

Lobe X of the Cerebellum: A Natural Neuro-Resistant Region


Abstract
:1. Introduction. The Cerebellum
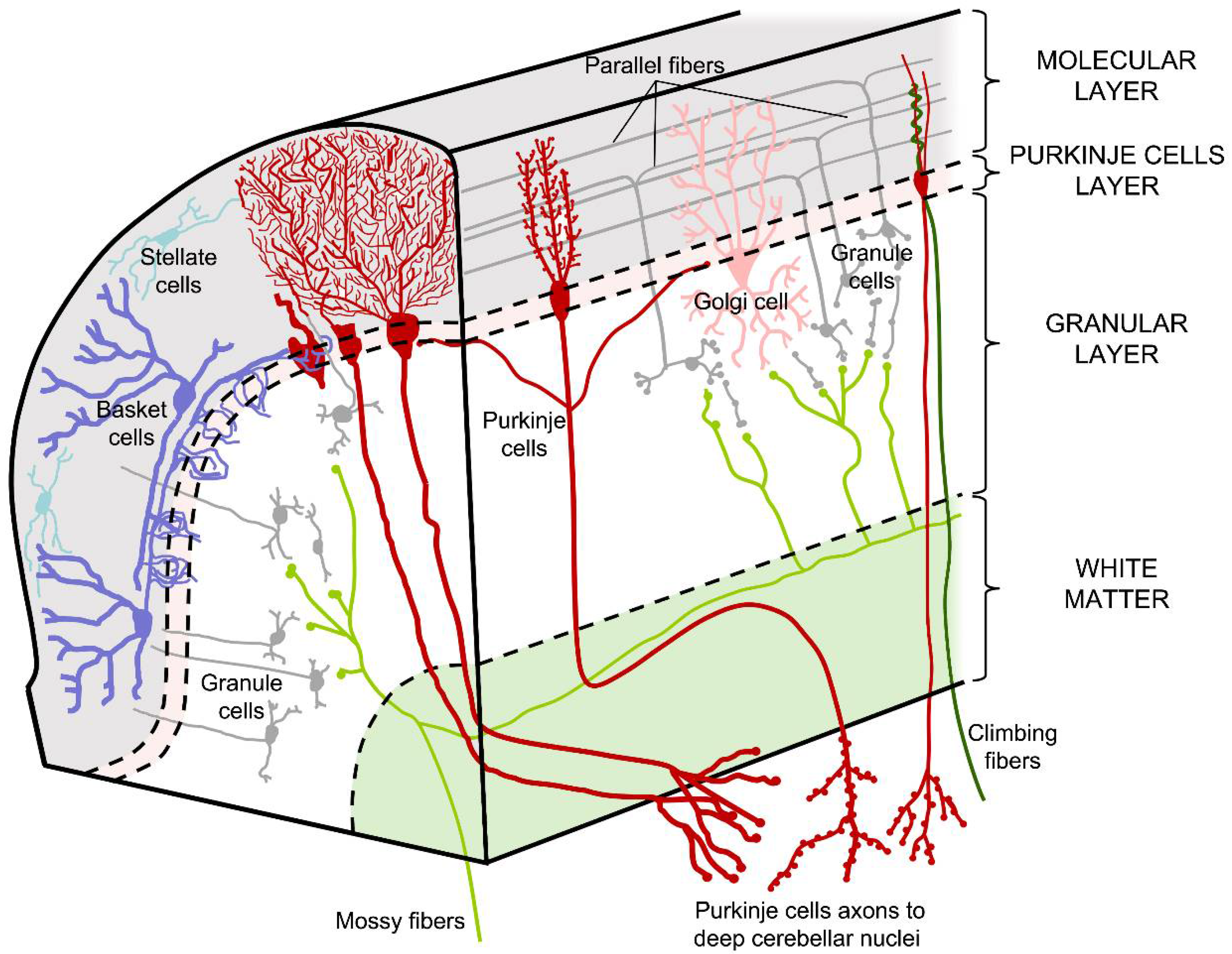
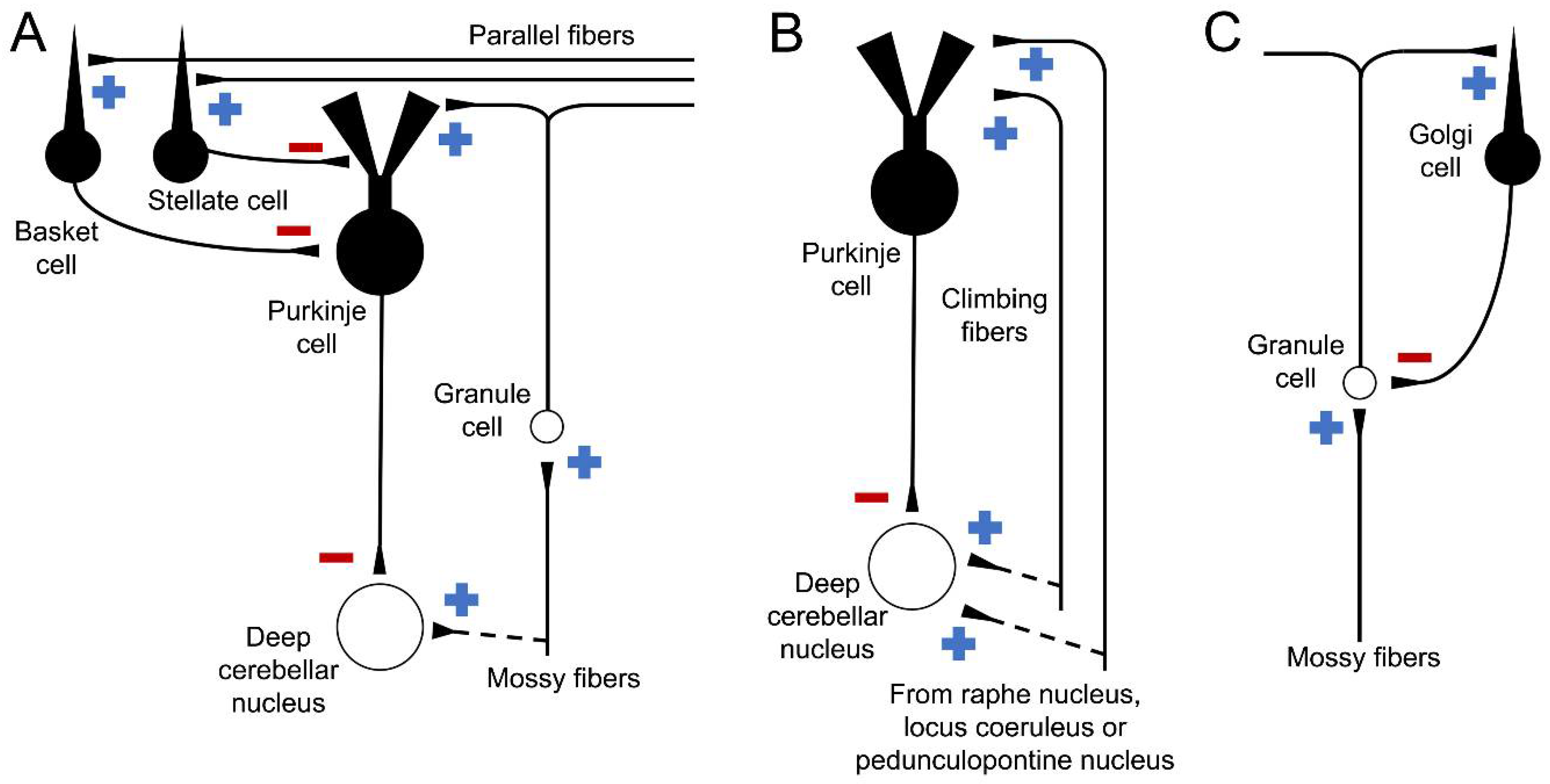
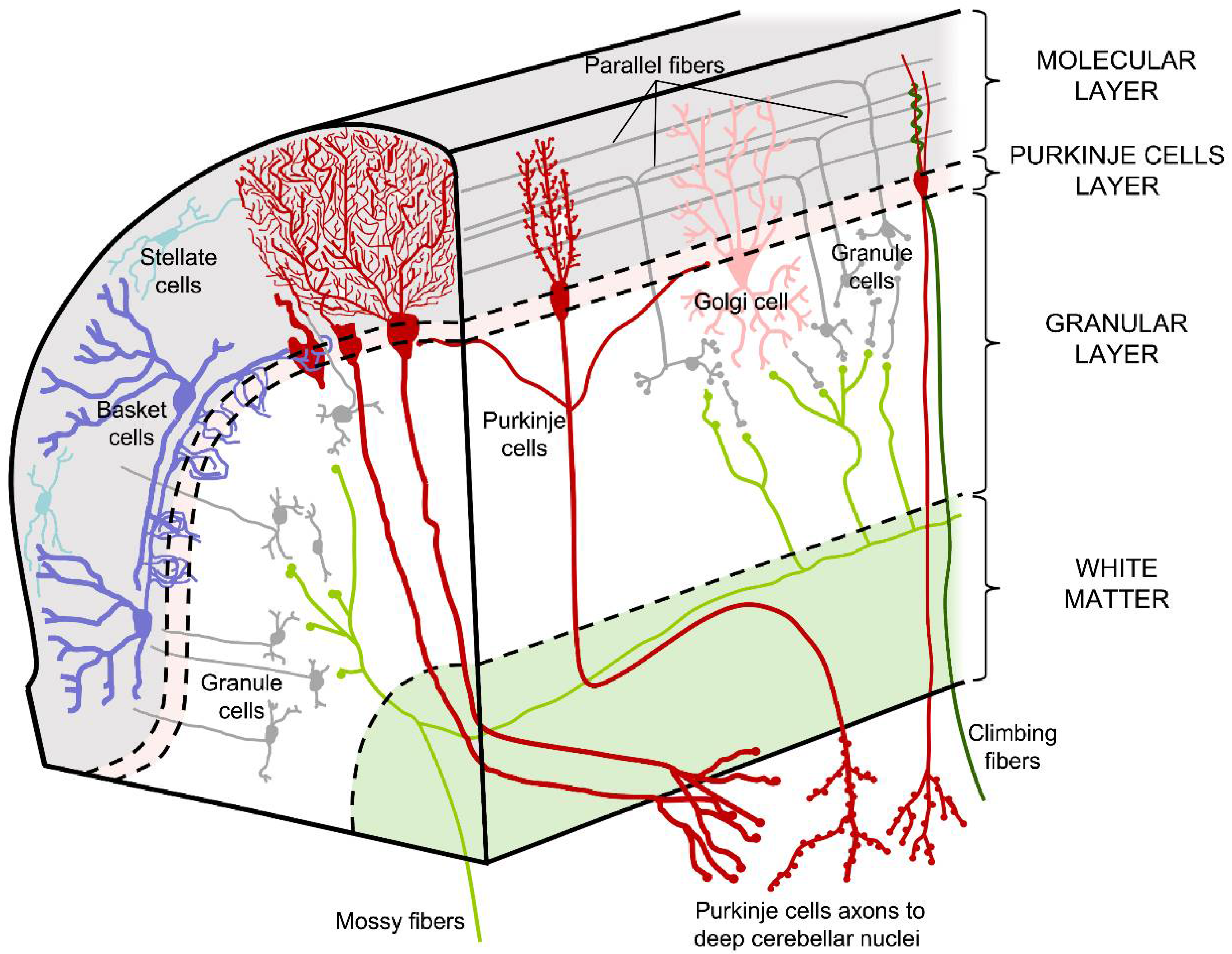
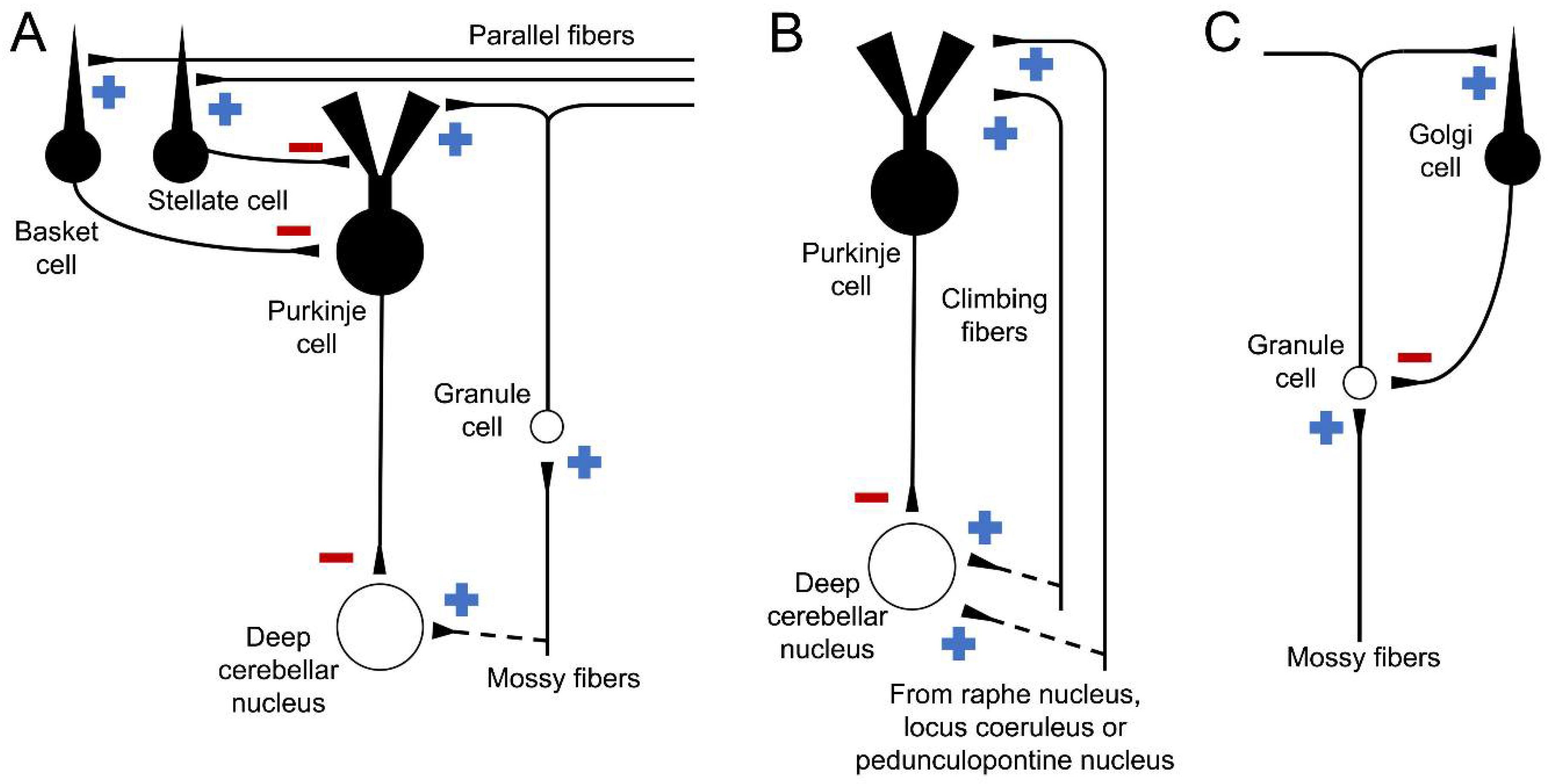
1.1. Cell Types and Cerebellar Pathways
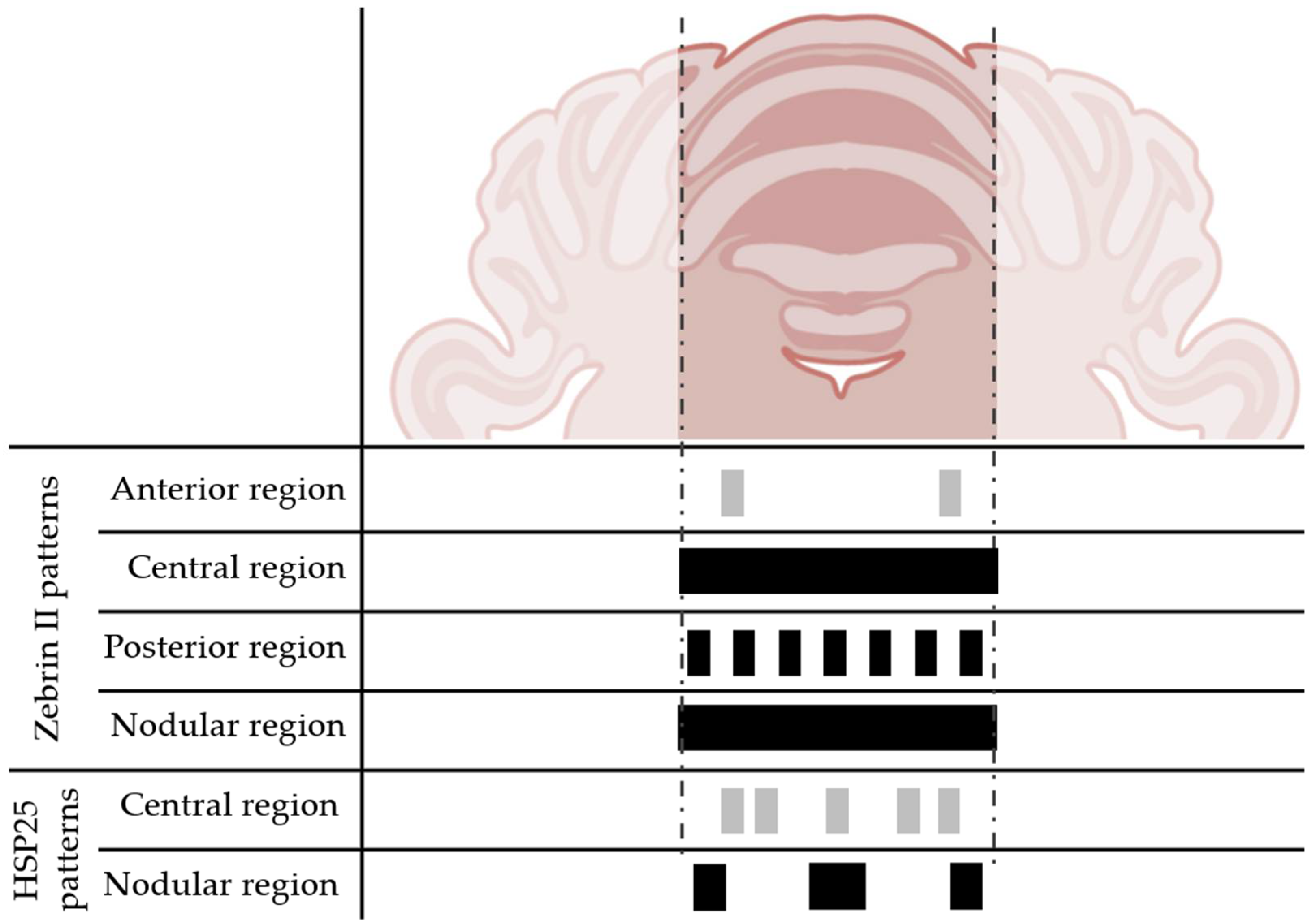
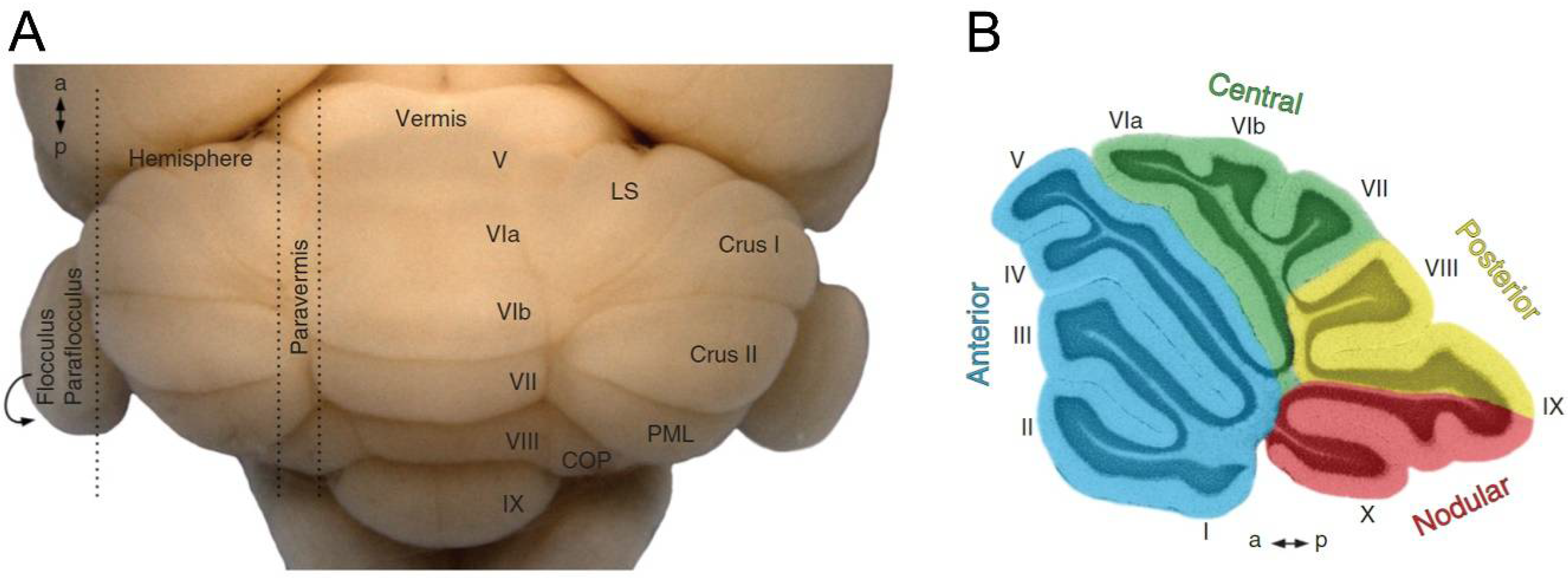
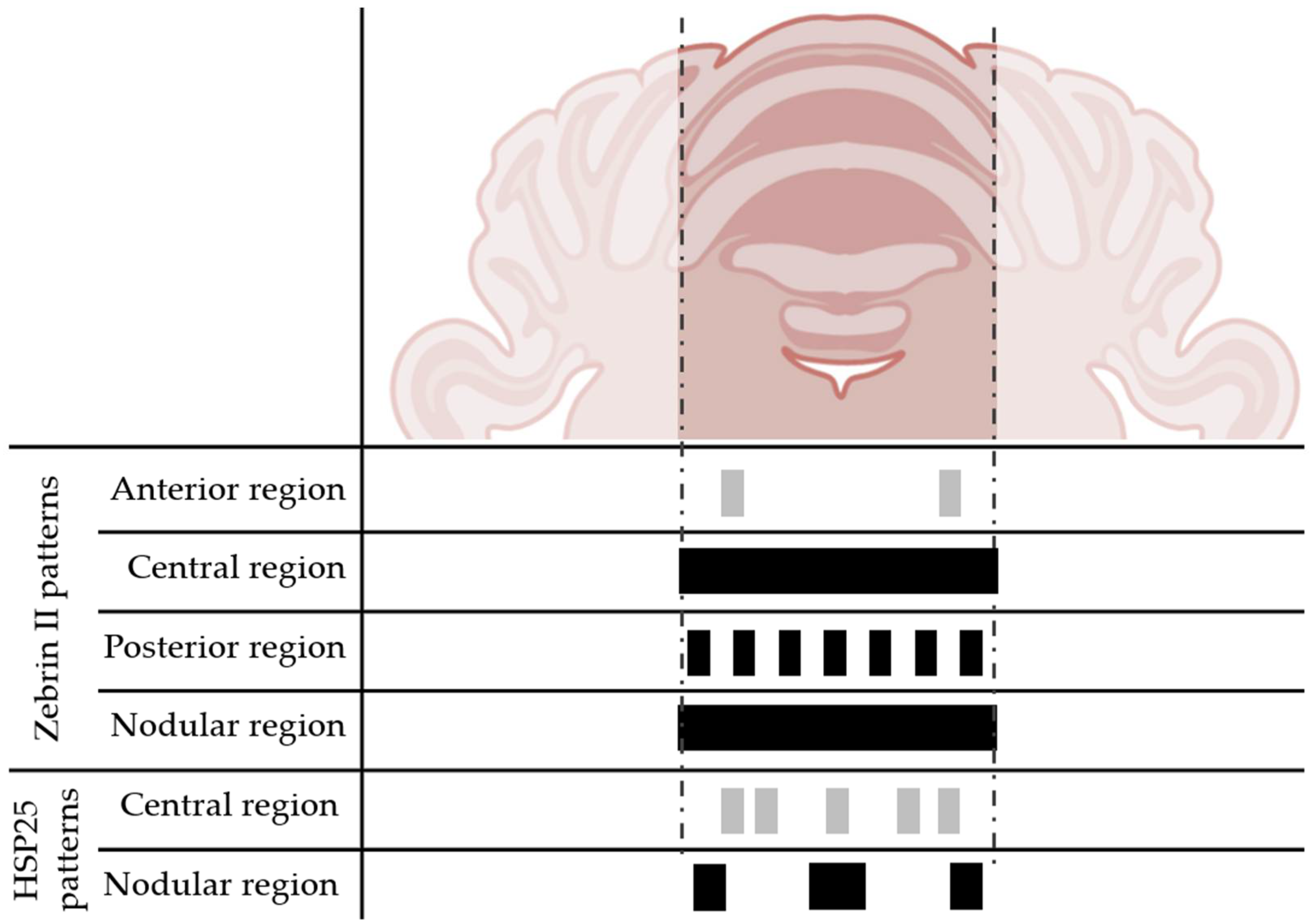
1.2. Regions of the Cerebellar Cortex
2. Objective
3. Models of Cerebellar Degeneration
3.1. Tottering, Leaner, and Nagoya Models
3.2. Toppler Model
3.3. Robotic Model
3.4. Shaker Model
3.5. Lurcher Model
3.6. NPC1 Model
3.7. Nervous Model
3.8. PCD Model
3.9. Tambaleante Model
3.10. Other Non-Genetic Models
4. Possible Causes of the Neuroresistance of Lobe X
5. Why Lobe X Is Different from Other Lobes?
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Freeland, K.R.; Anderson, G.H.; Thomas, T.W. Chapter 42. The Cerebellum. In Principles of Neural Science, 5th ed.; Springer: Berlin/Heidelberg, Germany, 2000. [Google Scholar]
- Voogd, J.; Glickstein, M. The anatomy of the cerebellum. Trends Cogn. Sci. 1998, 2, 307–313. [Google Scholar] [CrossRef] [PubMed]
- White, J.J.; Sillitoe, R.V. Development of the cerebellum: From gene expression patterns to circuit maps. Wiley Interdiscip. Rev. Dev. Biol. 2012, 2, 149–164. [Google Scholar] [CrossRef] [PubMed]
- Delgado-García, J.M. Structure and function of the cerebellum. Rev. Neurol. 2002, 33, 635–642. [Google Scholar]
- Kandel, E.R. Principles of Neural Science; McGraw-Hill: New York, NY, USA, 2013. [Google Scholar]
- Brooks, V.B.; Thach, W.T. Cerebellar Control of Posture and Movement. Compr. Physiol. 1981, 10, 877–946. [Google Scholar] [CrossRef]
- Galliano, E.; Potters, J.-W.; Elgersma, Y.; Wisden, W.; Kushner, S.A.; De Zeeuw, C.I.; Hoebeek, F.E. Synaptic Transmission and Plasticity at Inputs to Murine Cerebellar Purkinje Cells Are Largely Dispensable for Standard Nonmotor Tasks. J. Neurosci. 2013, 33, 12599–12618. [Google Scholar] [CrossRef] [Green Version]
- Newman, P.P.; Reza, H. Functional relationships between the hippocampus and the cerebellum: An electrophysiological study of the cat. J. Physiol. 1979, 287, 405–426. [Google Scholar] [CrossRef] [PubMed]
- Petersen, S.E.; Fox, P.T.; Posner, M.I.; Mintun, M.; Raichle, M.E. Positron emission tomographic studies of the cortical anatomy of single-word processing. Nature 1988, 331, 585–589. [Google Scholar] [CrossRef]
- Sillitoe, R.V.; Joyner, A.L. Morphology, Molecular Codes, and Circuitry Produce the Three-Dimensional Complexity of the Cerebellum. Annu. Rev. Cell Dev. Biol. 2007, 23, 549–577. [Google Scholar] [CrossRef]
- Mugnaini, E.; Dio, M.R.; Jaarsma, D. Chapter 8 The unipolar brush cells of the mammalian cerebellum and cochlear nucleus: Cytology and microcircuitry. In Progress in Brain Research; De Zeeuw, C.I., Strata, P., Voogd, J., Eds.; Elsevier: Amsterdam, The Netherlands, 1997. [Google Scholar]
- Allin, M.; Matsumoto, H.; Santhouse, A.M.; Nosarti, C.; AlAsady, M.H.S.; Stewart, A.L.; Rifkin, L.; Murray, R.M. Cognitive and motor function and the size of the cerebellum in adolescents born very pre-term. Brain 2001, 124, 60–66. [Google Scholar] [CrossRef] [Green Version]
- Levitt, P.; Rakic, P. Immunoperoxidase localization of glial fibrillary acidic protein in radial glial cells and astrocytes of the developing rhesus monkey brain. J. Comp. Neurol. 1980, 193, 815–840. [Google Scholar] [CrossRef]
- Beckinghausen, J.; Sillitoe, R.V. Insights into cerebellar development and connectivity. Neurosci. Lett. 2018, 688, 2–13. [Google Scholar] [CrossRef]
- Ito, M. Cerebellar circuitry as a neuronal machine. Prog. Neurobiol. 2006, 78, 272–303. [Google Scholar] [CrossRef] [PubMed]
