
Epidermal Growth Factor Receptor Expression and Resistance Patterns to Targeted Therapy in Non-Small Cell Lung Cancer: A Review


Abstract
:1. Introduction
2. EGFR Molecular Biology
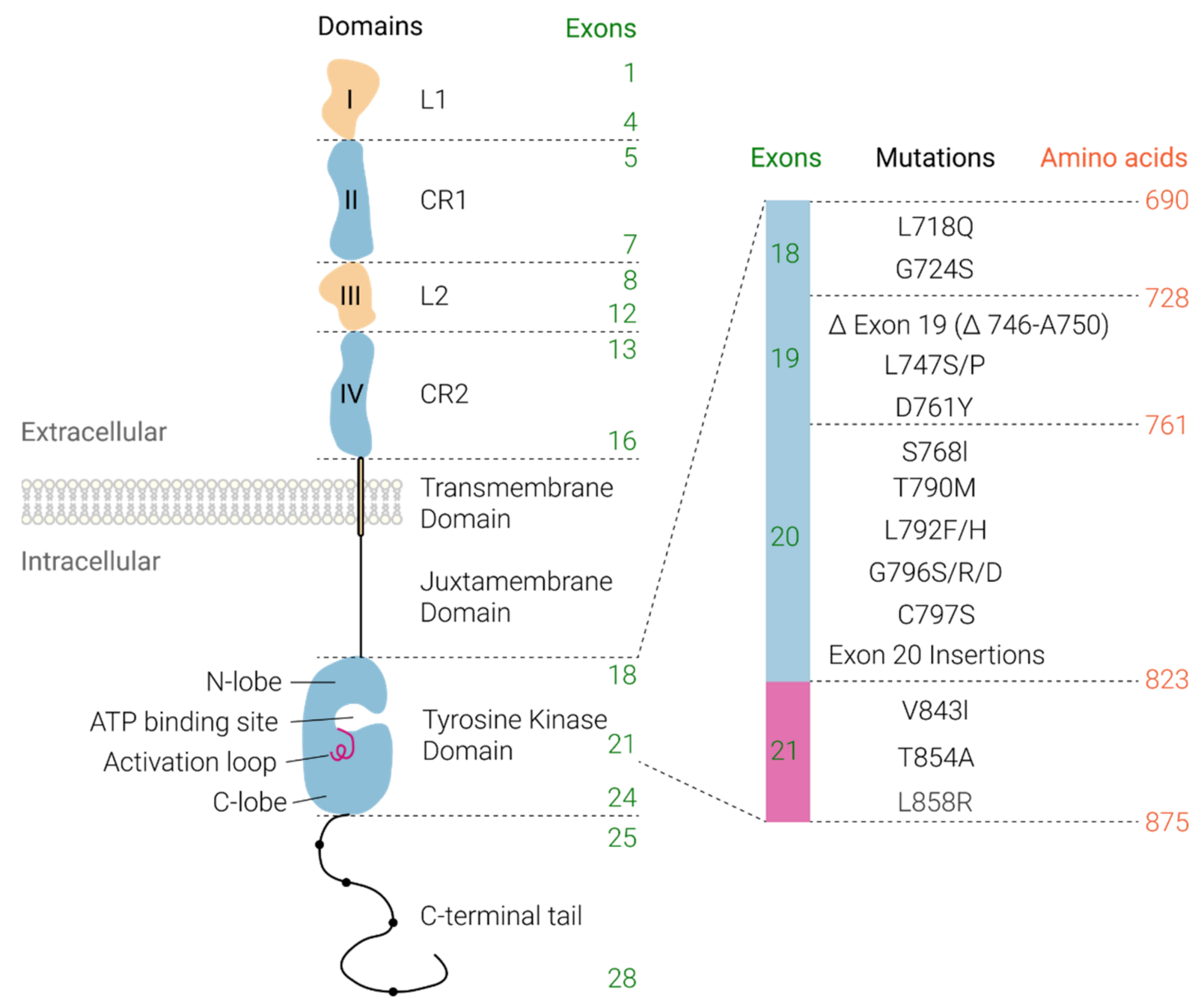
Receptor Structure and Activation
3. EGFR in NSCLC
4. EGFR-Targeted Therapy in NSCLC and Mechanisms of Resistance
4.1. Tyrosine Kinase Inhibitors (TKIs)
4.1.1. Acquired Resistance to First Generation TKIs
4.1.2. Acquired Resistance to Third-Generation TKIs
4.1.3. Acquired Resistance via Downstream Activation
4.1.4. Acquired Resistance via Alternative Pathways
4.1.5. Acquired Resistance via Other Mechanisms
4.2. Monoclonal Antibody (mAb) Therapy
5. Conclusions
6. Patents
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| Acronym | Definition |
| AGAP3 | ArfGAP with GTPase Domain, Ankyrin Repeat and PH Domain 3 |
| AGK | Acylglycerol kinase |
| Akt | Protein kinase B |
| AKT1 | AKT Serine/Threonine Kinase 1 |
| ALK | Anaplastic lymphoma kinase |
| ARMC10 | Armadillo Repeat Containing 10 |
| ATP | Adenosine triphosphate |
| BIM | B cell lymphoma-2-like |
| BRAF | B-Raf Proto-Oncogene |
| CCDC6 | Coiled-Coil Domain Containing 6 |
| CDC123 | Cell Division Cycle 123 |
| CEBPZ | CCAAT Enhancer Binding Protein Zeta |
| CRKL | CRK like proto-oncogene |
| DCBLD1 | Discoidin, CUB and LCCL Domain Containing 1 |
| DOCK4 | Dedicator of cytokinesis 4 |
| EGFR | Epidermal growth factor receptor |
| EIF4G2 | Eukaryotic translation initiation factor 4 gamma 2 |
| EMT | Epithelial to mesenchymal transition |
| EPS15 | Epidermal growth factor receptor substrate 15 |
| ERK | Extracellular-signal-regulated kinase |
| FGFR | Fibroblast growth factor receptor |
| GAB1 | Grb2 associated binding protein 1 |
| GAS6 | Growth arrest specific 6 |
| HER | Human epidermal growth factor receptor 2 |
| IGF1R | Insulin like growth factor 1 receptor |
| KIF5B | Kinesin Family Member 5B |
| KRAS | Kirsten rate sarcoma viral oncogene |
| LRRC71 | Leucine rich repeat containing 71 |
| MAPK | Mitogen-activated protein kinase |
| MEK | Mitogen-activated protein kinase kinase |
| miR-21 | mircoRNA 21 |
| mTOR | Mechanistic target of rapamycin |
| NF-κB | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| NTRK1 | Neurotrophic Receptor Tyrosine Kinase 1 |
| PDCD4 | Programmed Cell Death 4 |
| PFS | Progression free survival |
| PI3K | Phosphoinositide 3-kinase |
| PI3KCA | PI3K catalytic subunit alpha |
| PLEKHA6 | Pleckstrin homology domain containing A6 |
| PTEN | Phosphatase and tensin homolog |
| OS | Overall survival |
| RET | Rearranged during transfection |
| ROS1 | C-ros oncogene 1 |
| SALL2 | Spalt like transcription factor 2 |
| SCC | Squamous cell carcinoma |
| SCLC | Small cell lung cancer |
| STRN | Striatin |
| TACC3 | Transforming acidic coiled-coil containing protein 3 |
| TKI | Tyrosine kinase inhibitor |
| TRIM24 | Tripartite Motif Containing 24 |
References
- Fitzmaurice, C.; Dicker, D.; Pain, A.; Hamavid, H.; Moradi-Lakeh, M.; MacIntyre, M.F.; Allen, C.; Hansen, G.; Woodbrook, R.; Wolfe, C. The global burden of cancer 2013. JAMA Oncol. 2015, 1, 505–527. [Google Scholar] [CrossRef] [PubMed]
- Sher, T.; Dy, G.K.; Adjei, A.A. Small cell lung cancer. Mayo Clin. Proc. 2008, 83, 355–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Travis, W.D.; Brambilla, E.; Burke, A.P.; Marx, A.; Nicholson, A.G. Introduction to the 2015 World Health Organization classification of tumors of the lung, pleura, thymus, and heart. J. Thorac. Oncol. 2015, 10, 1240–1242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Travis, W.D.; Brambilla, E.; Nicholson, A.G.; Yatabe, Y.; Austin, J.H.; Beasley, M.B.; Chirieac, L.R.; Dacic, S.; Duhig, E.; Flieder, D.B. The 2015 World Health Organization classification of lung tumors: Impact of genetic, clinical and radiologic advances since the 2004 classification. J. Thorac. Oncol. 2015, 10, 1243–1260. [Google Scholar] [CrossRef] [Green Version]
- Zhou, F.; Zhou, C. Lung cancer in never smokers-the East Asian experience. Transl. Lung Cancer Res. 2018, 7, 450–463. [Google Scholar] [CrossRef]
- Garinet, S.; Laurent-Puig, P.; Blons, H.; Oudart, J.-B. Current and Future Molecular Testing in NSCLC, What Can We Expect from New Sequencing Technologies? J. Clin. Med. 2018, 7, 144. [Google Scholar] [CrossRef] [Green Version]
- Barlesi, F.; Mazieres, J.; Merlio, J.-P.; Debieuvre, D.; Mosser, J.; Lena, H.; Ouafik, L.H.; Besse, B.; Rouquette, I.; Westeel, V. Routine molecular profiling of patients with advanced non-small-cell lung cancer: Results of a 1-year nationwide programme of the French Cooperative Thoracic Intergroup (IFCT). Lancet 2016, 387, 1415–1426. [Google Scholar] [CrossRef]
- Kris, M.G.; Johnson, B.E.; Berry, L.D.; Kwiatkowski, D.J.; Iafrate, A.J.; Wistuba, I.I.; Varella-Garcia, M.; Franklin, W.A.; Aronson, S.L.; Su, P.-F. Using multiplexed assays of oncogenic drivers in lung cancers to select targeted drugs. JAMA 2014, 311, 1998–2006. [Google Scholar] [CrossRef]
- da Cunha Santos, G.; Shepherd, F.A.; Tsao, M.S. EGFR mutations and lung cancer. Annu. Rev. Pathol. 2011, 6, 49–69. [Google Scholar] [CrossRef] [Green Version]
- Tseng, C.H.; Chiang, C.J.; Tseng, J.S.; Yang, T.Y.; Hsu, K.H.; Chen, K.C.; Wang, C.L.; Chen, C.Y.; Yen, S.H.; Tsai, C.M.; et al. EGFR mutation, smoking, and gender in advanced lung adenocarcinoma. Oncotarget 2017, 8, 98384–98393. [Google Scholar] [CrossRef] [Green Version]
- Thomas, R.; Weihua, Z. Rethink of EGFR in Cancer with Its Kinase Independent Function on Board. Front. Oncol. 2019, 9, 800. [Google Scholar] [CrossRef]
- Pelloski, C.E.; Ballman, K.V.; Furth, A.F.; Zhang, L.; Lin, E.; Sulman, E.P.; Bhat, K.; McDonald, J.M.; Yung, W.K.; Colman, H.; et al. Epidermal growth factor receptor variant III status defines clinically distinct subtypes of glioblastoma. J. Clin. Oncol. 2007, 25, 2288–2294. [Google Scholar] [CrossRef]
- Sugawa, N.; Ekstrand, A.J.; James, C.D.; Collins, V.P. Identical splicing of aberrant epidermal growth factor receptor transcripts from amplified rearranged genes in human glioblastomas. Proc. Natl. Acad. Sci. USA 1990, 87, 8602–8606. [Google Scholar] [CrossRef] [Green Version]
- Del Vecchio, C.; Giacomini, C.; Vogel, H.; Jensen, K.; Florio, T.; Merlo, A.; Pollack, J.; Wong, A. EGFRvIII gene rearrangement is an early event in glioblastoma tumorigenesis and expression defines a hierarchy modulated by epigenetic mechanisms. Oncogene 2013, 32, 2670–2681. [Google Scholar] [CrossRef] [Green Version]
- De Luca, A.; Normanno, N. Predictive biomarkers to tyrosine kinase inhibitors for the epidermal growth factor receptor in non-small-cell lung cancer. Curr. Drug Targets 2010, 11, 851–864. [Google Scholar] [CrossRef]
- Li, A.R.; Chitale, D.; Riely, G.J.; Pao, W.; Miller, V.A.; Zakowski, M.F.; Rusch, V.; Kris, M.G.; Ladanyi, M. EGFR mutations in lung adenocarcinomas: Clinical testing experience and relationship to EGFR gene copy number and immunohistochemical expression. J. Mol. Diagn. 2008, 10, 242–248. [Google Scholar] [CrossRef] [Green Version]
- Jung, M.J.; Woo, C.G.; Lee, S.; Chin, S.; Kim, H.K.; Kwak, J.J.; Koh, E.S.; Lee, B.; Jang, K.-T.; Moon, A. Gene copy number variation and protein overexpression of EGFR and HER2 in distal extrahepatic cholangiocarcinoma. Pathology 2017, 49, 582–588. [Google Scholar] [CrossRef]
- Birkman, E.-M.; Ålgars, A.; Lintunen, M.; Ristamäki, R.; Sundström, J.; Carpén, O. EGFR gene amplification is relatively common and associates with outcome in intestinal adenocarcinoma of the stomach, gastro-oesophageal junction and distal oesophagus. BMC Cancer 2016, 16, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Wang, W.; Wang, C.; Wang, L.; Yang, M.; Qi, M.; Su, H.; Sun, X.; Liu, Z.; Zhang, J. Characterization of EGFR family gene aberrations in cholangiocarcinoma. Oncol. Rep. 2014, 32, 700–708. [Google Scholar] [CrossRef] [Green Version]
- Rossi, E.; Villanacci, V.; Danesino, C.; Donato, F.; Nascimbeni, R.; Bassotti, G. Epidermal growth factor receptor overexpression/amplification in adenocarcinomas arising in the gastrointestinal tract. Rev. Esp. Enferm. Dig. 2011, 103, 632–639. [Google Scholar] [CrossRef] [Green Version]
- Huang, S.-F.; Cheng, S.-D.; Chien, H.-T.; Liao, C.-T.; Chen, I.-H.; Wang, H.-M.; Chuang, W.-Y.; Wang, C.-Y.; Hsieh, L.-L. Relationship between epidermal growth factor receptor gene copy number and protein expression in oral cavity squamous cell carcinoma. Oral Oncol. 2012, 48, 67–72. [Google Scholar] [CrossRef] [PubMed]
- Itakura, Y.; Sasano, H.; Shiga, C.; Furukawa, Y.; Shiga, K.; Mori, S.; Nagura, H. Epidermal growth factor receptor overexpression in esophageal carcinoma. An immunohistochemical study correlated with clinicopathologic findings and DNA amplification. Cancer 1994, 74, 795–804. [Google Scholar] [CrossRef]
- Lemmon, M.A.; Schlessinger, J. Cell signaling by receptor tyrosine kinases. Cell 2010, 141, 1117–1134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oda, K.; Matsuoka, Y.; Funahashi, A.; Kitano, H. A comprehensive pathway map of epidermal growth factor receptor signaling. Mol. Syst. Biol. 2005, 1, 2005.0010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yarden, Y.; Sliwkowski, M.X. Untangling the ErbB signalling network. Nat. Rev. Mol. Cell Biol. 2001, 2, 127–137. [Google Scholar] [CrossRef]
- Wang, Z. ErbB Receptors and Cancer. Methods Mol. Biol. 2017, 1652, 3–35. [Google Scholar] [CrossRef]
- Normanno, N.; De Luca, A.; Bianco, C.; Strizzi, L.; Mancino, M.; Maiello, M.R.; Carotenuto, A.; De Feo, G.; Caponigro, F.; Salomon, D.S. Epidermal growth factor receptor (EGFR) signaling in cancer. Gene 2006, 366, 2–16. [Google Scholar] [CrossRef]
- Jorissen, R.N.; Walker, F.; Pouliot, N.; Garrett, T.P.; Ward, C.W.; Burgess, A.W. Epidermal growth factor receptor: Mechanisms of activation and signalling. Exp. Cell Res. 2003, 284, 31–53. [Google Scholar] [CrossRef]
- Wee, P.; Wang, Z. Epidermal Growth Factor Receptor Cell Proliferation Signaling Pathways. Cancers (Basel) 2017, 9. [Google Scholar] [CrossRef] [Green Version]
