1. Introduction
Lithium-ion batteries (LIBs) have many advantages over other energy storage devices, such as high energy density, low discharging rate, long cycling life, portability, and light weight, which makes them a promising technology for future energy storage applications [
1,
2,
3,
4]. The overall performance of LIBs is significantly influenced by the properties of electrode materials; hence, anode materials play a vital role in accomplishing high energy and power densities [
5,
6]. Commercial graphite is a conventional anode material for LIBs, owing to its high abundance, structural stability, low working potential, and long-term cyclability. However, its low theoretical capacity of 372 mAh g
−1 is incapable of meeting current high energy demands, such as automotive applications, electric vehicles, energy storage devices, etc. [
7,
8]. Therefore, alternative anode materials with high theoretical capacities with low working potentials are necessary to meet the high energy and power density demands. Silicon (Si) is considered one of the promising anode materials owing to its high theoretical capacity of ~4200 mAh g
−1 and low working potential of ~0.4 V vs. Li/Li
+. Its environmentally friendly nature and high abundance further boost its practical applicability as an efficient anode material for LIBs [
9,
10,
11]. However, massive volume expansion, low intrinsic conductivity, and an unstable solid electrolyte interphase (SEI) layer limit its practical applicability. Active Si undergoes severe structural and mechanical stress during Li
+ alloying/dealloying reactions and leads to the loss of contact with the correct collector and causes a rapid decline in capacities [
12,
13].
To address these issues, several strategies have been employed to improve the Si-based anodes, including reducing the Si particle size from bulk to nanosize [
14,
15], and synthesizing different Si nanostructures to control the cracking and pulverization of Si [
16,
17]. Nanosize active materials benefit in alleviating structural breakdowns affected by the large strain. The large surface area provided by nanomaterials could afford a large reaction site for Li
+ ions and thus retain superior specific capacities [
18]. However, the large-scale material production of these nanostructures is highly impractical due to the requirement of sophisticated equipment and high synthesis expenditures. Preparation of nano/micro/sub-micron Si particles from bulk Si by high-energy ball milling is an affordable and cost-effective approach, which greatly reduces the production cost of Si [
19,
20]. Merging nanosized anode materials with carbon/carbonaceous materials greatly improves the electrochemical performance by relieving the volume changes and holding the electrical contact during the lithiation/delithiation process [
18]. Introducing carbon materials as composites with Si and using protective carbon coatings are also effective ways to improve the performance of Si anodes [
21,
22,
23]. The amount of carbon in the composite material plays a crucial role in stabilizing the Si anode performance by affording the required electrical conductivity and mechanical stability [
24,
25]. However, adding large amounts of carbon significantly reduces the overall specific capacity of the composite material by forming a dead layer on the surface of the electrode. Impurity doping such as boron (B) [
26,
27] and phosphorous (P) [
28,
29] is another contemporary way to overcome these disadvantages and advance the conductivity and ionic mobility of Si anodes. Ke et al. synthesized thin carbon shell and nitrogen/phosphorus co-doped two-dimensional (2D) carbon sheet dual encapsulate Si nanoparticles, which showed high reversible capacities of 592 mAh g
−1 after 100 cycles at 200 mA g
−1, indicating the excellent conductive support of nitrogen and phosphorous dopants along with carbon [
30].
Recent reports reveal that transition metal alloying/doping/composites with Si improve its electrical conductivity and structural stability and are favorable in enhancing the overall electrochemical performance [
31,
32,
33]. Hou et al. prepared ultrafine cobalt sulfide encapsulated multi-channel carbon nanofibers as anodes, which showed excellent electrochemical properties with 737 mAh g
−1 capacities after 100 cycles at 200 mA g
−1 [
18]. Previous reports also suggested that the transition metal-doped Si nanoparticles showed improved electronic and magnetic properties [
34,
35]. Kim et al. prepared Al-doped Si film and studied it as anode material for LIBs. The Al-doped Si films showed better electrochemical properties than pristine Si [
36]. Huang et al. prepared a NiSi
x intermetallic skin-coated Si anode, where NiSi
x acted as a protective layer, which exhibited improved cyclability with rate capabilities [
37]. Lin et al. fabricated Cu-induced porous Si composite films as an anode, which showed excellent electrochemical properties [
38]. Lee et al. prepared a graphite–FeSi alloy composite anode, where FeSi
2 acted as a buffer matrix for active Si and exhibited good cyclability with high reversible capacities [
39]. Bimodal nanoporous NiO@Ni-Si networks prepared by Wang et al. delivered excellent long-term cyclability with 1387 mAh g
−1 capacities at 500 mA g
−1 after 1000 cycles, where the Ni-Si network plays a crucial role in accommodating the large volume changes during cycling [
40]. The composite of dual-shell Si/TiO
2/CF prepared by Zeng et al. delivered 583 mAh g
−1 specific capacity with 87.4% retention at 100 mA g
−1 after 180 cycles, where the rigid TiO
2 gives the required mechanical strength, and carbon affords excellent electrical conductivity to Si particles [
41]. From our previous report, Co-doped Si nanoparticles also showed favorable electrochemical properties, along with improved Li
+ diffusion properties [
30]. Apart from Si anodes, other anode materials doped with Cu, Fe, and Mn such as Li
3VO
4, SnO
2, TiO
2, ZnFe
2O
4, etc., showed improved electrochemical properties compared to their pristine material [
42,
43,
44,
45,
46]. All these results suggested that transition metal doping encourages Li
+ kinetics, intrinsic conductivity, and structural stability, which favors the overall electrochemical properties of the host electrode.