- Barmack, N.H.; Baughman, R.W.; Eckenstein, F.P. Cholinergic innervation of the cerebellum of rat, rabbit, cat, and monkey as revealed by choline acetyltransferase activity and immunohistochemistry. J. Comp. Neurol. 1992, 317, 233–249. [Google Scholar] [CrossRef]
- Miall, R.C. Cerebellum: Anatomy and Function. Neuroscience in the 21st Century: From Basic to Clinical; Springer: Berlin/Haidelberg, Germany, 2013; pp. 1149–1167. [Google Scholar]
- Larsell, O. The morphogenesis and adult pattern of the lobules and fissures of the cerebellum of the white rat. J. Comp. Neurol. 1952, 97, 281–356. [Google Scholar] [CrossRef] [PubMed]
- Hawkes, R. Chapter 3 An anatomical model of cerebellar modules. Prog. Brain Res. 1997, 114, 39–52. [Google Scholar] [CrossRef] [PubMed]
- Apps, R.; Hawkes, R. Cerebellar cortical organization: A one-map hypothesis. Nat. Rev. Neurosci. 2009, 10, 670–681. [Google Scholar] [CrossRef]
- Brochu, G.; Maler, L.; Hawkes, R. Zebrin II: A polypeptide antigen expressed selectively by purkinje cells reveals compartments in rat and fish cerebellum. J. Comp. Neurol. 1990, 291, 538–552. [Google Scholar] [CrossRef] [PubMed]
- Sarna, J.R.; Larouche, M.; Marzban, H.; Sillitoe, R.V.; Rancourt, D.E.; Hawkes, R. Patterned Purkinje cell degeneration in mouse models of Niemann-Pick type C disease. J. Comp. Neurol. 2003, 456, 279–291. [Google Scholar] [CrossRef] [PubMed]
- Larouche, M.; Che, P.M.; Hawkes, R. Neurogranin expression identifies a novel array of Purkinje cell parasagittal stripes during mouse cerebellar development. J. Comp. Neurol. 2005, 494, 215–227. [Google Scholar] [CrossRef]
- Marzban, H.; Chung, S.; Watanabe, M.; Hawkes, R. Phospholipase Cbeta4 expression reveals the continuity of cerebellar topography through development. J. Comp. Neurol. 2007, 502, 857–871. [Google Scholar] [CrossRef]
- Hawkes, R.; Turner, R.W. Compartmentation of NADPH-diaphorase activity in the mouse cerebellar cortex. J. Comp. Neurol. 1994, 346, 499–516. [Google Scholar] [CrossRef] [PubMed]
- Ozol, K.O.; Hawkes, R. Compartmentation of the granular layer of the cerebellum. Histol. Histopathol. 1997, 12, 171–184. [Google Scholar] [PubMed]
- Sillitoe, R.V.; Chung, S.H.; Fritschy, J.M.; Hoy, M.; Hawkes, R. Golgi cell dendrites are restricted by Purkinje cell stripe boundaries in the adult mouse cerebellar cortex. J. Neurosci. 2008, 28, 2820–2826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, C.; Elrick, M.J.; Dell’Orco, J.M.; Qin, Z.S.; Kalyana-Sundaram, S.; Chinnaiyan, A.M.; Shakkottai, V.G.; Lieberman, A.P. Heat Shock Protein Beta-1 Modifies Anterior to Posterior Purkinje Cell Vulnerability in a Mouse Model of Niemann-Pick Type C Disease. PLoS Genet. 2016, 12, e1006042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armstrong, C.L.; Chung, S.H.; Armstrong, J.N.; Hochgeschwender, U.; Jeong, Y.G.; Hawkes, R. A novel somatostatin-immunoreactive mossy fiber pathway associated with HSP25-immunoreactive purkinje cell stripes in the mouse cerebellum. J. Comp. Neurol. 2009, 517, 524–538. [Google Scholar] [CrossRef]
- Voogd, J.; Pardoe, J.; Ruigrok, T.J.H.; Apps, R. The Distribution of Climbing and Mossy Fiber Collateral Branches from the Copula Pyramidis and the Paramedian Lobule: Congruence of Climbing Fiber Cortical Zones and the Pattern of Zebrin Banding within the Rat Cerebellum. J. Neurosci. 2003, 23, 4645–4656. [Google Scholar] [CrossRef]
- Marzban, H.; Hawkes, R. On the Architecture of the Posterior Zone of the Cerebellum. Cerebellum 2010, 10, 422–434. [Google Scholar] [CrossRef]
- Armstrong, C.L.; Krueger-Naug, A.M.; Currie, R.W.; Hawkes, R. Expression of heat-shock proteina Hsp25 in mouse Purkinje cells during development reveals novel features of cerebellar compartmentation. J. Comp. Neurol. 2001, 429, 7–21. [Google Scholar] [CrossRef]