- Quesnelle, K.M.; Boehm, A.L.; Grandis, J.R. STAT-mediated EGFR signaling in cancer. J. Cell. Biochem. 2007, 102, 311–319. [Google Scholar] [CrossRef]
- Kumar, A.; Petri, E.T.; Halmos, B.; Boggon, T.J. Structure and clinical relevance of the epidermal growth factor receptor in human cancer. J. Clin. Oncol. 2008, 26, 1742–1751. [Google Scholar] [CrossRef] [Green Version]
- Chan, B.A.; Hughes, B.G. Targeted therapy for non-small cell lung cancer: Current standards and the promise of the future. Transl. Lung Cancer Res. 2015, 4, 36–54. [Google Scholar] [CrossRef]
- Pao, W.; Miller, V.; Zakowski, M.; Doherty, J.; Politi, K.; Sarkaria, I.; Singh, B.; Heelan, R.; Rusch, V.; Fulton, L. EGF receptor gene mutations are common in lung cancers from “never smokers” and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc. Natl. Acad. Sci. USA 2004, 101, 13306–13311. [Google Scholar] [CrossRef] [Green Version]
- Ohsaki, Y.; Tanno, S.; Fujita, Y.; Toyoshima, E.; Fujiuchi, S.; Nishigaki, Y.; Ishida, S.; Nagase, A.; Miyokawa, N.; Hirata, S. Epidermal growth factor receptor expression correlates with poor prognosis in non-small cell lung cancer patients with p53 overexpression. Oncol. Rep. 2000, 7, 603–610. [Google Scholar] [CrossRef]
- Pastorino, U.; Andreola, S.; Tagliabue, E.; Pezzella, F.; Incarbone, M.; Sozzi, G.; Buyse, M.; Menard, S.; Pierotti, M.; Rilke, F. Immunocytochemical markers in stage I lung cancer: Relevance to prognosis. J. Clin. Oncol. 1997, 15, 2858–2865. [Google Scholar] [CrossRef]
- Pfeiffer, P.; Clausen, P.; Andersen, K.; Rose, C. Lack of prognostic significance of epidermal growth factor receptor and the oncoprotein p185 HER-2 in patients with systemically untreated non-small-cell lung cancer: An immunohistochemical study on cryosections. Br. J. Cancer 1996, 74, 86–91. [Google Scholar] [CrossRef] [Green Version]
- Cox, G.; Jones, J.L.; O’Byrne, K.J. Matrix metalloproteinase 9 and the epidermal growth factor signal pathway in operable non-small cell lung cancer. Clin. Cancer Res. 2000, 6, 2349–2355. [Google Scholar]
- Hirsch, F.R.; Scagliotti, G.V.; Langer, C.J.; Varella-Garcia, M.; Franklin, W.A. Epidermal growth factor family of receptors in preneoplasia and lung cancer: Perspectives for targeted therapies. Lung Cancer 2003, 41, 29–42. [Google Scholar] [CrossRef]
- Rusch, V.; Klimstra, D.; Venkatraman, E.; Pisters, P.; Langenfeld, J.; Dmitrovsky, E. Overexpression of the epidermal growth factor receptor and its ligand transforming growth factor alpha is frequent in resectable non-small cell lung cancer but does not predict tumor progression. Clin. Cancer Res. 1997, 3, 515–522. [Google Scholar]
- Selvaggi, G.; Novello, S.; Torri, V.; Leonardo, E.; De Giuli, P.; Borasio, P.; Mossetti, C.; Ardissone, F.; Lausi, P.; Scagliotti, G.V. Epidermal growth factor receptor overexpression correlates with a poor prognosis in completely resected non-small-cell lung cancer. Ann. Oncol. 2004, 15, 28–32. [Google Scholar] [CrossRef]
- Fontanini, G.; De Laurentiis, M.; Vignati, S.; Chinè, S.; Lucchi, M.; Silvestri, V.; Mussi, A.; De Placido, S.; Tortora, G.; Bianco, A.R. Evaluation of epidermal growth factor-related growth factors and receptors and of neoangiogenesis in completely resected stage I-IIIA non-small-cell lung cancer: Amphiregulin and microvessel count are independent prognostic indicators of survival. Clin. Cancer Res. 1998, 4, 241–249. [Google Scholar] [PubMed]
- Volm, M.; Rittgen, W.; Drings, P. Prognostic value of ERBB-1, VEGF, cyclin A, FOS, JUN and MYC in patients with squamous cell lung carcinomas. Br. J. Cancer 1998, 77, 663–669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greatens, T.M.; Niehans, G.A.; Rubins, J.B.; Jessurun, J.; Kratzke, R.A.; Maddaus, M.A.; Niewoehner, D.E. Do molecular markers predict survival in non–small-cell lung cancer? Am. J. Respir. Crit. Care Med. 1998, 157, 1093–1097. [Google Scholar] [CrossRef]
- Nakamura, H.; Kawasaki, N.; Taguchi, M.; Kabasawa, K. Survival impact of epidermal growth factor receptor overexpression in patients with non-small cell lung cancer: A meta-analysis. Thorax 2006, 61, 140–145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- AACR Project GENIE: Powering Precision Medicine through an International Consortium. Cancer Discov. 2017, 7, 818–831. [CrossRef] [PubMed] [Green Version]
- Spindler, K.L.; Lindebjerg, J.; Nielsen, J.N.; Olsen, D.A.; Bisgård, C.; Brandslund, I.; Jakobsen, A. Epidermal growth factor receptor analyses in colorectal cancer: A comparison of methods. Int. J. Oncol. 2006, 29, 1159–1165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scartozzi, M.; Bearzi, I.; Berardi, R.; Mandolesi, A.; Fabris, G.; Cascinu, S. Epidermal growth factor receptor (EGFR) status in primary colorectal tumors does not correlate with EGFR expression in related metastatic sites: Implications for treatment with EGFR-targeted monoclonal antibodies. J. Clin. Oncol. 2004, 22, 4772–4778. [Google Scholar] [CrossRef]
- Saltz, L.B.; Meropol, N.J.; Loehrer, P.J., Sr.; Needle, M.N.; Kopit, J.; Mayer, R.J. Phase II trial of cetuximab in patients with refractory colorectal cancer that expresses the epidermal growth factor receptor. J. Clin. Oncol. 2004, 22, 1201–1208. [Google Scholar] [CrossRef]
- Goldstein, N.S.; Armin, M. Epidermal growth factor receptor immunohistochemical reactivity in patients with American Joint Committee on Cancer Stage IV colon adenocarcinoma. Cancer 2001, 92, 1331–1346. [Google Scholar] [CrossRef]
- Hashmi, A.A.; Hussain, Z.F.; Aijaz, S.; Irfan, M.; Khan, E.Y.; Naz, S.; Faridi, N.; Khan, A.; Edhi, M.M. Immunohistochemical expression of epidermal growth factor receptor (EGFR) in South Asian head and neck squamous cell carcinoma: Association with various risk factors and clinico-pathologic and prognostic parameters. World J. Surg. Oncol. 2018, 16, 118. [Google Scholar] [CrossRef]
- Grandis, J.R.; Tweardy, D.J. Elevated levels of transforming growth factor alpha and epidermal growth factor receptor messenger RNA are early markers of carcinogenesis in head and neck cancer. Cancer Res. 1993, 53, 3579–3584. [Google Scholar]
- Boeck, S.; Jung, A.; Laubender, R.P.; Neumann, J.; Egg, R.; Goritschan, C.; Vehling-Kaiser, U.; Winkelmann, C.; Fischer von Weikersthal, L.; Clemens, M.R.; et al. EGFR pathway biomarkers in erlotinib-treated patients with advanced pancreatic cancer: Translational results from the randomised, crossover phase 3 trial AIO-PK0104. Br. J. Cancer 2013, 108, 469–476. [Google Scholar] [CrossRef]
- Dancer, J.; Takei, H.; Ro, J.Y.; Lowery-Nordberg, M. Coexpression of EGFR and HER-2 in pancreatic ductal adenocarcinoma: A comparative study using immunohistochemistry correlated with gene amplification by fluorescencent in situ hybridization. Oncol. Rep. 2007, 18, 151–155. [Google Scholar] [CrossRef] [Green Version]
- Bloomston, M.; Bhardwaj, A.; Ellison, E.C.; Frankel, W.L. Epidermal growth factor receptor expression in pancreatic carcinoma using tissue microarray technique. Dig. Surg. 2006, 23, 74–79. [Google Scholar] [CrossRef]
- Gonzalez-Conchas, G.A.; Rodriguez-Romo, L.; Hernandez-Barajas, D.; Gonzalez-Guerrero, J.F.; Rodriguez-Fernandez, I.A.; Verdines-Perez, A.; Templeton, A.J.; Ocana, A.; Seruga, B.; Tannock, I.F. Epidermal growth factor receptor overexpression and outcomes in early breast cancer: A systematic review and a meta-analysis. Cancer Treat. Rev. 2018, 62, 1–8. [Google Scholar] [CrossRef]
- Di Lorenzo, G.; Tortora, G.; D’Armiento, F.P.; De Rosa, G.; Staibano, S.; Autorino, R.; D’Armiento, M.; De Laurentiis, M.; De Placido, S.; Catalano, G. Expression of epidermal growth factor receptor correlates with disease relapse and progression to androgen-independence in human prostate cancer. Clin. Cancer Res. 2002, 8, 3438–3444. [Google Scholar]
- de Muga, S.; Hernández, S.; Agell, L.; Salido, M.; Juanpere, N.; Lorenzo, M.; Lorente, J.A.; Serrano, S.; Lloreta, J. Molecular alterations of EGFR and PTEN in prostate cancer: Association with high-grade and advanced-stage carcinomas. Mod. Pathol. 2010, 23, 703–712. [Google Scholar] [CrossRef]
- Lin, G.; Sun, X.-J.; Han, Q.-B.; Wang, Z.; Xu, Y.-P.; Gu, J.-L.; Wu, W.; Zhang, G.U.; Hu, J.-L.; Sun, W.-Y.; et al. Epidermal growth factor receptor protein overexpression and gene amplification are associated with aggressive biological behaviors of esophageal squamous cell carcinoma. Oncol. Lett. 2015, 10, 901–906. [Google Scholar] [CrossRef] [Green Version]
- Abedi-Ardekani, B.; Dar, N.A.; Mir, M.M.; Zargar, S.A.; Lone, M.M.; Martel-Planche, G.; Villar, S.; Mounawar, M.; Saidi, F.; Malekzadeh, R.; et al. Epidermal growth factor receptor (EGFR) mutations and expression in squamous cell carcinoma of the esophagus in central Asia. BMC Cancer 2012, 12, 602. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.A.; Lee, H.S.; Lee, H.E.; Jeon, Y.K.; Yang, H.K.; Kim, W.H. EGFR in gastric carcinomas: Prognostic significance of protein overexpression and high gene copy number. Histopathology 2008, 52, 738–746. [Google Scholar] [CrossRef]
- Galizia, G.; Lieto, E.; Orditura, M.; Castellano, P.; Mura, A.L.; Imperatore, V.; Pinto, M.; Zamboli, A.; De Vita, F.; Ferraraccio, F. Epidermal growth factor receptor (EGFR) expression is associated with a worse prognosis in gastric cancer patients undergoing curative surgery. World J. Surg. 2007, 31, 1458–1468. [Google Scholar] [CrossRef] [PubMed]
- Kira, S.; Nakanishi, T.; Suemori, S.; Kitamoto, M.; Watanabe, Y.; Kajiyama, G. Expression of transforming growth factor alpha and epidermal growth factor receptor in human hepatocellular carcinoma. Liver 1997, 17, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Ito, Y.; Takeda, T.; Sakon, M.; Tsujimoto, M.; Higashiyama, S.; Noda, K.; Miyoshi, E.; Monden, M.; Matsuura, N. Expression and clinical significance of erb-B receptor family in hepatocellular carcinoma. Br. J. Cancer 2001, 84, 1377–1383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watanabe, K.; Tachibana, O.; Sata, K.; Yonekawa, Y.; Kleihues, P.; Ohgaki, H. Overexpression of the EGF receptor and p53 mutations are mutually exclusive in the evolution of primary and secondary glioblastomas. Brain Pathol. 1996, 6, 217–223, discussion 223–214. [Google Scholar] [CrossRef]