In our previous work, the influence of metal doping on Si NPs was studied, where low doping concentrations of transition metals (Mn, Ni) were used to dope into Si nanoparticles [
47]. However, the usage of Si NPs (commercially purchased) increases the production cost, which is a drawback for industrial applications. In this study, bulk Si is taken as precursor material instead of Si NPs, which is cost-effective, greatly reduces expenses, and encourages large-scale manufacture. The addition of graphite powder significantly enhances the stability of bulk Si in advancing a long and stable cycle life. Based on the above findings, considering a relatively cost-effective and scalable approach, we proposed a metal-doped Si/graphite composite as an anode material for LIBs. Here, three different transition metals (Cu, Fe, Mn) with low doping concentrations (0.5 at %) were used to dope into the Si host, and subsequently, construct a composite with graphite. The metal dopants in the composite enhanced the intrinsic conductivity of Si, as the graphite provides the required electrical conductivity and structural stability to Si. All prepared anodes delivered favorable electrochemical properties attributed to enriched electrical conductivity and structural stability provided by metal dopants and graphite in the composite material.
2. Experimental
2.1. Materials and Methods
Si powder (325 mesh, 99% trace metals basis, Sigma-Aldrich,Burlington, MA, USA), copper nitrate trihydrate (Cu(NO3)2·3 H2O—Sigma-Aldrich, Burlington, MA, USA), iron nitrate nonahydrate (Fe(NO3)3·9 H2O—Sigma-Aldrich, Burlington, MA, USA), manganese nitrate hexahydrate (Mn(NO3)2·6H2O—Sigma-Aldrich, Burlington, MA, USA), graphite powder (TIMCAL TIMREX® SFG6, TIMCAL Ltd. Bodio, Leventina, Switzerland), and an ethanol solvent (≥99.5%, Sigma-Aldrich, Burlington, MA, USA) were purchased and used without further purification.
The metal-doped Si/graphite composite was prepared by a three-step process. Firstly, nano/micro Si particles were prepared from bulk Si (325 mesh). Later, the prepared nano/micro Si was doped with transition metals, followed by making a composite with graphite powder, yielding a transition metal-doped Si/graphite composite.
2.2. Preparation of Nano/Micro Si
To prepare nano/micro Si, 5 g of Si powder (325 mesh) was taken into a high-density polyethylene (HDPE) vial along with a few zirconium balls, with a ball-to-powder ratio of 50:1. Then, the vial was tightly closed with Teflon tape and placed in a ball mill. The milling was carried out for 48 h at the speed of 150 rpm at room temperature. The obtained Si powder with zirconium balls was sieved for about 2 h and the powder was collected, referred to as Si-48.
2.3. Fabrication of Transition Metal-Doped Nano/Micro Si
The prepared Si-48 powder was taken into a zirconium sample holder of a mini-mill and added with the desired amount (0.5 at%) of Cu precursor along with ~1 mL of ethanol. Then, the lid was placed and the milling was carried out for 1 h at a speed of 30 oscillations sec−1 to make a uniform slurry. The obtained slurry was taken into an alumina crucible and placed in a tubular furnace and heat-treated at 600 °C for 2 h at a heat rate of 3 °C min−1 under an argon atmosphere. When the furnace reached room temperature, the sample was collected and signed as SiCu by its metal dopant. Using this same approach, Fe-doped and Mn-doped Si materials were prepared and named SiFe and SiMn, respectively. All the prepared materials were used for further analysis.