- Sillitoe, R.V.; Hawkes, R. Whole-mount Immunohistochemistry: A High-throughput Screen for Patterning Defects in the Mouse Cerebellum. J. Histochem. Cytochem. 2002, 50, 235–244. [Google Scholar] [CrossRef] [Green Version]
- Double, K.L.; Reyes, S.; Werry, E.L.; Halliday, G.M. Selective cell death in neurodegeneration: Why are some neurons spared in vulnerable regions? Prog. Neurobiol. 2010, 92, 316–329. [Google Scholar] [CrossRef]
- Sarna, J.R.; Hawkes, R. Patterned Purkinje cell death in the cerebellum. Prog. Neurobiol. 2003, 70, 473–507. [Google Scholar] [CrossRef] [PubMed]
- Clark, H.B.; Burright, E.N.; Yunis, W.S.; Larson, S.; Wilcox, C.; Hartman, B.; Matilla, A.; Zoghbi, H.Y.; Orr, H.T. Purkinje cell expression of a mutant allele of SCA1 in transgenic mice leads to disparate effects on motor behaviors, followed by a progressive cerebellar dysfunction and histological alterations. J. Neurosci. 1997, 17, 7385–7395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoneshige, A.; Suzuki, K.; Suzuki, K.; Matsuda, J. A mutation in the saposin C domain of the sphingolipid activator protein (Prosaposin) gene causes neurodegenerative disease in mice. J. Neurosci. Res. 2010, 88, 2118–2134. [Google Scholar] [CrossRef] [PubMed]
- Tavani, F.; Zimmerman, R.A.; Berry, G.; Sullivan, K.; Gatti, R.; Bingham, P. Ataxia-telangiectasia: The pattern of cerebellar atrophy on MRI. Neuroradiology 2003, 45, 315–319. [Google Scholar] [CrossRef] [PubMed]
- Sarna, J.; Miranda, S.R.P.; Schuchman, E.H.; Hawkes, R. Patterned cerebellar Purkinje cell death in a transgenic mouse model of Niemann Pick type A/B disease. Eur. J. Neurosci. 2001, 13, 1873–1880. [Google Scholar] [CrossRef]
- Kume, A.; Takahashi, A.; Hashizume, Y.; Asai, J. A histometrical and comparative study on Purkinje cell loss and olivary nucleus cell loss in multiple system atrophy. J. Neurol. Sci. 1991, 101, 178–186. [Google Scholar] [CrossRef] [PubMed]
- Torvik, A.; Torp, S. The prevalence of alcoholic cerebellar atrophy: A morphometric and histological study of an autopsy material. J. Neurol. Sci. 1986, 75, 43–51. [Google Scholar] [CrossRef]
- Biran, V.; Heine, V.M.; Verney, C.; Sheldon, R.A.; Spadafora, R.; Vexler, Z.S.; Rowitch, D.H.; Ferriero, D.M. Cerebellar abnormalities following hypoxia alone compared to hypoxic–ischemic forebrain injury in the developing rat brain. Neurobiol. Dis. 2011, 41, 138–146. [Google Scholar] [CrossRef] [Green Version]
- Andersen, B.B.; Pakkenberg, B. Aging of the human cerebellum: A stereological study. J. Comp. Neurol. 2003, 466, 356–365. [Google Scholar] [CrossRef]
- Heckroth, J.A.; Abbott, L.C. Purkinje cell loss from alternating sagittal zones in the cerebellum of leaner mutant mice. Brain Res. 1994, 658, 93–104. [Google Scholar] [CrossRef]
- Duchala, C.S.; Shick, H.E.; Garcia, J.; Deweese, D.M.; Sun, X.; Stewart, V.J.; Macklin, W.B. The toppler mouse: A novel mutant exhibiting loss of Purkinje cells. J. Comp. Neurol. 2004, 476, 113–129. [Google Scholar] [CrossRef] [PubMed]
- Isaacs, A.M.; Oliver, P.L.; Jones, E.L.; Jeans, A.; Potter, A.; Hovik, B.H.; Nolan, P.M.; Vizor, L.; Glenister, P.; Simon, A.K.; et al. A Mutation inAf4Is Predicted to Cause Cerebellar Ataxia and Cataracts in the Robotic Mouse. J. Neurosci. 2003, 23, 1631–1637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tolbert, D.L.; Ewald, M.; Gutting, J.; La Regina, M.C. Spatial and temporal pattern of Purkinje cell degeneration in shaker mutant rats with hereditary cerebellar ataxia. J. Comp. Neurol. 1995, 355, 490–507. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, C.L.; Duffin, C.A.; McFarland, R.; Vogel, M.W. Mechanisms of Compartmental Purkinje Cell Death and Survival in the Lurcher Mutant Mouse. Cerebellum 2010, 10, 504–514. [Google Scholar] [CrossRef]
- Wang, T.; Morgan, J.I. The Purkinje cell degeneration (pcd) mouse: An unexpected molecular link between neuronal degeneration and regeneration. Brain Res. 2007, 1140, 26–40. [Google Scholar] [CrossRef]