- Stadlmann, S.; Gueth, U.; Reiser, U.; Diener, P.A.; Zeimet, A.G.; Wight, E.; Mirlacher, M.; Sauter, G.; Mihatsch, M.J.; Singer, G. Epithelial growth factor receptor status in primary and recurrent ovarian cancer. Mod. Pathol. 2006, 19, 607–610. [Google Scholar] [CrossRef]
- Hashmi, A.A.; Hussain, Z.F.; Irfan, M.; Khan, E.Y.; Faridi, N.; Naqvi, H.; Khan, A.; Edhi, M.M. Prognostic significance of epidermal growth factor receptor (EGFR) over expression in urothelial carcinoma of urinary bladder. BMC Urol. 2018, 18, 59. [Google Scholar] [CrossRef] [Green Version]
- Carlsson, J.; Wester, K.; De La Torre, M.; Malmström, P.-U.; Gårdmark, T. EGFR-expression in primary urinary bladder cancer and corresponding metastases and the relation to HER2-expression. On the possibility to target these receptors with radionuclides. Radiol. Oncol. 2015, 49, 50–58. [Google Scholar] [CrossRef] [Green Version]
- Uhlman, D.L.; Nguyen, P.; Manivel, J.C.; Zhang, G.; Hagen, K.; Fraley, E.; Aeppli, D.; Niehans, G.A. Epidermal growth factor receptor and transforming growth factor alpha expression in papillary and nonpapillary renal cell carcinoma: Correlation with metastatic behavior and prognosis. Clin. Cancer Res. 1995, 1, 913–920. [Google Scholar]
- Moch, H.; Sauter, G.; Buchholz, N.; Gasser, T.C.; Bubendorf, L.; Waldman, F.M.; Mihatsch, M.J. Epidermal growth factor receptor expression is associated with rapid tumor cell proliferation in renal cell carcinoma. Hum. Pathol. 1997, 28, 1255–1259. [Google Scholar] [CrossRef]
- Cossu-Rocca, P.; Muroni, M.R.; Sanges, F.; Sotgiu, G.; Asunis, A.; Tanca, L.; Onnis, D.; Pira, G.; Manca, A.; Dore, S.; et al. EGFR kinase-dependent and kinase-independent roles in clear cell renal cell carcinoma. Am. J. Cancer Res. 2016, 6, 71–83. [Google Scholar]
- Rosell, R.; Moran, T.; Queralt, C.; Porta, R.; Cardenal, F.; Camps, C.; Majem, M.; Lopez-Vivanco, G.; Isla, D.; Provencio, M. Screening for epidermal growth factor receptor mutations in lung cancer. N. Engl. J. Med. 2009, 361, 958–967. [Google Scholar] [CrossRef] [Green Version]
- Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y.; Au, J.S.-K.; Thongprasert, S.; Srinivasan, S.; Tsai, C.-M.; Khoa, M.T.; Heeroma, K.; Itoh, Y.; Cornelio, G.; Yang, P.-C. A prospective, molecular epidemiology study of EGFR mutations in Asian patients with advanced non–small-cell lung cancer of adenocarcinoma histology (PIONEER). J. Thorac. Oncol. 2014, 9, 154–162. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.-H.; Richert, N.; Ito, S.; Merlino, G.T.; Pastan, I. Characterization of epidermal growth factor receptor gene expression in malignant and normal human cell lines. Proc. Natl. Acad. Sci. USA 1984, 81, 7308–7312. [Google Scholar] [CrossRef] [Green Version]
- King, C.R.; Kraus, M.H.; Williams, L.T.; Merlino, G.T.; Pastan, I.H.; Aaronson, S.A. Human tumor cell lines with EGF receptor gene amplification in the absence of aberrant sized mRNAs. Nucleic Acids Res. 1985, 13, 8477–8486. [Google Scholar] [CrossRef] [Green Version]
- Merlino, G.; Ishii, S.; Whang-Peng, J.; Knutsen, T.; Xu, Y.; Clark, A.; Stratton, R.; Wilson, R.; Ma, D.P.; Roe, B. Structure and localization of genes encoding aberrant and normal epidermal growth factor receptor RNAs from A431 human carcinoma cells. Mol. Cell. Biol. 1985, 5, 1722–1734. [Google Scholar] [CrossRef] [Green Version]
- Merlino, G.; Xu, Y.; Richert, N.; Clark, A.; Ishii, S.; Banks-Schlegel, S.; Pastan, I. Elevated epidermal growth factor receptor gene copy number and expression in a squamous carcinoma cell line. J. Clin. Investig. 1985, 75, 1077–1079. [Google Scholar] [CrossRef]
- Harrison, P.T.; Vyse, S.; Huang, P.H. Rare epidermal growth factor receptor (EGFR) mutations in non-small cell lung cancer. Semin. Cancer Biol. 2020, 61, 167–179. [Google Scholar] [CrossRef]
- Kobayashi, Y.; Mitsudomi, T. Not all epidermal growth factor receptor mutations in lung cancer are created equal: Perspectives for individualized treatment strategy. Cancer Sci. 2016, 107, 1179–1186. [Google Scholar] [CrossRef]
- Mitsudomi, T.; Morita, S.; Yatabe, Y.; Negoro, S.; Okamoto, I.; Tsurutani, J.; Seto, T.; Satouchi, M.; Tada, H.; Hirashima, T.; et al. Gefitinib versus cisplatin plus docetaxel in patients with non-small-cell lung cancer harbouring mutations of the epidermal growth factor receptor (WJTOG3405): An open label, randomised phase 3 trial. Lancet Oncol. 2010, 11, 121–128. [Google Scholar] [CrossRef]
- Sequist, L.V.; Yang, J.C.; Yamamoto, N.; O’Byrne, K.; Hirsh, V.; Mok, T.; Geater, S.L.; Orlov, S.; Tsai, C.M.; Boyer, M.; et al. Phase III study of afatinib or cisplatin plus pemetrexed in patients with metastatic lung adenocarcinoma with EGFR mutations. J. Clin. Oncol. 2013, 31, 3327–3334. [Google Scholar] [CrossRef] [Green Version]
- Rosell, R.; Carcereny, E.; Gervais, R.; Vergnenegre, A.; Massuti, B.; Felip, E.; Palmero, R.; Garcia-Gomez, R.; Pallares, C.; Sanchez, J.M.; et al. Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): A multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 2012, 13, 239–246. [Google Scholar] [CrossRef]
- Wu, Y.L.; Cheng, Y.; Zhou, X.; Lee, K.H.; Nakagawa, K.; Niho, S.; Tsuji, F.; Linke, R.; Rosell, R.; Corral, J.; et al. Dacomitinib versus gefitinib as first-line treatment for patients with EGFR-mutation-positive non-small-cell lung cancer (ARCHER 1050): A randomised, open-label, phase 3 trial. Lancet Oncol. 2017, 18, 1454–1466. [Google Scholar] [CrossRef]
- Fukuoka, M.; Yano, S.; Giaccone, G.; Tamura, T.; Nakagawa, K.; Douillard, J.Y.; Nishiwaki, Y.; Vansteenkiste, J.; Kudoh, S.; Rischin, D.; et al. Multi-institutional randomized phase II trial of gefitinib for previously treated patients with advanced non-small-cell lung cancer (The IDEAL 1 Trial) [corrected]. J. Clin. Oncol. 2003, 21, 2237–2246. [Google Scholar] [CrossRef] [PubMed]
- Kris, M.G.; Natale, R.B.; Herbst, R.S.; Lynch, T.J., Jr.; Prager, D.; Belani, C.P.; Schiller, J.H.; Kelly, K.; Spiridonidis, H.; Sandler, A.; et al. Efficacy of gefitinib, an inhibitor of the epidermal growth factor receptor tyrosine kinase, in symptomatic patients with non-small cell lung cancer: A randomized trial. JAMA 2003, 290, 2149–2158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, V.A.; Kris, M.G.; Shah, N.; Patel, J.; Azzoli, C.; Gomez, J.; Krug, L.M.; Pao, W.; Rizvi, N.; Pizzo, B.; et al. Bronchioloalveolar pathologic subtype and smoking history predict sensitivity to gefitinib in advanced non-small-cell lung cancer. J. Clin. Oncol. 2004, 22, 1103–1109. [Google Scholar] [CrossRef] [PubMed]
- Giaccone, G. Epidermal growth factor receptor inhibitors in the treatment of non-small-cell lung cancer. J. Clin. Oncol. 2005, 23, 3235–3242. [Google Scholar] [CrossRef]
- Kim, E.S.; Hirsh, V.; Mok, T.; Socinski, M.A.; Gervais, R.; Wu, Y.L.; Li, L.Y.; Watkins, C.L.; Sellers, M.V.; Lowe, E.S.; et al. Gefitinib versus docetaxel in previously treated non-small-cell lung cancer (INTEREST): A randomised phase III trial. Lancet 2008, 372, 1809–1818. [Google Scholar] [CrossRef]
- Maruyama, R.; Nishiwaki, Y.; Tamura, T.; Yamamoto, N.; Tsuboi, M.; Nakagawa, K.; Shinkai, T.; Negoro, S.; Imamura, F.; Eguchi, K.; et al. Phase III study, V-15-32, of gefitinib versus docetaxel in previously treated Japanese patients with non-small-cell lung cancer. J. Clin. Oncol. 2008, 26, 4244–4252. [Google Scholar] [CrossRef]
- Lee, D.H.; Park, K.; Kim, J.H.; Lee, J.S.; Shin, S.W.; Kang, J.H.; Ahn, M.J.; Ahn, J.S.; Suh, C.; Kim, S.W. Randomized Phase III trial of gefitinib versus docetaxel in non-small cell lung cancer patients who have previously received platinum-based chemotherapy. Clin. Cancer Res. 2010, 16, 1307–1314. [Google Scholar] [CrossRef] [Green Version]
- Ciuleanu, T.; Stelmakh, L.; Cicenas, S.; Miliauskas, S.; Grigorescu, A.C.; Hillenbach, C.; Johannsdottir, H.K.; Klughammer, B.; Gonzalez, E.E. Efficacy and safety of erlotinib versus chemotherapy in second-line treatment of patients with advanced, non-small-cell lung cancer with poor prognosis (TITAN): A randomised multicentre, open-label, phase 3 study. Lancet Oncol. 2012, 13, 300–308. [Google Scholar] [CrossRef]
- Massarelli, E.; Johnson, F.M.; Erickson, H.S.; Wistuba, I.I.; Papadimitrakopoulou, V. Uncommon Epidermal Growth Factor Receptor mutations in non-small cell lung cancer and their mechanisms of EGFR tyrosine kinase inhibitors sensitivity and resistance. Lung Cancer 2013, 80, 235–241. [Google Scholar] [CrossRef]
- Lynch, T.J.; Bell, D.W.; Sordella, R.; Gurubhagavatula, S.; Okimoto, R.A.; Brannigan, B.W.; Harris, P.L.; Haserlat, S.M.; Supko, J.G.; Haluska, F.G. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non–small-cell lung cancer to gefitinib. N. Engl. J. Med. 2004, 350, 2129–2139. [Google Scholar] [CrossRef]
- Paez, J.G.; Jänne, P.A.; Lee, J.C.; Tracy, S.; Greulich, H.; Gabriel, S.; Herman, P.; Kaye, F.J.; Lindeman, N.; Boggon, T.J. EGFR mutations in lung cancer: Correlation with clinical response to gefitinib therapy. Science 2004, 304, 1497–1500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mok, T.S.; Wu, Y.L.; Thongprasert, S.; Yang, C.H.; Chu, D.T.; Saijo, N.; Sunpaweravong, P.; Han, B.; Margono, B.; Ichinose, Y.; et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N. Engl. J. Med. 2009, 361, 947–957. [Google Scholar] [CrossRef]
- Han, J.Y.; Park, K.; Kim, S.W.; Lee, D.H.; Kim, H.Y.; Kim, H.T.; Ahn, M.J.; Yun, T.; Ahn, J.S.; Suh, C.; et al. First-SIGNAL: First-line single-agent iressa versus gemcitabine and cisplatin trial in never-smokers with adenocarcinoma of the lung. J. Clin. Oncol. 2012, 30, 1122–1128. [Google Scholar] [CrossRef]
- Maemondo, M.; Inoue, A.; Kobayashi, K.; Sugawara, S.; Oizumi, S.; Isobe, H.; Gemma, A.; Harada, M.; Yoshizawa, H.; Kinoshita, I.; et al. Gefitinib or chemotherapy for non-small-cell lung cancer with mutated EGFR. N. Engl. J. Med. 2010, 362, 2380–2388. [Google Scholar] [CrossRef] [Green Version]
