cammisotto, V.; Peruzzi, M.; De Castro, G.; Duse, M.; et al. Enhanced NOX-2 derived oxidative stress in offspring of patients with early myocardial infarction. Int. J. Cardiol. 2019, 293, 56–59. [Google Scholar] [CrossRef] [PubMed]
- Imai, Y.; Kuba, K.; Neely, G.G.; Yaghubian-Malhami, R.; Perkmann, T.; van Loo, G.; Ermolaeva, M.; Veldhuizen, R.; Leung, Y.H.; Wang, H.; et al. Identification of oxidative stress and Toll-like receptor 4 signaling as a key pathway of acute lung injury. Cell 2008, 133, 235–249. [Google Scholar] [CrossRef]
- Waxman, D.A.; Kanzaria, H.K.; Schriger, D.L. Acute Myocardial Infarction after Laboratory-Confirmed Influenza Infection. N. Engl. J. Med. 2018, 378, 2538–2539. [Google Scholar] [CrossRef]
- Shi, Y.; Wang, Y.; Shao, C.; Huang, J.; Gan, J.; Huang, X.; Bucci, E.; Piacentini, M.; Ippolito, G.; Melino, G. COVID-19 infection: The perspectives on immune responses. Cell Death Differ. 2020, 27, 1451–1454. [Google Scholar] [CrossRef] [Green Version]
- Jain, S.K.; Parsanathan, R.; Levine, S.N.; Bocchini, J.A.; Holick, M.F.; Vanchiere, J.A. The potential link between inherited G6PD deficiency, oxidative stress, and vitamin D deficiency and the racial inequities in mortality associated with COVID-19. Free Radic. Biol. Med. 2020, 161, 84–91. [Google Scholar] [CrossRef]
- Polonikov, A. Endogenous Deficiency of Glutathione as the Most Likely Cause of Serious Manifestations and Death in COVID-19 Patients. ACS Infect. Dis. 2020, 6, 1558–1562. [Google Scholar] [CrossRef]
- Fauci, A.S.; Lane, H.C.; Redfield, R.R. Covid-19-Navigating the Uncharted. N. Engl. J. Med. 2020, 382, 1268–1269. [Google Scholar] [CrossRef] [PubMed]
- Kornfeld, O.S.; Hwang, S.; Disatnik, M.H.; Chen, C.H.; Qvit, N.; Mochly-Rosen, D. Mitochondrial reactive oxygen species at the heart of the matter: New therapeutic approaches for cardiovascular diseases. Circ. Res. 2015, 116, 1783–1799. [Google Scholar] [CrossRef] [PubMed]
- Kassi, E.N.; Papavassiliou, K.A.; Papavassiliou, A.G. Defective Anti-oxidant System: An Aggravating Factor for COVID-19 Patients Outcome? Arch. Med. Res. 2020, 51, 726–727. [Google Scholar] [CrossRef] [PubMed]
- Channappanavar, R.; Perlman, S. Pathogenic human coronavirus infections: Causes and consequences of cytokine storm and immunopathology. Semin. Immunopathol. 2017, 39, 529–539. [Google Scholar] [CrossRef]
- Peiris, J.S.; Chu, C.M.; Cheng, V.C.; Chan, K.S.; Hung, I.F.; Poon, L.L.; Law, K.I.; Tang, B.S.; Hon, T.Y.; Chan, C.S.; et al. Clinical progression and viral load in a community outbreak of coronavirus-associated SARS pneumonia: A prospective study. Lancet 2003, 361, 1767–1772. [Google Scholar] [CrossRef] [Green Version]
- Hu, H.; Ma, F.; Wei, X.; Fang, Y. Coronavirus fulminant myocarditis saved with glucocorticoid and human immunoglobulin. Eur. Heart J. 2020, in press. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.; Gao, X. Immunological responses against SARS-coronavirus infection in humans. Cell. Mol. Immunol. 2004, 1, 119–122. [Google Scholar]
- Wang, Y.D.; Sin, W.Y.; Xu, G.B.; Yang, H.H.; Wong, T.Y.; Pang, X.W.; He, X.Y.; Zhang, H.G.; Ng, J.N.; Cheng, C.S.; et al. T-cell epitopes in severe acute respiratory syndrome (SARS) coronavirus spike protein elicit a specific T-cell immune response in patients who recover from SARS. J. Virol. 2004, 78, 5612–5618. [Google Scholar] [CrossRef] [Green Version]