- Baltanás, F.C.; Berciano, M.T.; Santos, E.; Lafarga, M. The Childhood-Onset Neurodegeneration with Cerebellar Atrophy (CONDCA) Disease Caused by AGTPBP1 Gene Mutations: The Purkinje Cell Degeneration Mouse as an Animal Model for the Study of this Human Disease. Biomedicines 2021, 9, 1157. [Google Scholar] [CrossRef]
- Fletcher, C.F.; Lutz, C.M.; O’Sullivan, T.; Shaughnessy, J.D.; Hawkes, R.; Frankel, W.N.; Copeland, N.G.; Jenkins, N.A. Absence Epilepsy in Tottering Mutant Mice Is Associated with Calcium Channel Defects. Cell 1996, 87, 607–617. [Google Scholar] [CrossRef] [Green Version]
- Herrup, K.; Wilczynski, S. Cerebellar cell degeneration in the leaner mutant mouse. Neuroscience 1982, 7, 2185–2196. [Google Scholar] [CrossRef]
- Sawada, K.; Haga, H.; Fukui, Y. Ataxic mutant mice with defects in Ca2+ channel α1A subunit gene: Morphological and functional abnormalities in cerebellar cortical neurons. Congenit. Anom. 2000, 40, 99–107. [Google Scholar] [CrossRef]
- Doyle, J.; Ren, X.; Lennon, G.; Stubbs, L. Mutations in the Cacnl1a4 calcium channel gene are associated with seizures, cerebellar degeneration, and ataxia in tottering and leaner mutant mice. Mamm. Genome 1997, 8, 113–120. [Google Scholar] [CrossRef]
- Lorenzon, N.M.; Lutz, C.M.; Frankel, W.N.; Beam, K.G. Altered calcium channel currents in Purkinje cells of the neurological mutant mouse leaner. J. Neurosci. 1998, 18, 4482–4489. [Google Scholar] [CrossRef] [PubMed]
- Sawada, K.; Azad, A.K.; Sakata-Haga, H.; Lee, N.-S.; Jeong, Y.-G.; Fukui, Y. Striking pattern of Purkinje cell loss in cerebellum of an ataxic mutant mouse, tottering. Acta Neurobiol. Exp. 2009, 69, 138–145. [Google Scholar]
- Jeong, Y.-G.; Hyun, B.-H.; Hawkes, R. Abnormalities in cerebellar Purkinje cells in the novel ataxic mutant mouse, pogo. Dev. Brain Res. 2000, 125, 61–67. [Google Scholar] [CrossRef]
- Duffin, C.A.; Mcfarland, R.; Sarna, J.R.; Vogel, M.W.; Armstrong, C.L. Heat shock protein 25 expression and preferential Purkinje cell survival in the lurcher mutant mouse cerebellum. J. Comp. Neurol. 2010, 518, 1892–1907. [Google Scholar] [CrossRef] [PubMed]
- Praggastis, M.; Tortelli, B.; Zhang, J.; Fujiwara, H.; Sidhu, R.; Chacko, A.; Chen, Z.; Chung, C.; Lieberman, A.P.; Sikora, J.; et al. A murine Niemann-Pick C1 I1061T knock-in model recapitulates the pathological features of the most prevalent human disease allele. J. Neurosci. 2015, 35, 8091–8106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Angelis, M.H.; Flaswinkel, H.; Fuchs, H.; Rathkolb, B.; Soewarto, D.; Marschall, S.; Heffner, S.; Pargent, W.; Wuensch, K.; Jung, M.; et al. Genome-wide, large-scale production of mutant mice by ENU mutagenesis. Nat. Genet. 2000, 25, 444–447. [Google Scholar] [CrossRef] [PubMed]
- Isnard, P.; Coré, N.; Naquet, P.; Djabali, M. Altered lymphoid development in mice deficient for the mAF4 proto-oncogene. Blood 2000, 96, 705–710. [Google Scholar] [CrossRef] [PubMed]
- Bitoun, E.; Davies, K.E. The Robotic Mouse: Understanding the Role of AF4, a Cofactor of Transcriptional Elongation and Chromatin Remodelling, in Purkinje Cell Function. Cerebellum 2009, 8, 175–183. [Google Scholar] [CrossRef]
- La Regina, M.C.; Yates-Siilata, K.; Woods, L.; Tolbert, D. Preliminary characterization of hereditary cerebellar ataxia in rats. Lab. Anim. Sci. 1992, 42, 19–26. [Google Scholar]
- Wolf, L.W.; LaRegina, M.C.; Tolbert, D.L. A behavioral study of the development of hereditary cerebellar ataxia in the shaker rat mutant. Behav. Brain Res. 1996, 75, 67–81. [Google Scholar] [CrossRef]
- Phillips, R.J.S. ‘Lurcher’, a new gene in linkage group XI of the house mouse. J. Genet. 1960, 57, 35–42. [Google Scholar] [CrossRef]