- Zhou, C.; Wu, Y.L.; Chen, G.; Feng, J.; Liu, X.Q.; Wang, C.; Zhang, S.; Wang, J.; Zhou, S.; Ren, S.; et al. Erlotinib versus chemotherapy as first-line treatment for patients with advanced EGFR mutation-positive non-small-cell lung cancer (OPTIMAL, CTONG-0802): A multicentre, open-label, randomised, phase 3 study. Lancet Oncol. 2011, 12, 735–742. [Google Scholar] [CrossRef]
- Wu, Y.L.; Zhou, C.; Hu, C.P.; Feng, J.; Lu, S.; Huang, Y.; Li, W.; Hou, M.; Shi, J.H.; Lee, K.Y.; et al. Afatinib versus cisplatin plus gemcitabine for first-line treatment of Asian patients with advanced non-small-cell lung cancer harbouring EGFR mutations (LUX-Lung 6): An open-label, randomised phase 3 trial. Lancet Oncol. 2014, 15, 213–222. [Google Scholar] [CrossRef]
- Gridelli, C.; Butts, C.; Ciardiello, F.; Feld, R.; Gallo, C.; Perrone, F. An international, multicenter, randomized phase III study of first-line erlotinib followed by second-line cisplatin/gemcitabine versus first-line cisplatin/gemcitabine followed by second-line erlotinib in advanced non-small-cell lung cancer: Treatment rationale and protocol dynamics of the TORCH trial. Clin. Lung Cancer 2008, 9, 235–238. [Google Scholar] [CrossRef]
- Gridelli, C.; Ciardiello, F.; Gallo, C.; Feld, R.; Butts, C.; Gebbia, V.; Maione, P.; Morgillo, F.; Genestreti, G.; Favaretto, A.; et al. First-line erlotinib followed by second-line cisplatin-gemcitabine chemotherapy in advanced non-small-cell lung cancer: The TORCH randomized trial. J. Clin. Oncol. 2012, 30, 3002–3011. [Google Scholar] [CrossRef] [PubMed]
- Peters, S.; Adjei, A.A.; Gridelli, C.; Reck, M.; Kerr, K.; Felip, E. Metastatic non-small-cell lung cancer (NSCLC): ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2012, 23 (Suppl. 7), vii56–vii64. [Google Scholar] [CrossRef]
- Ettinger, D.S.; Akerley, W.; Borghaei, H.; Chang, A.C.; Cheney, R.T.; Chirieac, L.R.; D’Amico, T.A.; Demmy, T.L.; Ganti, A.K.; Govindan, R.; et al. Non-small cell lung cancer. J. Natl. Compr. Canc. Netw. 2012, 10, 1236–1271. [Google Scholar] [CrossRef]
- Garassino, M.C.; Martelli, O.; Broggini, M.; Farina, G.; Veronese, S.; Rulli, E.; Bianchi, F.; Bettini, A.; Longo, F.; Moscetti, L.; et al. Erlotinib versus docetaxel as second-line treatment of patients with advanced non-small-cell lung cancer and wild-type EGFR tumours (TAILOR): A randomised controlled trial. Lancet Oncol. 2013, 14, 981–988. [Google Scholar] [CrossRef]
- Inoue, A.; Suzuki, T.; Fukuhara, T.; Maemondo, M.; Kimura, Y.; Morikawa, N.; Watanabe, H.; Saijo, Y.; Nukiwa, T. Prospective phase II study of gefitinib for chemotherapy-naive patients with advanced non-small-cell lung cancer with epidermal growth factor receptor gene mutations. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2006, 24, 3340–3346. [Google Scholar] [CrossRef] [PubMed]
- Asahina, H.; Yamazaki, K.; Kinoshita, I.; Sukoh, N.; Harada, M.; Yokouchi, H.; Ishida, T.; Ogura, S.; Kojima, T.; Okamoto, Y.; et al. A phase II trial of gefitinib as first-line therapy for advanced non-small cell lung cancer with epidermal growth factor receptor mutations. Br. J. Cancer 2006, 95, 998–1004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sutani, A.; Nagai, Y.; Udagawa, K.; Uchida, Y.; Koyama, N.; Murayama, Y.; Tanaka, T.; Miyazawa, H.; Nagata, M.; Kanazawa, M.; et al. Gefitinib for non-small-cell lung cancer patients with epidermal growth factor receptor gene mutations screened by peptide nucleic acid-locked nucleic acid PCR clamp. Br. J. Cancer 2006, 95, 1483–1489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshida, K.; Yatabe, Y.; Park, J.Y.; Shimizu, J.; Horio, Y.; Matsuo, K.; Kosaka, T.; Mitsudomi, T.; Hida, T. Prospective validation for prediction of gefitinib sensitivity by epidermal growth factor receptor gene mutation in patients with non-small cell lung cancer. J. Thorac. Oncol. 2007, 2, 22–28. [Google Scholar] [CrossRef]
- Sunaga, N.; Tomizawa, Y.; Yanagitani, N.; Iijima, H.; Kaira, K.; Shimizu, K.; Tanaka, S.; Suga, T.; Hisada, T.; Ishizuka, T.; et al. Phase II prospective study of the efficacy of gefitinib for the treatment of stage III/IV non-small cell lung cancer with EGFR mutations, irrespective of previous chemotherapy. Lung Cancer 2007, 56, 383–389. [Google Scholar] [CrossRef]
- Tamura, K.; Okamoto, I.; Kashii, T.; Negoro, S.; Hirashima, T.; Kudoh, S.; Ichinose, Y.; Ebi, N.; Shibata, K.; Nishimura, T.; et al. Multicentre prospective phase II trial of gefitinib for advanced non-small cell lung cancer with epidermal growth factor receptor mutations: Results of the West Japan Thoracic Oncology Group trial (WJTOG0403). Br. J. Cancer 2008, 98, 907–914. [Google Scholar] [CrossRef] [Green Version]
- Sequist, L.V.; Martins, R.G.; Spigel, D.; Grunberg, S.M.; Spira, A.; Jänne, P.A.; Joshi, V.A.; McCollum, D.; Evans, T.L.; Muzikansky, A.; et al. First-line gefitinib in patients with advanced non-small-cell lung cancer harboring somatic EGFR mutations. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2008, 26, 2442–2449. [Google Scholar] [CrossRef]
- Paz-Ares, L.; Sanchez, J.; Garcia-Velasco, A.; Massuti, B.; Lopez-Vivanco, G.; Provencio, M.; Montes, A.; Isla, D.; Amador, M.; Rosell, R.J.J.O.C.O. A prospective phase II trial of erlotinib in advanced non-small cell lung cancer (NSCLC) patients (p) with mutations in the tyrosine kinase (TK) domain of the epidermal growth factor receptor (EGFR). J. Clin. Oncol. 2006, 24, 7020. [Google Scholar] [CrossRef]
- Porta, R.; Queralt, C.; Cardenal, F.; Mayo, C.; Provencio, M.; Camps, C.; Isla, D.; González-Larriba, J.; Tarón, M.; Rosell, R.J.J.O.C.O. Erlotinib customization based on epidermal growth factor receptor (EGFR) mutations in stage IV non-small-cell lung cancer (NSCLC) patients (p). J. Clin. Oncol. 2008, 26, 8038. [Google Scholar] [CrossRef]
- Costa, D.B.; Kobayashi, S.; Tenen, D.G.; Huberman, M.S. Pooled analysis of the prospective trials of gefitinib monotherapy for EGFR-mutant non-small cell lung cancers. Lung Cancer 2007, 58, 95–103. [Google Scholar] [CrossRef] [Green Version]
- Ahn, M.J.; Tsai, C.M.; Shepherd, F.A.; Bazhenova, L.; Sequist, L.V.; Hida, T.; Yang, J.C.H.; Ramalingam, S.S.; Mitsudomi, T.; Jänne, P.A.; et al. Osimertinib in patients with T790M mutation-positive, advanced non-small cell lung cancer: Long-term follow-up from a pooled analysis of 2 phase 2 studies. Cancer 2019, 125, 892–901. [Google Scholar] [CrossRef]
- Hata, A.N.; Niederst, M.J.; Archibald, H.L.; Gomez-Caraballo, M.; Siddiqui, F.M.; Mulvey, H.E.; Maruvka, Y.E.; Ji, F.; Bhang, H.E.; Krishnamurthy Radhakrishna, V.; et al. Tumor cells can follow distinct evolutionary paths to become resistant to epidermal growth factor receptor inhibition. Nat. Med. 2016, 22, 262–269. [Google Scholar] [CrossRef]
- Ikeda, K.; Nomori, H.; Mori, T.; Sasaki, J.; Kobayashi, T. Novel germline mutation: EGFR V843I in patient with multiple lung adenocarcinomas and family members with lung cancer. Ann. Thorac. Surg. 2008, 85, 1430–1432. [Google Scholar] [CrossRef] [PubMed]
- Ohtsuka, K.; Ohnishi, H.; Kurai, D.; Matsushima, S.; Morishita, Y.; Shinonaga, M.; Goto, H.; Watanabe, T. Familial lung adenocarcinoma caused by the EGFR V843I germ-line mutation. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2011, 29, e191–e192. [Google Scholar] [CrossRef]
- Demierre, N.; Zoete, V.; Michielin, O.; Stauffer, E.; Zimmermann, D.R.; Betticher, D.C.; Peters, S. A dramatic lung cancer course in a patient with a rare EGFR germline mutation exon 21 V843I: Is EGFR TKI resistance predictable? Lung Cancer 2013, 80, 81–84. [Google Scholar] [CrossRef]
- Matsushima, S.; Ohtsuka, K.; Ohnishi, H.; Fujiwara, M.; Nakamura, H.; Morii, T.; Kishino, T.; Goto, H.; Watanabe, T. V843I, a lung cancer predisposing EGFR mutation, is responsible for resistance to EGFR tyrosine kinase inhibitors. J. Thorac. Oncol. 2014, 9, 1377–1384. [Google Scholar] [CrossRef] [Green Version]
- Nakagawa, T.; Takeuchi, S.; Yamada, T.; Ebi, H.; Sano, T.; Nanjo, S.; Ishikawa, D.; Sato, M.; Hasegawa, Y.; Sekido, Y.; et al. EGFR-TKI resistance due to BIM polymorphism can be circumvented in combination with HDAC inhibition. Cancer Res. 2013, 73, 2428–2434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costa, C.; Molina, M.A.; Drozdowskyj, A.; Giménez-Capitán, A.; Bertran-Alamillo, J.; Karachaliou, N.; Gervais, R.; Massuti, B.; Wei, J.; Moran, T.; et al. The impact of EGFR T790M mutations and BIM mRNA expression on outcome in patients with EGFR-mutant NSCLC treated with erlotinib or chemotherapy in the randomized phase III EURTAC trial. Clin. Cancer Res. 2014, 20, 2001–2010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costa, D.B.; Halmos, B.; Kumar, A.; Schumer, S.T.; Huberman, M.S.; Boggon, T.J.; Tenen, D.G.; Kobayashi, S. BIM mediates EGFR tyrosine kinase inhibitor-induced apoptosis in lung cancers with oncogenic EGFR mutations. PLoS Med. 2007, 4, 1669–1679, discussion 1680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cragg, M.S.; Kuroda, J.; Puthalakath, H.; Huang, D.C.; Strasser, A. Gefitinib-induced killing of NSCLC cell lines expressing mutant EGFR requires BIM and can be enhanced by BH3 mimetics. PLoS Med. 2007, 4, 1681–1689, discussion 1690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, Y.; Somwar, R.; Politi, K.; Balak, M.; Chmielecki, J.; Jiang, X.; Pao, W. Induction of BIM is essential for apoptosis triggered by EGFR kinase inhibitors in mutant EGFR-dependent lung adenocarcinomas. PLoS Med. 2007, 4, e294. [Google Scholar] [CrossRef]
- Faber, A.C.; Corcoran, R.B.; Ebi, H.; Sequist, L.V.; Waltman, B.A.; Chung, E.; Incio, J.; Digumarthy, S.R.; Pollack, S.F.; Song, Y.; et al. BIM expression in treatment-naive cancers predicts responsiveness to kinase inhibitors. Cancer Discov. 2011, 1, 352–365. [Google Scholar] [CrossRef] [Green Version]
- Takezawa, K.; Okamoto, I.; Nishio, K.; Jänne, P.A.; Nakagawa, K. Role of ERK-BIM and STAT3-Survivin Signaling Pathways in ALK Inhibitor–Induced Apoptosis in EML4-ALK–Positive Lung Cancer. Clin. Cancer Res. 2011, 17, 2140. [Google Scholar] [CrossRef] [Green Version]