- Ruan, Q.; Yang, K.; Wang, W.; Jiang, L.; Song, J. Clinical predictors of mortality due to COVID-19 based on an analysis of data of 150 patients from Wuhan, China. Intensiv. Care Med. 2020, 46, 846–848. [Google Scholar] [CrossRef] [Green Version]
- Mehta, P.; McAuley, D.F.; Brown, M.; Sanchez, E.; Tattersall, R.S.; Manson, J.J. Hlh across speciality collaboration, U.K. COVID-19: Consider cytokine storm syndromes and immunosuppression. Lancet 2020, 395, 1033–1034. [Google Scholar] [CrossRef]
- Xu, X.; Han, M.; Li, T.; Sun, W.; Wang, D.; Fu, B.; Zhou, Y.; Zheng, X.; Yang, Y.; Li, X.; et al. Effective treatment of severe COVID-19 patients with tocilizumab. Proc. Natl. Acad. Sci. USA 2020, 117, 10970–10975. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zhang, A.; Wan, Y.; Liu, X.; Qiu, C.; Xi, X.; Ren, Y.; Wang, J.; Dong, Y.; Bao, M.; et al. Early hypercytokinemia is associated with interferon-induced transmembrane protein-3 dysfunction and predictive of fatal H7N9 infection. Proc. Natl. Acad. Sci. USA 2014, 111, 769–774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, C.K.; Lam, C.W.; Wu, A.K.; Ip, W.K.; Lee, N.L.; Chan, I.H.; Lit, L.C.; Hui, D.S.; Chan, M.H.; Chung, S.S.; et al. Plasma inflammatory cytokines and chemokines in severe acute respiratory syndrome. Clin. Exp. Immunol. 2004, 136, 95–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, E.S.; Choe, P.G.; Park, W.B.; Oh, H.S.; Kim, E.J.; Nam, E.Y.; Na, S.H.; Kim, M.; Song, K.H.; Bang, J.H.; et al. Clinical Progression and Cytokine Profiles of Middle East Respiratory Syndrome Coronavirus Infection. J. Korean Med. Sci. 2016, 31, 1717–1725. [Google Scholar] [CrossRef] [PubMed]
- Mann, D.L. Innate immunity and the failing heart: The cytokine hypothesis revisited. Circ. Res. 2015, 116, 1254–1268. [Google Scholar] [CrossRef] [Green Version]
- Jamal, F.A.; Khaled, S.K. The Cardiovascular Complications of Chimeric Antigen Receptor T Cell Therapy. Curr. Hematol. Malig. Rep. 2020, 15, 130–132. [Google Scholar] [CrossRef]
- Gu, J.; Gong, E.; Zhang, B.; Zheng, J.; Gao, Z.; Zhong, Y.; Zou, W.; Zhan, J.; Wang, S.; Xie, Z.; et al. Multiple organ infection and the pathogenesis of SARS. J. Exp. Med. 2005, 202, 415–424. [Google Scholar] [CrossRef]
- Cheung, C.Y.; Poon, L.L.; Ng, I.H.; Luk, W.; Sia, S.F.; Wu, M.H.; Chan, K.H.; Yuen, K.Y.; Gordon, S.; Guan, Y.; et al. Cytokine responses in severe acute respiratory syndrome coronavirus-infected macrophages in vitro: Possible relevance to pathogenesis. J. Virol. 2005, 79, 7819–7826. [Google Scholar] [CrossRef] [Green Version]
- Law, H.K.; Cheung, C.Y.; Ng, H.Y.; Sia, S.F.; Chan, Y.O.; Luk, W.; Nicholls, J.M.; Peiris, J.S.; Lau, Y.L. Chemokine up-regulation in SARS-coronavirus-infected, monocyte-derived human dendritic cells. Blood 2005, 106, 2366–2374. [Google Scholar] [CrossRef] [Green Version]
- Bao, L.; Deng, W.; Huang, B.; Gao, H.; Liu, J.; Ren, L.; Wei, Q.; Yu, P.; Xu, Y.; Qi, F.; et al. The pathogenicity of SARS-CoV-2 in hACE2 transgenic mice. Nature 2020, 583, 830–833. [Google Scholar] [CrossRef]