- Cheng, S.S.-W.; Heintz, N. Massive Loss of Mid- and Hindbrain Neurons during Embryonic Development of Homozygous Lurcher Mice. J. Neurosci. 1997, 17, 2400–2407. [Google Scholar] [CrossRef] [PubMed]
- Vogel, M.W.; Caston, J.; Yuzaki, M.; Mariani, J. The Lurcher mouse: Fresh insights from an old mutant. Brain Res. 2007, 1140, 4–18. [Google Scholar] [CrossRef] [PubMed]
- MCfarland, R.; Blokhin, A.; Sydnor, J.; Mariani, J.; Vogel, M.W. Oxidative stress, nitric oxide, and the mechanisms of cell death in Lurcher Purkinje cells. Dev. Neurobiol. 2007, 67, 1032–1046. [Google Scholar] [CrossRef] [PubMed]
- Pentchev, P.G.; Comly, M.E.; Kruth, H.S.; Patel, S.; Proestel, M.; Weintroub, H. The cholesterol storage disorder of the mutant BALB/c mouse. A primary genetic lesion closely linked to defective esterification of exogenously derived cholesterol and its relationship to human type C Niemann-Pick disease. J. Biol. Chem. 1986, 261, 2772–2777. [Google Scholar] [CrossRef] [PubMed]
- Vanier, M.T.; Rodriguez-Lafrasse, C.; Rousson, R.; Duthel, S.; Harzer, K.; Pentchev, P.G.; Revol, A.; Louisot, P. Type C Niemann-Pick Disease: Biochemical Aspects and Phenotypic Heterogeneity. Dev. Neurosci. 1991, 13, 307–314. [Google Scholar] [CrossRef]
- Akaboshi, S.; Yano, T.; Miyawaki, S.; Ohno, K.; Takeshita, K. A C57BL/KsJ mouse model of Niemann-Pick disease (spm) belongs to the same complementation group as the major childhood type of Niemann-Pick disease type C. Hum. Genet. 1997, 99, 350–353. [Google Scholar] [CrossRef]
- Sidman, R.L.; Green, M.C. “Nervous,” a new mutant mouse with cerebellar disease. In Les Mutants Pathologiques chez l’Animal; Sabourdy, M., Ed.; Centre National de la Reserche Scientifique: Paris, France, 1970; pp. 69–79. [Google Scholar]
- Mullen, R.J.; Lavail, M.M. Two new types of retinal degeneration in cerebellar mutant mice. Nature 1975, 258, 528–530. [Google Scholar] [CrossRef]
- Mallet, J.; Huchet, M.; Pougeois, R.; Changeux, J.-P. Anatomical, physiological and biochemical studies on the cerebellum from mutant mice. III. Protein differences associated with the weaver, staggerer and nervous mutations. Brain Res. 1976, 103, 291–312. [Google Scholar] [CrossRef]
- Mikoshiba, K.; Okano, H.; Tsukada, Y. P400 Protein Characteristic to Purkinje Cells and Related Proteins in Cerebella from Neuropathological Mutant Mice: Autoradiographic Study by 14C-Leucine and Phosphorylation. Dev. Neurosci. 1985, 7, 179–187. [Google Scholar] [CrossRef]
- Sotelo, C.; Triller, A. Fate of presynaptic afferents to Purkinje cells in the adult nervous mutant mouse: A model to study presynaptic stabilization. Brain Res. 1979, 175, 11–36. [Google Scholar] [CrossRef] [PubMed]
- Wassef, M.; Sotelo, C.; Cholley, B.; Brehier, A.; Thomasset, M. Cerebellar mutations affecting the postnatal survival of Purkinje cells in the mouse disclose a longitudinal pattern of differentially sensitive cells. Dev. Biol. 1987, 124, 379–389. [Google Scholar] [CrossRef] [PubMed]
- Edwards, M.A.; Crandall, J.E.; Leclerc, N.; Yamamoto, M. Effects of nervous mutation on purkinje cell compartments defined by Zebrin II and 9-O-acetylated gangliosides expression. Neurosci. Res. 1994, 19, 167–174. [Google Scholar] [CrossRef]
- Shashi, V.; Magiera, M.M.; Klein, D.; Zaki, M.; Schoch, K.; Rudnik-Schöneborn, S.; Norman, A.; Neto, O.L.A.; Dusl, M.; Yuan, X.; et al. Loss of tubulin deglutamylaseCCP1 causes infantile-onset neurodegeneration. EMBO J. 2018, 37, e100540. [Google Scholar] [CrossRef] [PubMed]
- Karakaya, M.; Paketci, C.; Altmueller, J.; Thiele, H.; Hoelker, I.; Yis, U.; Wirth, B. Biallelic variant in AGTPBP1 causes infantile lower motor neuron degeneration and cerebellar atrophy. Am. J. Med. Genet. Part A 2019, 179, 1580–1584. [Google Scholar] [CrossRef] [PubMed]