- Leventakos, K.; Kipp, B.R.; Rumilla, K.M.; Winters, J.L.; Yi, E.S.; Mansfield, A.S. S768I Mutation in EGFR in Patients with Lung Cancer. J. Thorac. Oncol. 2016, 11, 1798–1801. [Google Scholar] [CrossRef] [Green Version]
- Balak, M.N.; Gong, Y.; Riely, G.J.; Somwar, R.; Li, A.R.; Zakowski, M.F.; Chiang, A.; Yang, G.; Ouerfelli, O.; Kris, M.G.; et al. Novel D761Y and common secondary T790M mutations in epidermal growth factor receptor-mutant lung adenocarcinomas with acquired resistance to kinase inhibitors. Clin. Cancer Res. 2006, 12, 6494–6501. [Google Scholar] [CrossRef] [Green Version]
- Toyooka, S.; Date, H.; Uchida, A.; Kiura, K.; Takata, M. The epidermal growth factor receptor D761Y mutation and effect of tyrosine kinase inhibitor. Clin. Cancer Res. 2007, 13, 3431, author reply 3431–3432. [Google Scholar] [CrossRef] [Green Version]
- Bean, J.; Riely, G.J.; Balak, M.; Marks, J.L.; Ladanyi, M.; Miller, V.A.; Pao, W. Acquired resistance to epidermal growth factor receptor kinase inhibitors associated with a novel T854A mutation in a patient with EGFR-mutant lung adenocarcinoma. Clin. Cancer Res. 2008, 14, 7519–7525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, S.-K.; Ko, J.-C.; Yang, J.C.-H.; Shih, J.-Y. Afatinib is effective in the treatment of lung adenocarcinoma with uncommon EGFR p.L747P and p.L747S mutations. Lung Cancer 2019, 133, 103–109. [Google Scholar] [CrossRef] [PubMed]
- Chiba, M.; Togashi, Y.; Bannno, E.; Kobayashi, Y.; Nakamura, Y.; Hayashi, H.; Terashima, M.; De Velasco, M.A.; Sakai, K.; Fujita, Y.; et al. Efficacy of irreversible EGFR-TKIs for the uncommon secondary resistant EGFR mutations L747S, D761Y, and T854A. BMC Cancer 2017, 17, 281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, H.A.; Arcila, M.E.; Rekhtman, N.; Sima, C.S.; Zakowski, M.F.; Pao, W.; Kris, M.G.; Miller, V.A.; Ladanyi, M.; Riely, G.J. Analysis of tumor specimens at the time of acquired resistance to EGFR-TKI therapy in 155 patients with EGFR-mutant lung cancers. Clin. Cancer Res. 2013, 19, 2240–2247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oxnard, G.R.; Arcila, M.E.; Chmielecki, J.; Ladanyi, M.; Miller, V.A.; Pao, W. New strategies in overcoming acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in lung cancer. Clin. Cancer Res. 2011, 17, 5530–5537. [Google Scholar] [CrossRef] [Green Version]
- Tu, H.Y.; Ke, E.E.; Yang, J.J.; Sun, Y.L.; Yan, H.H.; Zheng, M.Y.; Bai, X.Y.; Wang, Z.; Su, J.; Chen, Z.H.; et al. A comprehensive review of uncommon EGFR mutations in patients with non-small cell lung cancer. Lung Cancer 2017, 114, 96–102. [Google Scholar] [CrossRef]
- Kuiper, J.L.; Hashemi, S.M.; Thunnissen, E.; Snijders, P.J.; Grünberg, K.; Bloemena, E.; Sie, D.; Postmus, P.E.; Heideman, D.A.; Smit, E.F. Non-classic EGFR mutations in a cohort of Dutch EGFR-mutated NSCLC patients and outcomes following EGFR-TKI treatment. Br. J. Cancer 2016, 115, 1504–1512. [Google Scholar] [CrossRef]
- Wang, F.; Li, C.; Wu, Q.; Lu, H.J.T.C.R. EGFR exon 20 insertion mutations in non-small cell lung cancer. Transl. Cancer Res. 2020, 9, 2982–2991. [Google Scholar] [CrossRef]
- Papadimitrakopoulou, V.; Wu, Y.-L.; Han, J.-Y.; Ahn, M.-J.; Ramalingam, S.; John, T.; Okamoto, I.; Yang, J.-H.; Bulusu, K.; Laus, G.J.A.O.O. Analysis of resistance mechanisms to osimertinib in patients with EGFR T790M advanced NSCLC from the AURA3 study. Ann. Oncol. 2018, 29, viii741. [Google Scholar] [CrossRef]
- Zhou, T.; Zhou, X.; Li, P.; Qi, C.; Ling, Y.J.J.O.T.D. EGFR L747P mutation in one lung adenocarcinoma patient responded to afatinib treatment: A case report. J. Thorac. Dis. 2018, 10, E802–E805. [Google Scholar] [CrossRef]
- Huang, J.; Wang, Y.; Zhai, Y.; Wang, J. Non-small cell lung cancer harboring a rare EGFR L747P mutation showing intrinsic resistance to both gefitinib and osimertinib (AZD9291): A case report. Thorac. Cancer 2018, 9, 745–749. [Google Scholar] [CrossRef] [Green Version]
- Ercan, D.; Zejnullahu, K.; Yonesaka, K.; Xiao, Y.; Capelletti, M.; Rogers, A.; Lifshits, E.; Brown, A.; Lee, C.; Christensen, J.G.; et al. Amplification of EGFR T790M causes resistance to an irreversible EGFR inhibitor. Oncogene 2010, 29, 2346–2356. [Google Scholar] [CrossRef] [Green Version]
- Thress, K.S.; Paweletz, C.P.; Felip, E.; Cho, B.C.; Stetson, D.; Dougherty, B.; Lai, Z.; Markovets, A.; Vivancos, A.; Kuang, Y.; et al. Acquired EGFR C797S mutation mediates resistance to AZD9291 in non-small cell lung cancer harboring EGFR T790M. Nat. Med. 2015, 21, 560–562. [Google Scholar] [CrossRef] [Green Version]
- Jia, Y.; Yun, C.H.; Park, E.; Ercan, D.; Manuia, M.; Juarez, J.; Xu, C.; Rhee, K.; Chen, T.; Zhang, H.; et al. Overcoming EGFR(T790M) and EGFR(C797S) resistance with mutant-selective allosteric inhibitors. Nature 2016, 534, 129–132. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.; Yang, N.; Ou, Q.; Xiang, Y.; Jiang, T.; Wu, X.; Bao, H.; Tong, X.; Wang, X.; Shao, Y.W.; et al. Investigating Novel Resistance Mechanisms to Third-Generation EGFR Tyrosine Kinase Inhibitor Osimertinib in Non-Small Cell Lung Cancer Patients. Clin. Cancer Res. 2018, 24, 3097–3107. [Google Scholar] [CrossRef] [Green Version]
- Zheng, D.; Hu, M.; Bai, Y.; Zhu, X.; Lu, X.; Wu, C.; Wang, J.; Liu, L.; Wang, Z.; Ni, J.; et al. EGFR G796D mutation mediates resistance to osimertinib. Oncotarget 2017, 8, 49671–49679. [Google Scholar] [CrossRef] [Green Version]
- Chen, K.; Zhou, F.; Shen, W.; Jiang, T.; Wu, X.; Tong, X.; Shao, Y.W.; Qin, S.; Zhou, C. Novel Mutations on EGFR Leu792 Potentially Correlate to Acquired Resistance to Osimertinib in Advanced NSCLC. J. Thorac. Oncol. 2017, 12, e65–e68. [Google Scholar] [CrossRef] [Green Version]
- Bersanelli, M.; Minari, R.; Bordi, P.; Gnetti, L.; Bozzetti, C.; Squadrilli, A.; Lagrasta, C.A.; Bottarelli, L.; Osipova, G.; Capelletto, E.; et al. L718Q Mutation as New Mechanism of Acquired Resistance to AZD9291 in EGFR-Mutated NSCLC. J. Thorac. Oncol. 2016, 11, e121–e123. [Google Scholar] [CrossRef] [Green Version]
- Fassunke, J.; Müller, F.; Keul, M.; Michels, S.; Dammert, M.A.; Schmitt, A.; Plenker, D.; Lategahn, J.; Heydt, C.; Brägelmann, J.; et al. Overcoming EGFRG724S-mediated osimertinib resistance through unique binding characteristics of second-generation EGFR inhibitors. Nat. Commun. 2018, 9, 4655. [Google Scholar] [CrossRef]
- Leonetti, A.; Sharma, S.; Minari, R.; Perego, P.; Giovannetti, E.; Tiseo, M. Resistance mechanisms to osimertinib in EGFR-mutated non-small cell lung cancer. Br. J. Cancer 2019, 121, 725–737. [Google Scholar] [CrossRef]
- Suda, K.; Mizuuchi, H.; Murakami, I.; Uramoto, H.; Tanaka, F.; Sato, K.; Takemoto, T.; Iwasaki, T.; Sekido, Y.; Yatabe, Y.; et al. CRKL amplification is rare as a mechanism for acquired resistance to kinase inhibitors in lung cancers with epidermal growth factor receptor mutation. Lung Cancer 2014, 85, 147–151. [Google Scholar] [CrossRef] [PubMed]
- Cheung, H.W.; Du, J.; Boehm, J.S.; He, F.; Weir, B.A.; Wang, X.; Butaney, M.; Sequist, L.V.; Luo, B.; Engelman, J.A.; et al. Amplification of CRKL induces transformation and epidermal growth factor receptor inhibitor resistance in human non-small cell lung cancers. Cancer Discov. 2011, 1, 608–625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ercan, D.; Xu, C.; Yanagita, M.; Monast, C.S.; Pratilas, C.A.; Montero, J.; Butaney, M.; Shimamura, T.; Sholl, L.; Ivanova, E.V.; et al. Reactivation of ERK signaling causes resistance to EGFR kinase inhibitors. Cancer Discov. 2012, 2, 934–947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guix, M.; Faber, A.C.; Wang, S.E.; Olivares, M.G.; Song, Y.; Qu, S.; Rinehart, C.; Seidel, B.; Yee, D.; Arteaga, C.L.; et al. Acquired resistance to EGFR tyrosine kinase inhibitors in cancer cells is mediated by loss of IGF-binding proteins. J. Clin. Investig. 2008, 118, 2609–2619. [Google Scholar] [CrossRef]
- Cortot, A.B.; Repellin, C.E.; Shimamura, T.; Capelletti, M.; Zejnullahu, K.; Ercan, D.; Christensen, J.G.; Wong, K.K.; Gray, N.S.; Jänne, P.A. Resistance to irreversible EGF receptor tyrosine kinase inhibitors through a multistep mechanism involving the IGF1R pathway. Cancer Res. 2013, 73, 834–843. [Google Scholar] [CrossRef] [Green Version]
- Engelman, J.A.; Zejnullahu, K.; Mitsudomi, T.; Song, Y.; Hyland, C.; Park, J.O.; Lindeman, N.; Gale, C.M.; Zhao, X.; Christensen, J.; et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science 2007, 316, 1039–1043. [Google Scholar] [CrossRef]
- Ortiz-Cuaran, S.; Scheffler, M.; Plenker, D.; Dahmen, L.; Scheel, A.H.; Fernandez-Cuesta, L.; Meder, L.; Lovly, C.M.; Persigehl, T.; Merkelbach-Bruse, S.; et al. Heterogeneous Mechanisms of Primary and Acquired Resistance to Third-Generation EGFR Inhibitors. Clin. Cancer Res. 2016, 22, 4837–4847. [Google Scholar] [CrossRef] [Green Version]
- Planchard, D.; Loriot, Y.; Andre, F.; Gobert, A.; Auger, N.; Lacroix, L.; Soria, J.C. EGFR-independent mechanisms of acquired resistance to AZD9291 in EGFR T790M-positive NSCLC patients. Ann. Oncol. 2015, 26, 2073–2078. [Google Scholar] [CrossRef]
- Takezawa, K.; Pirazzoli, V.; Arcila, M.E.; Nebhan, C.A.; Song, X.; de Stanchina, E.; Ohashi, K.; Janjigian, Y.Y.; Spitzler, P.J.; Melnick, M.A.; et al. HER2 amplification: A potential mechanism of acquired resistance to EGFR inhibition in EGFR-mutant lung cancers that lack the second-site EGFRT790M mutation. Cancer Discov. 2012, 2, 922–933. [Google Scholar] [CrossRef] [Green Version]
- Piotrowska, Z.; Thress, K.S.; Mooradian, M.; Heist, R.S.; Azzoli, C.G.; Temel, J.S.; Rizzo, C.; Nagy, R.J.; Lanman, R.B.; Gettinger, S.N.; et al. MET amplification (amp) as a resistance mechanism to osimertinib. J. Clin. Oncol. 2017, 35, 9020. [Google Scholar] [CrossRef]
- Xu, J.; Wang, J.; Zhang, S. Mechanisms of resistance to irreversible epidermal growth factor receptor tyrosine kinase inhibitors and therapeutic strategies in non-small cell lung cancer. Oncotarget 2017, 8, 90557–90578. [Google Scholar] [CrossRef] [Green Version]