- Sun, J.; Zhuang, Z.; Zheng, J.; Li, K.; Wong, R.L.; Liu, D.; Huang, J.; He, J.; Zhu, A.; Zhao, J.; et al. Generation of a Broadly Useful Model for COVID-19 Pathogenesis, Vaccination, and Treatment. Cell 2020, 182, 734–743.e5. [Google Scholar] [CrossRef] [PubMed]
- Ettayebi, K.; Crawford, S.E.; Murakami, K.; Broughman, J.R.; Karandikar, U.; Tenge, V.R.; Neill, F.H.; Blutt, S.E.; Zeng, X.L.; Qu, L.; et al. Replication of human noroviruses in stem cell-derived human enteroids. Science 2016, 353, 1387–1393. [Google Scholar] [CrossRef] [PubMed]
- Lamers, M.M.; Beumer, J.; van der Vaart, J.; Knoops, K.; Puschhof, J.; Breugem, T.I.; Ravelli, R.B.G.; van Schayck, J.P.; Mykytyn, A.Z.; Duimel, H.Q.; et al. SARS-CoV-2 productively infects human gut enterocytes. Science 2020, 369, 50–54. [Google Scholar] [CrossRef] [PubMed]
- Mahalingam, R.; Dharmalingam, P.; Santhanam, A.; Kotla, S.; Davuluri, G.; Karmouty-Quintana, H.; Ashrith, G.; Thandavarayan, R.A. Single-cell RNA sequencing analysis of SARS-CoV-2 entry receptors in human organoids. J. Cell. Physiol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Monteil, V.; Kwon, H.; Prado, P.; Hagelkruys, A.; Wimmer, R.A.; Stahl, M.; Leopoldi, A.; Garreta, E.; Del Pozo, C.H.; Prosper, F.; et al. Inhibition of SARS-CoV-2 Infections in Engineered Human Tissues Using Clinical-Grade Soluble Human ACE2. Cell 2020, 181, 905–913.e907. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Renner, D.M.; Comar, C.E.; Whelan, J.N.; Reyes, H.M.; Cardenas-Diaz, F.L.; Truitt, R.; Tan, L.H.; Dong, B.; Alysandratos, K.D.; et al. SARS-CoV-2 induces double-stranded RNA-mediated innate immune responses in respiratory epithelial derived cells and cardiomyocytes. bioRxiv Prepr. Serv. Biol. 2020. [Google Scholar] [CrossRef]
- Perez-Bermejo, J.A.; Kang, S.; Rockwood, S.J.; Simoneau, C.R.; Joy, D.A.; Ramadoss, G.N.; Silva, A.C.; Flanigan, W.R.; Li, H.; Nakamura, K.; et al. SARS-CoV-2 infection of human iPSC-derived cardiac cells predicts novel cytopathic features in hearts of COVID-19 patients. bioRxiv Prepr. Serv. Biol. 2020. [Google Scholar] [CrossRef]
- Lu, D.; Chatterjee, S.; Xiao, K.; Riedel, I.; Wang, Y.; Foo, R.; Bar, C.; Thum, T. MicroRNAs targeting the SARS-CoV-2 entry receptor ACE2 in cardiomyocytes. J. Mol. Cell. Cardiol. 2020, 148, 46–49. [Google Scholar] [CrossRef]
- Kwon, Y.; Nukala, S.B.; Srivastava, S.; Miyamoto, H.; Ismail, N.I.; Rehman, J.; Ong, S.B.; Lee, W.H.; Ong, S.G. Detection of Viral RNA Fragments in Human iPSC-Cardiomyocytes following Treatment with Extracellular Vesicles from SARS-CoV-2 Coding-Sequence-Overexpressing Lung Epithelial Cells. bioRxiv Prepr. Serv. Biol. 2020. [Google Scholar] [CrossRef]
- Yang, L.; Han, Y.; Nilsson-Payant, B.E.; Gupta, V.; Wang, P.; Duan, X.; Tang, X.; Zhu, J.; Zhao, Z.; Jaffre, F.; et al. A Human Pluripotent Stem Cell-based Platform to Study SARS-CoV-2 Tropism and Model Virus Infection in Human Cells and Organoids. Cell Stem. Cell 2020, 27, 125–136.e127. [Google Scholar] [CrossRef]
- Yang, J.; Wang, W.; Chen, Z.; Lu, S.; Yang, F.; Bi, Z.; Bao, L.; Mo, F.; Li, X.; Huang, Y.; et al. A vaccine targeting the RBD of the S protein of SARS-CoV-2 induces protective immunity. Nature 2020, 586, 572–577. [Google Scholar] [CrossRef] [PubMed]