- Sheffer, R.; Gur, M.; Brooks, R.; Salah, S.; Daana, M.; Fraenkel, N.; Eisenstein, E.; Rabie, M.; Nevo, Y.; Jalas, C.; et al. Biallelic variants in AGTPBP1, involved in tubulin deglutamylation, are associated with cerebellar degeneration and motor neuropathy. Eur. J. Hum. Genet. 2019, 27, 1419–1426. [Google Scholar] [CrossRef]
- Mullen, R.J.; Eicher, E.M.; Sidman, R.L. Purkinje cell degeneration, a new neurological mutation in the mouse. Proc. Natl. Acad. Sci. USA 1976, 73, 208–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Gorman, S.; Sidman, R.L. Degeneration of thalamic neurons in “Purkinje cell degeneration” mutant mice. I. Distribution of neuron loss. J. Comp. Neurol. 1985, 234, 277–297. [Google Scholar] [CrossRef] [PubMed]
- Greer, C.A.; Shepherd, G.M. Mitral cell degeneration and sensory function in the neurological mutant mouse Purkinje cell degeneration (PCD). Brain Res. 1982, 235, 156–161. [Google Scholar] [CrossRef]
- Muñoz-Castañeda, R.; Díaz, D.; Peris, L.; Andrieux, A.; Bosc, C.; Muñoz-Castañeda, J.M.; Janke, C.; Alonso, J.R.; Moutin, M.-J.; Weruaga, E. Cytoskeleton stability is essential for the integrity of the cerebellum and its motor- and affective-related behaviors. Sci. Rep. 2018, 8, 3072. [Google Scholar] [CrossRef] [Green Version]
- Baltanás, F.C.; Casafont, I.; Weruaga, E.; Alonso, J.R.; Berciano, M.T.; Lafarga, M. Nucleolar Disruption and Cajal Body Disassembly are Nuclear Hallmarks of DNA Damage-Induced Neurodegeneration in Purkinje Cells. Brain Pathol. 2010, 21, 374–388. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Gonzalez, A.; La Spada, A.R.; Treadaway, J.; Higdon, J.C.; Harris, B.S.; Sidman, R.L.; Morgan, J.I.; Zuo, J. Purkinje cell degeneration (pcd) phenotypes caused by mutations in the axotomy-induced gene, Nna1. Science 2002, 295, 1904–1906. [Google Scholar] [CrossRef]
- Chakrabarti, L.; Neal, J.T.; Miles, M.; Martinez, R.A.; Smith, A.C.; Sopher, B.L.; La Spada, A.R. The Purkinje cell degeneration 5J mutation is a single amino acid insertion that destabilizes Nna1 protein. Mamm. Genome 2006, 17, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Harris, A.; Morgan, J.I.; Pecot, M.; Soumare, A.; Osborne, A.; Soares, H.D. Regenerating motor neurons express Nna1, a novel ATP/GTP-binding protein related to zinc carboxypeptidases. Mol. Cell Neurosci. 2000, 16, 578–596. [Google Scholar] [CrossRef] [PubMed]
- Rogowski, K.; Van Dijk, J.; Magiera, M.M.; Bosc, C.; Deloulme, J.C.; Bosson, A.; Peris, L.; Gold, N.D.; Lacroix, B.; Bosch Grau, M.; et al. A family of protein-deglutamylating enzymes associated with neurodegeneration. Cell 2010, 143, 564–578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baltanás, F.C.; Berciano, M.T.; Valero, J.; Gómez, C.; Díaz, D.; Alonso, J.R.; Lafarga, M.; Weruaga, E. Differential glial activation during the degeneration of Purkinje cells and mitral cells in the PCD mutant mice. Glia 2012, 61, 254–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mashimo, T.; Hadjebi, O.; Amair-Pinedo, F.; Tsurumi, T.; Langa, F.; Serikawa, T.; Sotelo, C.; Guénet, J.-L.; Rosa, J.L. Progressive Purkinje Cell Degeneration in tambaleante Mutant Mice Is a Consequence of a Missense Mutation in HERC1 E3 Ubiquitin Ligase. PLoS Genet. 2009, 5, e1000784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruiz, R.; Pérez-Villegas, E.M.; Bachiller, S.; Rosa, J.L.; Armengol, J.A. HERC 1 Ubiquitin Ligase Mutation Affects Neocortical, CA3 Hippocampal and Spinal Cord Projection Neurons: An Ultrastructural Study. Front. Neuroanat. 2016, 10, 42. [Google Scholar] [CrossRef] [Green Version]
- Winkelman, M.D.; Hines, J.D. Cerebellar degeneration caused by high-dose cytosine arabinoside: A clinicopathological study. Ann. Neurol. 1983, 14, 520–527. [Google Scholar] [CrossRef]
- Ciesielski, K.T.; Yanofsky, R.; Ludwig, R.N.; Hill, D.E.; Hart, B.L.; Astur, R.S.; Snyder, T. Hypoplasia of the Cerebellar Vermis and Cognitive Deficits in Survivors of Childhood Leukemia. Arch. Neurol. 1994, 51, 985–993. [Google Scholar] [CrossRef]