- Tan, D.S.-W.; Kim, D.-W.; Leighl, N.B.; Riely, G.J.; Yang, J.C.-H.; Wolf, J.; Seto, T.; Felip, E.; Aix, S.P.; Jonnaert, M.; et al. Genomic profiling of resistant tumor samples following progression on EGF816, a third generation, mutant-selective EGFR tyrosine kinase inhibitor (TKI), in advanced non-small cell lung cancer (NSCLC). J. Clin. Oncol. 2017, 35, 11506. [Google Scholar] [CrossRef]
- Sos, M.L.; Koker, M.; Weir, B.A.; Heynck, S.; Rabinovsky, R.; Zander, T.; Seeger, J.M.; Weiss, J.; Fischer, F.; Frommolt, P.; et al. PTEN loss contributes to erlotinib resistance in EGFR-mutant lung cancer by activation of Akt and EGFR. Cancer Res. 2009, 69, 3256–3261. [Google Scholar] [CrossRef] [Green Version]
- Tan, D.S.-W.; Lim, K.H.; Tai, W.M.; Ahmad, A.; Pan, S.; Ng, Q.S.; Ang, M.-K.; Gogna, A.; Ng, Y.L.; Tan, B.S.; et al. A phase Ib safety and tolerability study of a pan class I PI3K inhibitor buparlisib (BKM120) and gefitinib (gef) in EGFR TKI-resistant NSCLC. J. Clin. Oncol. 2013, 31, 8107. [Google Scholar] [CrossRef]
- Fang, W.; Huang, Y.; Gu, W.; Gan, J.; Wang, W.; Zhang, S.; Wang, K.; Zhan, J.; Yang, Y.; Huang, Y.; et al. PI3K-AKT-mTOR pathway alterations in advanced NSCLC patients after progression on EGFR-TKI and clinical response to EGFR-TKI plus everolimus combination therapy. Transl. Lung Cancer Res. 2020, 9, 1258–1267. [Google Scholar] [CrossRef]
- Ho, C.C.; Liao, W.Y.; Lin, C.A.; Shih, J.Y.; Yu, C.J.; Yang, J.C. Acquired BRAF V600E Mutation as Resistant Mechanism after Treatment with Osimertinib. J. Thorac. Oncol. 2017, 12, 567–572. [Google Scholar] [CrossRef] [Green Version]
- Ohashi, K.; Sequist, L.V.; Arcila, M.E.; Moran, T.; Chmielecki, J.; Lin, Y.L.; Pan, Y.; Wang, L.; de Stanchina, E.; Shien, K.; et al. Lung cancers with acquired resistance to EGFR inhibitors occasionally harbor BRAF gene mutations but lack mutations in KRAS, NRAS, or MEK1. Proc. Natl. Acad. Sci. USA 2012, 109, E2127–E2133. [Google Scholar] [CrossRef] [Green Version]
- Pao, W.; Wang, T.Y.; Riely, G.J.; Miller, V.A.; Pan, Q.; Ladanyi, M.; Zakowski, M.F.; Heelan, R.T.; Kris, M.G.; Varmus, H.E. KRAS mutations and primary resistance of lung adenocarcinomas to gefitinib or erlotinib. PLoS Med. 2005, 2, e17. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Lee, J.C.; Lin, L.; Olivas, V.; Au, V.; LaFramboise, T.; Abdel-Rahman, M.; Wang, X.; Levine, A.D.; Rho, J.K.; et al. Activation of the AXL kinase causes resistance to EGFR-targeted therapy in lung cancer. Nat. Genet. 2012, 44, 852–860. [Google Scholar] [CrossRef]
- Sequist, L.V.; Waltman, B.A.; Dias-Santagata, D.; Digumarthy, S.; Turke, A.B.; Fidias, P.; Bergethon, K.; Shaw, A.T.; Gettinger, S.; Cosper, A.K.; et al. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci. Transl. Med. 2011, 3, 75ra26. [Google Scholar] [CrossRef] [Green Version]
- Ham, J.S.; Kim, S.; Kim, H.K.; Byeon, S.; Sun, J.M.; Lee, S.H.; Ahn, J.S.; Park, K.; Choi, Y.L.; Han, J.; et al. Two Cases of Small Cell Lung Cancer Transformation from EGFR Mutant Adenocarcinoma During AZD9291 Treatment. J. Thorac. Oncol. 2016, 11, e1–e4. [Google Scholar] [CrossRef] [Green Version]
- Levin, P.A.; Mayer, M.; Hoskin, S.; Sailors, J.; Oliver, D.H.; Gerber, D.E. Histologic Transformation from Adenocarcinoma to Squamous Cell Carcinoma as a Mechanism of Resistance to EGFR Inhibition. J. Thorac. Oncol. 2015, 10, e86–e88. [Google Scholar] [CrossRef] [Green Version]
- Scher, K.S.; Saldivar, J.S.; Fishbein, M.; Marchevsky, A.; Reckamp, K.L. EGFR-mutated lung cancer with T790M-acquired resistance in the brain and histologic transformation in the lung. J. Natl. Compr. Cancer Netw. JNCCN 2013, 11, 1040–1044. [Google Scholar] [CrossRef] [Green Version]
- Schoenfeld, A.J.; Chan, J.M.; Rizvi, H.; Rekhtman, N.; Daneshbod, Y.; Kubota, D.; Chang, J.C.; Arcila, M.E.; Ladanyi, M.; Somwar, R.; et al. Tissue-based molecular and histological landscape of acquired resistance to osimertinib given initially or at relapse in patients with EGFR-mutant lung cancers. J. Clin. Oncol. 2019, 37, 9028. [Google Scholar] [CrossRef]
- Bivona, T.G.; Hieronymus, H.; Parker, J.; Chang, K.; Taron, M.; Rosell, R.; Moonsamy, P.; Dahlman, K.; Miller, V.A.; Costa, C.; et al. FAS and NF-κB signalling modulate dependence of lung cancers on mutant EGFR. Nature 2011, 471, 523–526. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Ren, S.; Li, X.; Wang, Y.; Garfield, D.; Zhou, S.; Chen, X.; Su, C.; Chen, M.; Kuang, P.; et al. MiR-21 overexpression is associated with acquired resistance of EGFR-TKI in non-small cell lung cancer. Lung Cancer 2014, 83, 146–153. [Google Scholar] [CrossRef] [PubMed]
- Enrico, D.; Lacroix, L.; Chen, J.; Rouleau, E.; Scoazec, J.-Y.; Loriot, Y.; Tselikas, L.; Jovelet, C.; Planchard, D.; Gazzah, A.; et al. Oncogenic Fusions May Be Frequently Present at Resistance of EGFR Tyrosine Kinase Inhibitors in Patients With NSCLC: A Brief Report. JTO Clin. Res. Rep. 2020, 1, 100023. [Google Scholar] [CrossRef]
- Xu, H.; Shen, J.; Xiang, J.; Li, H.; Li, B.; Zhang, T.; Zhang, L.; Mao, X.; Jian, H.; Shu, Y. Characterization of acquired receptor tyrosine-kinase fusions as mechanisms of resistance to EGFR tyrosine-kinase inhibitors. Cancer Manag. Res. 2019, 11, 6343–6351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solomon, B.; Varella-Garcia, M.; Camidge, D.R. ALK gene rearrangements: A new therapeutic target in a molecularly defined subset of non-small cell lung cancer. J. Thorac. Oncol. 2009, 4, 1450–1454. [Google Scholar] [CrossRef] [Green Version]
- Schrock, A.B.; Zhu, V.W.; Hsieh, W.S.; Madison, R.; Creelan, B.; Silberberg, J.; Costin, D.; Bharne, A.; Bonta, I.; Bosemani, T.; et al. Receptor Tyrosine Kinase Fusions and BRAF Kinase Fusions are Rare but Actionable Resistance Mechanisms to EGFR Tyrosine Kinase Inhibitors. J. Thorac. Oncol. 2018, 13, 1312–1323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rich, T.A.; Reckamp, K.L.; Chae, Y.K.; Doebele, R.C.; Iams, W.T.; Oh, M.; Raymond, V.M.; Lanman, R.B.; Riess, J.W.; Stinchcombe, T.E.; et al. Analysis of Cell-Free DNA from 32,989 Advanced Cancers Reveals Novel Co-occurring Activating RET Alterations and Oncogenic Signaling Pathway Aberrations. Clin. Cancer Res. 2019, 25, 5832–5842. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, S.; Boggon, T.J.; Dayaram, T.; Jänne, P.A.; Kocher, O.; Meyerson, M.; Johnson, B.E.; Eck, M.J.; Tenen, D.G.; Halmos, B. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N. Engl. J. Med. 2005, 352, 786–792. [Google Scholar] [CrossRef]
- Pao, W.; Miller, V.A.; Politi, K.A.; Riely, G.J.; Somwar, R.; Zakowski, M.F.; Kris, M.G.; Varmus, H. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med. 2005, 2, e73. [Google Scholar] [CrossRef] [Green Version]
- Riely, G.J.; Pao, W.; Pham, D.; Li, A.R.; Rizvi, N.; Venkatraman, E.S.; Zakowski, M.F.; Kris, M.G.; Ladanyi, M.; Miller, V.A. Clinical course of patients with non-small cell lung cancer and epidermal growth factor receptor exon 19 and exon 21 mutations treated with gefitinib or erlotinib. Clin. Cancer Res. 2006, 12, 839–844. [Google Scholar] [CrossRef] [Green Version]
- Chmielecki, J.; Foo, J.; Oxnard, G.R.; Hutchinson, K.; Ohashi, K.; Somwar, R.; Wang, L.; Amato, K.R.; Arcila, M.; Sos, M.L.; et al. Optimization of dosing for EGFR-mutant non-small cell lung cancer with evolutionary cancer modeling. Sci. Transl. Med. 2011, 3, 90ra59. [Google Scholar] [CrossRef] [Green Version]
- Lim, S.M.; Syn, N.L.; Cho, B.C.; Soo, R.A. Acquired resistance to EGFR targeted therapy in non-small cell lung cancer: Mechanisms and therapeutic strategies. Cancer Treat. Rev. 2018, 65, 1–10. [Google Scholar] [CrossRef]
- Maheswaran, S.; Sequist, L.V.; Nagrath, S.; Ulkus, L.; Brannigan, B.; Collura, C.V.; Inserra, E.; Diederichs, S.; Iafrate, A.J.; Bell, D.W.; et al. Detection of mutations in EGFR in circulating lung-cancer cells. N. Engl. J. Med. 2008, 359, 366–377. [Google Scholar] [CrossRef] [Green Version]
- Toyooka, S.; Kiura, K.; Mitsudomi, T. EGFR mutation and response of lung cancer to gefitinib. N. Engl. J. Med. 2005, 352, 2136, author reply 2136. [Google Scholar] [CrossRef]
- Bell, D.W.; Gore, I.; Okimoto, R.A.; Godin-Heymann, N.; Sordella, R.; Mulloy, R.; Sharma, S.V.; Brannigan, B.W.; Mohapatra, G.; Settleman, J.; et al. Inherited susceptibility to lung cancer may be associated with the T790M drug resistance mutation in EGFR. Nat. Genet. 2005, 37, 1315–1316. [Google Scholar] [CrossRef]
- Mulloy, R.; Ferrand, A.; Kim, Y.; Sordella, R.; Bell, D.W.; Haber, D.A.; Anderson, K.S.; Settleman, J. Epidermal growth factor receptor mutants from human lung cancers exhibit enhanced catalytic activity and increased sensitivity to gefitinib. Cancer Res. 2007, 67, 2325–2330. [Google Scholar] [CrossRef] [Green Version]
- Settleman, J.; Kurie, J.M. Drugging the bad “AKT-TOR” to overcome TKI-resistant lung cancer. Cancer Cell 2007, 12, 6–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suda, K.; Onozato, R.; Yatabe, Y.; Mitsudomi, T. EGFR T790M mutation: A double role in lung cancer cell survival? J. Thorac. Oncol. 2009, 4, 1–4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yun, C.H.; Mengwasser, K.E.; Toms, A.V.; Woo, M.S.; Greulich, H.; Wong, K.K.; Meyerson, M.; Eck, M.J. The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc. Natl. Acad. Sci. USA 2008, 105, 2070–2075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thatcher, N.; Chang, A.; Parikh, P.; Rodrigues Pereira, J.; Ciuleanu, T.; von Pawel, J.; Thongprasert, S.; Tan, E.H.; Pemberton, K.; Archer, V.; et al. Gefitinib plus best supportive care in previously treated patients with refractory advanced non-small-cell lung cancer: Results from a randomised, placebo-controlled, multicentre study (Iressa Survival Evaluation in Lung Cancer). Lancet (Lond. Engl.) 2005, 366, 1527–1537. [Google Scholar] [CrossRef]
- Miller, V.A.; Hirsh, V.; Cadranel, J.; Chen, Y.M.; Park, K.; Kim, S.W.; Zhou, C.; Su, W.C.; Wang, M.; Sun, Y.; et al. Afatinib versus placebo for patients with advanced, metastatic non-small-cell lung cancer after failure of erlotinib, gefitinib, or both, and one or two lines of chemotherapy (LUX-Lung 1): A phase 2b/3 randomised trial. Lancet Oncol. 2012, 13, 528–538. [Google Scholar] [CrossRef]