- Zost, S.J.; Gilchuk, P.; Case, J.B.; Binshtein, E.; Chen, R.E.; Nkolola, J.P.; Schafer, A.; Reidy, J.X.; Trivette, A.; Nargi, R.S.; et al. Potently neutralizing and protective human antibodies against SARS-CoV-2. Nature 2020, 584, 443–449. [Google Scholar] [CrossRef] [PubMed]
- Gao, Q.; Bao, L.; Mao, H.; Wang, L.; Xu, K.; Yang, M.; Li, Y.; Zhu, L.; Wang, N.; Lv, Z.; et al. Development of an inactivated vaccine candidate for SARS-CoV-2. Science 2020, 369, 77–81. [Google Scholar] [CrossRef] [PubMed]
- Jackson, L.A.; Anderson, E.J.; Rouphael, N.G.; Roberts, P.C.; Makhene, M.; Coler, R.N.; McCullough, M.P.; Chappell, J.D.; Denison, M.R.; Stevens, L.J.; et al. An mRNA Vaccine against SARS-CoV-2—Preliminary Report. N. Engl. J. Med. 2020, 383, 1920–1931. [Google Scholar] [CrossRef] [PubMed]
- Zhu, F.C.; Guan, X.H.; Li, Y.H.; Huang, J.Y.; Jiang, T.; Hou, L.H.; Li, J.X.; Yang, B.F.; Wang, L.; Wang, W.J.; et al. Immunogenicity and safety of a recombinant adenovirus type-5-vectored COVID-19 vaccine in healthy adults aged 18 years or older: A randomised, double-blind, placebo-controlled, phase 2 trial. Lancet 2020, 396, 479–488. [Google Scholar] [CrossRef]
- Braun, J.; Loyal, L.; Frentsch, M.; Wendisch, D.; Georg, P.; Kurth, F.; Hippenstiel, S.; Dingeldey, M.; Kruse, B.; Fauchere, F.; et al. SARS-CoV-2-reactive T cells in healthy donors and patients with COVID-19. Nature 2020, 587, 270–274. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Sr. No. | Study | Sample Numbers | Death | CVD | Hypertension | Diabetes Mellitus | Obesity |
|---|---|---|---|---|---|---|---|
| 1 | Garg et al. | 178 | n/a | 27.80% | 49.70% | 28.30% | n/a |
| 2 | Shi et al. | 416 | n/a | 14.70% | 30.50% | 14% | n/a |
| 3 | Grasselli et al. | 1043 | n/a | 21.40% | 48.80% | 21.40% | n/a |
| 4 | Bello-Chavolla OY et al. | 51,633 | 5332 (10.3) | 1381 (2.7%) | 11,151 (21.6) | 9460 (18.3%) | 10708 (20.7%) |
| 5 | Argenziano et al. | 1000 | n/a | 233 (23.3) | 601 (60.1%) | 372 (37.2%) | 352 (35.2%) |
| 6 | Hajifathalian et al. | 770 | n/a | 162 (21.0%) | 432 (56.1%) | 238 (30.9%) | 277 (35.9%) |
| 7 | Price-Haywood et al. | 3481 | n/a | 267 (7.6%) | 1074 (30.8%) | 566 (16.2%) | 1727 (49.6%) |
| 8 | Huang et al. | 41 | n/a | 15% | 15% | 20% | n/a |
| 9 | Wang et al. | 138 | n/a | 15% | 31% | 10% | n/a |
| 10 | Zhou et al. | 191 | 54 | 8% | 30% | 19% | n/a |
| 11 | Liu et al. | 78 | n/a | 3% | 40% | 25% | n/a |
| 12 | Guan et al. | 1099 | n/a | 3% | 15% | 7% | n/a |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Magadum, A.; Kishore, R. Cardiovascular Manifestations of COVID-19 Infection. Cells 2020, 9, 2508. https://doi.org/10.3390/cells9112508
Magadum A, Kishore R. Cardiovascular Manifestations of COVID-19 Infection. Cells. 2020; 9(11):2508. https://doi.org/10.3390/cells9112508
Chicago/Turabian StyleMagadum, Ajit, and Raj Kishore. 2020. "Cardiovascular Manifestations of COVID-19 Infection" Cells 9, no. 11: 2508. https://doi.org/10.3390/cells9112508
APA StyleMagadum, A., & Kishore, R. (2020). Cardiovascular Manifestations of COVID-19 Infection. Cells, 9(11), 2508. https://doi.org/10.3390/cells9112508