- Butterworth, R.F.; Hamel, E.; Landreville, F.; Barbeau, A. Cerebellar ataxia produced by 3-acetyl pyridine in rat. Can. J. Neurol. Sci. 1978, 5, 131–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hess, B.H.; Krewet, J.A.; Tolbert, D.L. Olivocerebellar projections are necessary for exogenous trophic factors to delay heredo-Purkinje cell degeneration. Brain Res. 2003, 986, 54–62. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, C.; Hawkes, R. Selective Purkinje cell ectopia in the cerebellum of the Weaver mouse. J. Comp. Neurol. 2001, 439, 151–161. [Google Scholar] [CrossRef] [PubMed]
- Kostenko, S.; Moens, U. Heat shock protein 27 phosphorylation: Kinases, phosphatases, functions and pathology. Cells Mol. Life Sci. 2009, 66, 3289–3307. [Google Scholar] [CrossRef]
- Sawada, K.; Haga, H.; Fukui, Y. Alternating array of tyrosine hydroxylase and heat shock protein 25 immunopositive Purkinje cell stripes in zebrin II-defined transverse zoneof the cerebellum of rolling mouse Nagoya. Brain Res. 2010, 9, 46–53. [Google Scholar] [CrossRef]
- Kim, C.-H.; Shin, J.J.; Kim, J.; Kim, S.J. Reduced spike frequency adaptation in Purkinje cells of the vestibulocerebellum. Neurosci. Lett. 2013, 535, 45–50. [Google Scholar] [CrossRef]
- Martin, K.B.; Williams, I.M.; Cluzeau, C.V.; Cougnoux, A.; Dale, R.K.; Iben, J.R.; Cawley, N.X.; Wassif, C.A.; Porter, F.D. Identification of Novel Pathways Associated with Patterned Cerebellar Purkinje Neuron Degeneration in Niemann-Pick Disease, Type C1. Int. J. Mol. Sci. 2019, 21, 292. [Google Scholar] [CrossRef] [Green Version]
- Jakob, U.; Gaestel, M.; Engel, K.; Buchner, J. Small heat shock proteins are molecular chaperones. J. Biol. Chem. 1993, 268, 1517–1520. [Google Scholar] [CrossRef]
- Lavoie, J.N.; Hickey, E.; Weber, L.A.; Landry, J. Modulation of actin microfilament dynamics and fluid phase pinocytosis by phosphorylation of heat shock protein 27. J. Biol. Chem. 1993, 268, 24210–24214. [Google Scholar] [CrossRef]
- Huot, J.; Houle, F.; Spitz, D.; Landry, J. HSP27 phosphorylation-mediated resistance against actin fragmentation and cell death induced by oxidative stress. Cancer Res 1996, 56, 273–279. [Google Scholar]
- Carver, J.A.; Aquilina, J.A.; Cooper, P.G.; Williams, G.A.; Truscott, R.J. Alpha-crystallin: Molecular chaperone and protein surfactant. Biochim. Biophys. Acta 1994, 1204, 195–206. [Google Scholar] [CrossRef] [PubMed]
- Boelens, W.C.; De Jong, W.W. Alpha-Crystallins, versatile stress-proteins. Mol. Biol. Rep. 1995, 21, 75–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Y.; Macrae, T.H. Small heat shock proteins: Molecular structure and chaperone function. Cells Mol. Life Sci. 2005, 62, 2460–2476. [Google Scholar] [CrossRef]
- Armstrong, C.L.; Krueger-Naug, A.M.; Currie, R.W.; Hawkes, R. Constitutive expression of the 25-kDa heat shock protein Hsp25 reveals novel parasagittal bands of Purkinje cells in the adult mouse cerebellar cortex. J. Comp. Neurol. 2000, 416, 383–397. [Google Scholar] [CrossRef]
- Valero, J.; Berciano, M.T.; Weruaga, E.; Lafarga, M.; Alonso, J.R. Pre-neurodegeneration of mitral cells in the pcd mutant mouse is associated with DNA damage, transcriptional repression, and reorganization of nuclear speckles and Cajal bodies. Mol. Cells Neurosci. 2006, 33, 283–295. [Google Scholar] [CrossRef] [PubMed]
- Takata, H.; Hanafusa, T.; Mori, T.; Shimura, M.; Iida, Y.; Ishikawa, K.; Yoshikawa, K.; Yoshikawa, Y.; Maeshima, K. Chromatin Compaction Protects Genomic DNA from Radiation Damage. PLoS ONE 2013, 8, e75622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cann, K.L.; Dellaire, G. Heterochromatin and the DNA damage response: The need to relaxThis paper is one of a selection of papers in a Special Issue entitled 31st Annual International Asilomar Chromatin and Chromosomes Conference, and has undergone the Journal’s usual peer review process. Biochem. Cell Biol. 2011, 89, 45–60. [Google Scholar] [CrossRef]