- Li, D.; Ambrogio, L.; Shimamura, T.; Kubo, S.; Takahashi, M.; Chirieac, L.R.; Padera, R.F.; Shapiro, G.I.; Baum, A.; Himmelsbach, F.; et al. BIBW2992, an irreversible EGFR/HER2 inhibitor highly effective in preclinical lung cancer models. Oncogene 2008, 27, 4702–4711. [Google Scholar] [CrossRef] [Green Version]
- Mok, T.S.; Wu, Y.L.; Ahn, M.J.; Garassino, M.C.; Kim, H.R.; Ramalingam, S.S.; Shepherd, F.A.; He, Y.; Akamatsu, H.; Theelen, W.S.; et al. Osimertinib or Platinum-Pemetrexed in EGFR T790M-Positive Lung Cancer. N. Engl. J. Med. 2017, 376, 629–640. [Google Scholar] [CrossRef] [Green Version]
- Soria, J.C.; Ohe, Y.; Vansteenkiste, J.; Reungwetwattana, T.; Chewaskulyong, B.; Lee, K.H.; Dechaphunkul, A.; Imamura, F.; Nogami, N.; Kurata, T.; et al. Osimertinib in Untreated EGFR-Mutated Advanced Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 378, 113–125. [Google Scholar] [CrossRef]
- Brown, B.P.; Zhang, Y.K.; Westover, D.; Yan, Y.; Qiao, H.; Huang, V.; Du, Z.; Smith, J.A.; Ross, J.S.; Miller, V.A.; et al. On-target Resistance to the Mutant-Selective EGFR Inhibitor Osimertinib Can Develop in an Allele-Specific Manner Dependent on the Original EGFR-Activating Mutation. Clin. Cancer Res. 2019, 25, 3341–3351. [Google Scholar] [CrossRef] [Green Version]
- Le, X.; Puri, S.; Negrao, M.V.; Nilsson, M.B.; Robichaux, J.; Boyle, T.; Hicks, J.K.; Lovinger, K.L.; Roarty, E.; Rinsurongkawong, W.; et al. Landscape of EGFR-Dependent and -Independent Resistance Mechanisms to Osimertinib and Continuation Therapy Beyond Progression in EGFR-Mutant NSCLC. Clin. Cancer Res. 2018, 24, 6195–6203. [Google Scholar] [CrossRef] [Green Version]
- Ramalingam, S.S.; Cheng, Y.; Zhou, C.; Ohe, Y.; Imamura, F.; Cho, B.C.; Lin, M.C.; Majem, M.; Shah, R.; Rukazenkov, Y.; et al. LBA50—Mechanisms of acquired resistance to first-line osimertinib: Preliminary data from the phase III FLAURA study. Ann. Oncol. 2018, 29, viii740. [Google Scholar] [CrossRef]
- Ou, S.I.; Cui, J.; Schrock, A.B.; Goldberg, M.E.; Zhu, V.W.; Albacker, L.; Stephens, P.J.; Miller, V.A.; Ali, S.M. Emergence of novel and dominant acquired EGFR solvent-front mutations at Gly796 (G796S/R) together with C797S/R and L792F/H mutations in one EGFR (L858R/T790M) NSCLC patient who progressed on osimertinib. Lung Cancer 2017, 108, 228–231. [Google Scholar] [CrossRef] [Green Version]
- Arulananda, S.; Do, H.; Musafer, A.; Mitchell, P.; Dobrovic, A.; John, T. Combination Osimertinib and Gefitinib in C797S and T790M EGFR-Mutated Non-Small Cell Lung Cancer. J. Thorac. Oncol. 2017, 12, 1728–1732. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Yang, J.J.; Huang, J.; Ye, J.Y.; Zhang, X.C.; Tu, H.Y.; Han-Zhang, H.; Wu, Y.L. Lung Adenocarcinoma Harboring EGFR T790M and In Trans C797S Responds to Combination Therapy of First- and Third-Generation EGFR TKIs and Shifts Allelic Configuration at Resistance. J. Thorac. Oncol. 2017, 12, 1723–1727. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Gureasko, J.; Shen, K.; Cole, P.A.; Kuriyan, J. An allosteric mechanism for activation of the kinase domain of epidermal growth factor receptor. Cell 2006, 125, 1137–1149. [Google Scholar] [CrossRef] [Green Version]
- Dogruluk, T.; Tsang, Y.H.; Espitia, M.; Chen, F.; Chen, T.; Chong, Z.; Appadurai, V.; Dogruluk, A.; Eterovic, A.K.; Bonnen, P.E.; et al. Identification of Variant-Specific Functions of PIK3CA by Rapid Phenotyping of Rare Mutations. Cancer Res. 2015, 75, 5341–5354. [Google Scholar] [CrossRef] [Green Version]
- Imielinski, M.; Berger, A.H.; Hammerman, P.S.; Hernandez, B.; Pugh, T.J.; Hodis, E.; Cho, J.; Suh, J.; Capelletti, M.; Sivachenko, A.; et al. Mapping the hallmarks of lung adenocarcinoma with massively parallel sequencing. Cell 2012, 150, 1107–1120. [Google Scholar] [CrossRef] [Green Version]
- Sholl, L.M.; Aisner, D.L.; Varella-Garcia, M.; Berry, L.D.; Dias-Santagata, D.; Wistuba, I.I.; Chen, H.; Fujimoto, J.; Kugler, K.; Franklin, W.A.; et al. Multi-institutional Oncogenic Driver Mutation Analysis in Lung Adenocarcinoma: The Lung Cancer Mutation Consortium Experience. J. Thorac. Oncol. 2015, 10, 768–777. [Google Scholar] [CrossRef] [Green Version]
- Diaz, L.A., Jr.; Williams, R.T.; Wu, J.; Kinde, I.; Hecht, J.R.; Berlin, J.; Allen, B.; Bozic, I.; Reiter, J.G.; Nowak, M.A.; et al. The molecular evolution of acquired resistance to targeted EGFR blockade in colorectal cancers. Nature 2012, 486, 537–540. [Google Scholar] [CrossRef] [Green Version]
- Park, J.; Cho, Y.-H.; Shin, W.-J.; Lee, S.-K.; Lee, J.; Kim, T.; Cha, P.-H.; Yang, J.S.; Cho, J.; Min, D.S.; et al. A Ras destabilizer KYA1797K overcomes the resistance of EGFR tyrosine kinase inhibitor in KRAS-mutated non-small cell lung cancer. Sci. Rep. 2019, 9, 648. [Google Scholar] [CrossRef]
- Chabon, J.J.; Simmons, A.D.; Lovejoy, A.F.; Esfahani, M.S.; Newman, A.M.; Haringsma, H.J.; Kurtz, D.M.; Stehr, H.; Scherer, F.; Karlovich, C.A.; et al. Circulating tumour DNA profiling reveals heterogeneity of EGFR inhibitor resistance mechanisms in lung cancer patients. Nat. Commun. 2016, 7, 11815. [Google Scholar] [CrossRef]
- Wang, Q.; Yang, S.; Wang, K.; Sun, S.Y. MET inhibitors for targeted therapy of EGFR TKI-resistant lung cancer. J. Hematol. Oncol. 2019, 12, 63. [Google Scholar] [CrossRef]
- Noonan, S.A.; Berry, L.; Lu, X.; Gao, D.; Barón, A.E.; Chesnut, P.; Sheren, J.; Aisner, D.L.; Merrick, D.; Doebele, R.C.; et al. Identifying the Appropriate FISH Criteria for Defining MET Copy Number-Driven Lung Adenocarcinoma through Oncogene Overlap Analysis. J. Thorac. Oncol. 2016, 11, 1293–1304. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Li, L.; Han, R.; Jiao, L.; Zheng, J.; He, Y. Clinical analysis by next-generation sequencing for NSCLC patients with MET amplification resistant to osimertinib. Lung Cancer 2018, 118, 105–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Y.L.; Zhang, L.; Kim, D.W.; Liu, X.; Lee, D.H.; Yang, J.C.; Ahn, M.J.; Vansteenkiste, J.F.; Su, W.C.; Felip, E.; et al. Phase Ib/II Study of Capmatinib (INC280) Plus Gefitinib After Failure of Epidermal Growth Factor Receptor (EGFR) Inhibitor Therapy in Patients With EGFR-Mutated, MET Factor-Dysregulated Non-Small-Cell Lung Cancer. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2018, 36, 3101–3109. [Google Scholar] [CrossRef] [PubMed]
- Turke, A.B.; Zejnullahu, K.; Wu, Y.L.; Song, Y.; Dias-Santagata, D.; Lifshits, E.; Toschi, L.; Rogers, A.; Mok, T.; Sequist, L.; et al. Preexistence and clonal selection of MET amplification in EGFR mutant NSCLC. Cancer Cell 2010, 17, 77–88. [Google Scholar] [CrossRef] [Green Version]
- Yamada, T.; Matsumoto, K.; Wang, W.; Li, Q.; Nishioka, Y.; Sekido, Y.; Sone, S.; Yano, S. Hepatocyte Growth Factor Reduces Susceptibility to an Irreversible Epidermal Growth Factor Receptor Inhibitor in EGFR-T790M Mutant Lung Cancer. Clin. Cancer Res. 2010, 16, 174–183. [Google Scholar] [CrossRef] [Green Version]
- Mazières, J.; Barlesi, F.; Filleron, T.; Besse, B.; Monnet, I.; Beau-Faller, M.; Peters, S.; Dansin, E.; Früh, M.; Pless, M.; et al. Lung cancer patients with HER2 mutations treated with chemotherapy and HER2-targeted drugs: Results from the European EUHER2 cohort. Ann. Oncol. 2016, 27, 281–286. [Google Scholar] [CrossRef] [PubMed]
- Azuma, K.; Kawahara, A.; Sonoda, K.; Nakashima, K.; Tashiro, K.; Watari, K.; Izumi, H.; Kage, M.; Kuwano, M.; Ono, M.; et al. FGFR1 activation is an escape mechanism in human lung cancer cells resistant to afatinib, a pan-EGFR family kinase inhibitor. Oncotarget 2014, 5, 5908–5919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guibert, N.M.; Paweletz, C.; Hu, Y.; Feeney, N.B.; Plagnol, V.; Poole, V.; Jones, G.; Oxnard, G.R. Early detection of competing resistance mutations using plasma next-generation sequencing (NGS) in patients (pts) with EGFR-mutant NSCLC treated with osimertinib. J. Clin. Oncol. 2017, 35, 11529. [Google Scholar] [CrossRef]
- Sharma, S.V.; Lee, D.Y.; Li, B.; Quinlan, M.P.; Takahashi, F.; Maheswaran, S.; McDermott, U.; Azizian, N.; Zou, L.; Fischbach, M.A.; et al. A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell 2010, 141, 69–80. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.; Yamada, T.; Kita, K.; Taniguchi, H.; Arai, S.; Fukuda, K.; Terashima, M.; Ishimura, A.; Nishiyama, A.; Tanimoto, A.; et al. Transient IGF-1R inhibition combined with osimertinib eradicates AXL-low expressing EGFR mutated lung cancer. Nat. Commun. 2020, 11, 4607. [Google Scholar] [CrossRef]
- Niederst, M.J.; Sequist, L.V.; Poirier, J.T.; Mermel, C.H.; Lockerman, E.L.; Garcia, A.R.; Katayama, R.; Costa, C.; Ross, K.N.; Moran, T.; et al. RB loss in resistant EGFR mutant lung adenocarcinomas that transform to small-cell lung cancer. Nat. Commun. 2015, 6, 6377. [Google Scholar] [CrossRef]
- Jain, P.; Fierst, T.M.; Han, H.J.; Smith, T.E.; Vakil, A.; Storm, P.B.; Resnick, A.C.; Waanders, A.J. CRAF gene fusions in pediatric low-grade gliomas define a distinct drug response based on dimerization profiles. Oncogene 2017, 36, 6348–6358. [Google Scholar] [CrossRef] [Green Version]
- Lin, J.J.; Zhu, V.W.; Yoda, S.; Yeap, B.Y.; Schrock, A.B.; Dagogo-Jack, I.; Jessop, N.A.; Jiang, G.Y.; Le, L.P.; Gowen, K.; et al. Impact of EML4-ALK Variant on Resistance Mechanisms and Clinical Outcomes in ALK-Positive Lung Cancer. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2018, 36, 1199–1206. [Google Scholar] [CrossRef]
- Zahavi, D.; AlDeghaither, D.; O’Connell, A.; Weiner, L.M. Enhancing antibody-dependent cell-mediated cytotoxicity: A strategy for improving antibody-based immunotherapy. Antib. Ther. 2018, 1, 7–12. [Google Scholar] [CrossRef] [Green Version]
- Pirker, R.; Pereira, J.R.; Szczesna, A.; von Pawel, J.; Krzakowski, M.; Ramlau, R.; Vynnychenko, I.; Park, K.; Yu, C.T.; Ganul, V.; et al. Cetuximab plus chemotherapy in patients with advanced non-small-cell lung cancer (FLEX): An open-label randomised phase III trial. Lancet 2009, 373, 1525–1531. [Google Scholar] [CrossRef]