- Falk, M.; Lukásová, E.; Kozubek, S. Chromatin structure influences the sensitivity of DNA to gamma-radiation. Biochim. Biophys. Acta 2008, 1783, 2398–2414. [Google Scholar] [CrossRef] [Green Version]
- Friedman, D.; Strowbridge, B.W. Functional Role of NMDA Autoreceptors in Olfactory Mitral Cells. J. Neurophysiol. 2000, 84, 39–50. [Google Scholar] [CrossRef] [Green Version]
- Lowe, G. Electrical signaling in the olfactory bulb. Curr. Opin. Neurobiol. 2003, 13, 476–481. [Google Scholar] [CrossRef]
- Djurisic, M.; Antic, S.; Chen, W.R.; Zecevic, D. Voltage Imaging from Dendrites of Mitral Cells: EPSP Attenuation and Spike Trigger Zones. J. Neurosci. 2004, 24, 6703–6714. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Model | Mutation | Cause | Pathology | Degeneration Pattern |
|---|---|---|---|---|
| Leaner | Spontaneous tgla/tgla. | Ca2+ channel subunit damaged. | Ataxia starts at P10 and Purkinje cell loss observed at P40. | Posterior and nodular zones are less vulnerable. |
| Toppler | Spontaneous. Mutation located in chromosome 8. | Unknown. | Ataxia starts at 4–5 weeks of age. Major Purkinje cell death at P14–P30. | Purkinje cells of lobe X survive at P30. |
| Robotic | Induced by N-ethyl-N-nitrosourea. Af4 gene is altered. | Transcription cofactor coded by Af4 is truncated. | Ataxia starts at 3 weeks of age. Purkinje cell death starts at the 8th week. | Antero-posterior neurodegeneration. |
| Shaker (rat) | Spontaneous. Unknown. Two variants described: severe and mild. | Unknown. | Ataxia and body tremors at 3 months of age in the severe variant. Ataxia present only the in mild variant. | The anterior region is the most affected. Lobe X appears unaffected. |
| Lurcher | Spontaneous. +/Lc present ataxia. Lc/Lc is lethal. | δ2 glutamate receptor acts as a Ca2+ channel. | Purkinje cell death occurs from P10 to P65. | Antero-posterior neurodegeneration. Lobe X degeneration is delayed. |
| NPC1 | Spontaneous. Npc1 gene is mutated. | Intracellular cholesterol transport is altered. | Purkinje cell degeneration starts at P40. | Antero-posterior neurodegeneration. Lobe X is the most resistant region. |
| Nervous | Spontaneous. Located in chromosome 8 but unknown. | Unknown | Neurodegeneration starts at birth and slows down after two months. | Zebrin II positive Purkinje cells are more vulnerable. Lobe X shows some surviving cells. |
| PCD | Spontaneous. pcd/pcd. | Ccp1 gene affected. Hyper-glutamylation of microtubules. | Purkinje cell death starts around P18. | Lobe X is the last lobe to degenerate. Some survival cells detected at 9 months. |
| Tambaleante | Spontaneous. Glycine-to-glutamate substitution. | E3 ubiquitin ligase protein HERC1 overexpressed. | Some Purkinje cells remain alive at 2.5 months. | Random. Lobe X cells remain alive a few days longer. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hernández-Pérez, C.; Weruaga, E.; Díaz, D. Lobe X of the Cerebellum: A Natural Neuro-Resistant Region. Anatomia 2023, 2, 43-62. https://doi.org/10.3390/anatomia2010005
Hernández-Pérez C, Weruaga E, Díaz D. Lobe X of the Cerebellum: A Natural Neuro-Resistant Region. Anatomia. 2023; 2(1):43-62. https://doi.org/10.3390/anatomia2010005
Chicago/Turabian StyleHernández-Pérez, Carlos, Eduardo Weruaga, and David Díaz. 2023. "Lobe X of the Cerebellum: A Natural Neuro-Resistant Region" Anatomia 2, no. 1: 43-62. https://doi.org/10.3390/anatomia2010005
APA StyleHernández-Pérez, C., Weruaga, E., & Díaz, D. (2023). Lobe X of the Cerebellum: A Natural Neuro-Resistant Region. Anatomia, 2(1), 43-62. https://doi.org/10.3390/anatomia2010005