- Cho, J.; Chen, L.; Sangji, N.; Okabe, T.; Yonesaka, K.; Francis, J.M.; Flavin, R.J.; Johnson, W.; Kwon, J.; Yu, S.; et al. Cetuximab response of lung cancer-derived EGF receptor mutants is associated with asymmetric dimerization. Cancer Res 2013, 73, 6770–6779. [Google Scholar] [CrossRef] [Green Version]
- Vyse, S.; Huang, P.H. Targeting EGFR exon 20 insertion mutations in non-small cell lung cancer. Signal Transduct. Target. Ther. 2019, 4, 5. [Google Scholar] [CrossRef] [Green Version]
- van Veggel, B.; de Langen, A.J.; Hashemi, S.M.S.; Monkhorst, K.; Heideman, D.A.M.; Thunnissen, E.; Smit, E.F. Afatinib and Cetuximab in Four Patients With EGFR Exon 20 Insertion–Positive Advanced NSCLC. J. Thorac. Oncol. 2018, 13, 1222–1226. [Google Scholar] [CrossRef]
- Truong, T.H.; Ung, P.M.; Palde, P.B.; Paulsen, C.E.; Schlessinger, A.; Carroll, K.S. Molecular Basis for Redox Activation of Epidermal Growth Factor Receptor Kinase. Cell Chem. Biol. 2016, 23, 837–848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riess, J.W.; Groshen, S.G.; Reckamp, K.L.; Wakelee, H.A.; Oxnard, G.R.; Padda, S.K.; Koczywas, M.; Piotrowska, Z.; Sholl, L.M.; Paweletz, C.P.; et al. Osimertinib (Osi) plus necitumumab (Neci) in EGFR-mutant NSCLC: An ETCTN California cancer consortium phase I study. J. Clin. Oncol. 2019, 37, 9057. [Google Scholar] [CrossRef]


{kind=link}
| Cancer type | Frequency of Activating Mutations of EGFR | Study | Frequency of EGFR Overexpression | Sample Size | Method of Detection | Study |
|---|---|---|---|---|---|---|
| NSCLC | 50% of Asian patients 10–15% of Caucasian patients | [33] | 43–89% | 96–515 | IHC a | [34,35,36,37,38,39,40,41,42,43] |
| 31–58% | 2972 | Meta-analysis | [44] | |||
| Colorectal carcinoma (CRC) | Rare–2.33% | [45] | 51–75.5% | 99–193 | IHC | [46,47,48,49] |
| Head and neck SCC b | Rare–1.72% | [45] | 45% | 115 | IHC | [50] |
| 92% | 24 | Southern blot hybridisation | [51] | |||
| Pancreatic adenocarcinoma | Rare–0.78% | [45] | 49–69% | 32–181 | IHC | [52,53,54] |
| Breast cancer | Rare–1.31% | [45] | 27% | 21,418 | Meta-analysis | [55] |
| Prostate | Rare–0.82% | [45] | 31–100% | 74–98 | IHC | [56,57] |
| Oesophageal SCC | Rare–2.72% | [45] | 53.6–65% | 56–152 | IHC | [58,59] |
| Gastric | Rare–2.2% | [45] | 27–44% | 82–511 | IHC | [60,61] |
| Hepatocellular carcinoma | Rare–1.59% | [45] | 47–68% | 53–100 | IHC | [62,63] |
| Glioblastoma | 17.56% | [45] | 63% | 49 | IHC | [64] |
| Ovary | Rare–0.98% | [45] | 28–33% | 80 | IHC | [65] |
| Bladder | Rare–3.28% | [45] | 26.2–71% | 72–126 | IHC | [66,67] |
| Renal cell carcinoma | Rare–1.16% | [45] | 21–98% | 50–175 | IHC | [68,69,70] |
| Cancer Type | Tyrosine Kinase Inhibitors | Monoclonal Antibodies |
|---|---|---|
| NSCLC | 1st generation Gefitinib-metastatic NSCLC with EGFR exon 19 deletion or exon 21 mutation (L858R) Erlotinib-metastatic or locally advanced NSCLC with EGFR exon 19 deletion or exon 21 mutation (L858R) 2nd generation Dacomitinib-metastatic NSCLC with EGFR exon 19 deletion or exon 21 mutation (L858R) Afatinib-metastatic NSCLC with EGFR exon 19 deletion or exon 21 mutation (L858R) 3rd generation Osimertinib-metastatic EGFR T790M mutation-positive NSCLC, with progressive disease following first-line EGFR TKI therapy Olmutinib-second-line treatment of NSCLC with T790M mutation in EGFR | |
| Pancreatic cancer | Erlotinib-metastatic or advanced pancreatic cancer in combination with gemcitabine | |
| Breast cancer | Neratinib-HER2-overexpressing breast cancer Lapatinib-HER2-overexpressing breast cancer | |
| Thyroid cancer | Vandetanib-medullary thyroid carcinoma | |
| CRC | Cetuximab-metastatic KRAS-negative CRC; in combination with chemotherapy or as a single agent Panitumumab-metastatic KRAS-negative CRC; in combination with chemotherapy or as a monotherapy in patients who have failed chemotherapy | |
| Head and neck- SCC | Cetuximab-in combination with radiation therapy for locally advanced disease or in combination with chemotherapy for recurrent/metastatic disease |
| Effector | Prevalence | Resistance To | Mechanism |
|---|---|---|---|
| Germ Line Polymorphisms | |||
| EGFR-T790M | Preclinical [116] | 1st Gen. TKIs | Allosteric hindrance of ATP-binding pocket, increased ATP affinity |
| EGFR-V843I | 3/5 carriers developed disease [117] | 1st Gen. TKIs | Steric hindrance, associated with additional L858R mutation [118,119,120] |
| BIM | Deletion in 12.9% of East Asian individuals [121] | 1st/2nd/3rd TKIs | High BIM expression correlates with tumour apoptosis and enhanced PFS/OS [122,123,124,125,126,127] |
| Secondary EGFR Mutations | |||
| S768I | 9/1527 cases | 1st Gen. TKIs | Attenuated BIM to reduce apoptosis, often with concurrent G719 or L858R [123,128] |
| D761Y | 1/16 cases | 1st Gen. TKIs | Reduced EGFR phosphorylation with additional L858R mutation [129,130] |
| T854A | 1/48 cases | 1st Gen. TKIs | Steric hindrance [131] |
| L747S | 12/3648 [132] | 1st Gen. TKIs | Steric hindrance [133] |
| T790M | 98/155 cases [134] | 1st/2nd Gen. TKI | Allosteric hindrance of ATP-binding pocket, increased ATP affinity [135] |
| Exon20 Insertion | Asia: 67/218 [136], 23/186 Europe [137] | 1st/2nd/3rd Gen. TKIs | Conformational change inducing constitutive activation [136,137,138,139] |
| L747P | Case Report | 1st/3rd Gen. TKIs | Conformational change inducing constitutive activation [140,141] |
| T790M Amplification | Preclinical | 2nd Gen. TKIs | Reversible selection of amplified clone in response to TKI [142] |
| C797S | 6/15 cases [143] | 3rd Gen. TKIs | Mutation in EGFR prevents osimertinib binding, 84% co-occur with multiple resistance mechanisms [144] |
| G796S/R/D | 23/93 cases | 1st/3rd Gen. TKIs | Steric hindrance [145,146] |
| L792F/H | 10/93 cases | 3rd Gen. TKIs | Steric hindrance, arise in trans with T790M and cis with C797S [145,147] |
| L718Q | 9/93 cases [145,148] | 3rd Gen. TKIs | Steric hindrance [145] |
| G724S | 4/30 cases | 3rd Gen. TKIs | A glycine-rich loop conformation prevents initial reversible TKI binding. The mutation is associated with T790M loss, mutually exclusive to C979S, and afatinib-sensitive [149,150]. |
| Enhancement of Alternate Pathways | |||
| CRKL | 1/11 cases [151] | 1st/2nd Gen. TKIs | Amplification, leading to downstream activation of ERK and Akt [151,152] |
| MAPK | Case report | 1st Gen. TKIs | ERK overexpression [153] |
| IGF1R | Preclinical | 1st/2nd Gen. TKIs | Constitutive activation of PI3K/Akt pathway [154,155] |
| MET | 4/18 1st Gen-resistant cases [156], 3/5 3rd Gen-resistant cases [157] | 1st/3rd Gen. TKIs [156,157,158] | HER3-dependent PI3K activation [156] |
| HER2 | 3/26 cases [159], 2/5 3rd Gen-resistant cases [157] | 1st/2nd/3rd Gen. TKIs [157,159] | Alternative receptor amplification, mutually exclusive with the T790M mutation [159] |
| FGFR1 | 1/23 cases [160] | 3rd Gen. TKIs | Autocrine loop signalling [161] |
| Downstream Mutations | |||
| PTEN | 1/24 cases | 1st/2nd Gen. TKIs | PI3K/Akt activation via PIK3CA [162] |
| PIK3CA | 5/43 cases [163] | 1st/2nd/3rd Gen. TKIs | PI3K/Akt activation [164] |
| AKT1 | 3/49 cases | 1st/2nd/3rd Gen. TKIs | mTOR activation [165] |
| BRAF | 2/195 cases | 3rd Gen. TKIs | MEK and ERK overexpression [166,167] |
| KRAS | 3/43 cases [167], 9/38 adenocarcinoma cases [168] | 3rd Gen. TKIs | MAPK overexpression, mutually exclusive with the EGFR mutations [167,169,170] |
| EMT | |||
| AXL (and ligand GAS6) | 7/35 cases | 1st/2nd/3rd Gen. TKIs | EMT with vimentin overexpression [169] |
| Histologic Transformation | |||
| Chronic EGFR Inhibition | 5/37 1st Gen-resistance cases [170], 2 3rd Gen-resistant case studies [171] | 1st/3rd Gen. TKIs | Transformation to SCLC with EMT and potential retinoblastoma signalling [159,172,173] |
| Chronic EGFR Inhibition | 1st Gen-resistant case study [172,173], 5/71 3rd Gen-resistant cases [174] | 1st/3rd Gen. TKIs | Transformation from adenocarcinoma to SCC [172,173,174] |
| Transcriptional Regulation | |||
| NF-κB | Statistical significance across 52 cases, Preclinical | 1st Gen. TKIs | High NF-κB activity predicted resistance via survival signalling [175] |
| Epigenetics | Preclinical | 1st Gen. TKIs | miR-21 expression reducing PTEN and PDCD4 activity [176] |
| Oncogene Fusion | |||
| EIF4G2 | 1/32 | 1st/2nd Gen. TKIs | Treatment-resistance associated fusion to GAB1 [177] |
| ALK | 108/2835 | 2nd/3rd Gen. TKI | Treatment-resistance associated fusion to EML4, STRN, CEBPZ predominantly in adenocarcinoma cases in minimal smokers [178,179] |
| NTRK1 | 4/3875 cases | 2nd/3rd Gen. TKIs | Treatment-resistance associated fusion to LRRC71, PLEKHA6, RRL8, RP11 [178] |
| BRAF | 1/31 with KIF5A [177], 10/3595 [180] | 2nd/3rd Gen. TKIs | Treatment-resistance associated fusion to AGAP3, AGK, ARMC10, DOCK4, EPS15, KIAA1549, SALL2, TRIM24 [180] |
| RET | 6/3875 cases | 3rd Gen. TKIs | Treatment-resistance associated fusion to CCDC6, MCPA4, CDC123, KIF5B [178,181] |
| ROS1 | 3/3875 cases | 3rd Gen. TKIs | Treatment-resistance associated fusion to DCBLD1 [178] |
| FGFR3 | 5/32 | 3rd Gen. TKIs | Treatment-resistance associated fusion to TACC3 [177] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Karlsen, E.-A.; Kahler, S.; Tefay, J.; Joseph, S.R.; Simpson, F. Epidermal Growth Factor Receptor Expression and Resistance Patterns to Targeted Therapy in Non-Small Cell Lung Cancer: A Review. Cells 2021, 10, 1206. https://doi.org/10.3390/cells10051206
Karlsen E-A, Kahler S, Tefay J, Joseph SR, Simpson F. Epidermal Growth Factor Receptor Expression and Resistance Patterns to Targeted Therapy in Non-Small Cell Lung Cancer: A Review. Cells. 2021; 10(5):1206. https://doi.org/10.3390/cells10051206
Chicago/Turabian StyleKarlsen, Emma-Anne, Sam Kahler, Joan Tefay, Shannon R. Joseph, and Fiona Simpson. 2021. "Epidermal Growth Factor Receptor Expression and Resistance Patterns to Targeted Therapy in Non-Small Cell Lung Cancer: A Review" Cells 10, no. 5: 1206. https://doi.org/10.3390/cells10051206
APA StyleKarlsen, E.-A., Kahler, S., Tefay, J., Joseph, S. R., & Simpson, F. (2021). Epidermal Growth Factor Receptor Expression and Resistance Patterns to Targeted Therapy in Non-Small Cell Lung Cancer: A Review. Cells, 10(5), 1206. https://doi.org/10.3390/cells10051206