2.4. Preparation of Metal-Doped Si/Graphite Composite
The preparation of metal-doped Si/graphite composite was as follows. Firstly, 425 mg of metal (Cu/Fe/Mn)-doped Si particles was taken into a mini-mill and 75 mg of graphite powder was added and milled for 30 min for thorough mixing of the two materials. Then, the mixture was transferred to an alumina crucible of a tubular furnace and heat-treated at 400 °C for 2 h at a heat rate of 5 °C min−1 in an argon atmosphere. After the furnace was cooled to room temperature, the samples were collected and named SiCuG, SiFeG, and SiMnG, referring to the presence of metal dopant and graphite. Si-48 was also added with graphite powder and prepared composite with the same experimental conditions to analyze the effect of metal dopants, and named SiG. All the prepared materials were used for further analysis.
2.5. Material Characterization
The crystal structure of the prepared materials was investigated by an X-ray diffractometer (XRD) using the Rigaku D/MAX-2200 (Rigaku corp., Tokyo, Japan) Ultima instrument equipped with Cu@Kα radiation (λ = 1.54056 Å) in the range of 10° < 2θ < 80° operating at 30 kV and 40 mA. The surface morphology and microstructures were analyzed by scanning electron microscopy SEM, JSM7000F (JEOL Ltd., Tokyo, Japan) and transmission electron microscopy TEM, JEM-2100F (JEOL Ltd., Tokyo, Japan). Raman spectroscopy was used to analyze the chemical composition of the samples. Thermogravimetric analysis (TGA) was operated from 25 °C to 800 °C to investigate the amount of carbon in the materials. X-ray photoelectron spectroscopy (XPS) on a PHI Quantear SXM (ULVAC-PHI Inc., Kanagawa, Japan) was employed to investigate the surface chemical states of the prepared materials. The Brunauer–Emmett–Teller (BET) method was applied to analyze the specific surface area of the prepared composite materials.
2.6. Electrochemical Measurements
The CR 2032 button-type coin cells were used to evaluate the electrochemical properties of all prepared materials. The working electrodes (SiCuG, SiFeG, and SiMnG) were prepared by adding 80% of active materials with 20% of polyamide-imide (PAI-(Typolymer. Co., Ltd. Torlon(R) Amideimide, 4000T-HV, Seoul, Korea)) in N-methyl pyrrolidone (NMP) as a binder. No conductive carbon additive was added due to the presence of graphite in the active material. For a comparison study, the other working electrodes (Si-48, SiCu, SiFe, and SiMn) were prepared by adding 68% active material with 12% super P as conductive carbon and 20% of PAI binder in NMP to match the amounts of Si, carbon, and binder. All components were taken into a mini-mill and milled for 15 min to make a uniform slurry. The resultant slurry was uniformly coated on a copper foil current collected with a 20 µm thickness by using the traditional doctor blade approach (wet film thickness: 0–3000 μm, controllable accuracy: 5 μm). Then, the electrodes were dried in the oven at 80 °C to evaporate the excess solvent, subsequently transferred to the vacuum oven, and dried at 200 °C for 3 h to activate the binder. After that, the electrodes were punched into 14 mm disks and used as working electrodes. The mass loading of the active material was ~2.6 mg.·cm−1, with an average thickness of 145 μm. All coin cells were fabricated in an Ar-filled glove box (H2O < 0.1 ppm and O2 < 1 ppm). Metallic lithium (MTI KOREA Corp., Seoul, Korea) with thickness: 0.17 mm, diameter: 16 mm was used as a counter electrode without prior surface cleaning. Then, 1 M LiPF6 in ethylene carbonate (EC), diethyl carbonate (DEC), and fluoroethylene carbonate (FEC) were used in a v/v ratio of 5:70:25. Polypropylene (PP Wellcos Corporation, Separator 2400) with a thickness of 27 μm and a diameter of 19 mm was used as the separator. The cyclability tests were carried out at 200 mA g−1, and the rate capability tests were conducted between 200 and 3200 mA g−1 applied currents, in the voltage range of 0.01 V–2.0 V. The redox behavior of the electrodes was investigated by employing cyclic voltammetry (CV) in the voltage window of 0.01 V–2.0 V at a scan rate of 0.1 mV s−1. Electrochemical impedance spectroscopy (EIS) was employed within the frequency range of 10 mHz to 10 kHz. For full cell preparation, 16 mm diameter disks of the prepared anodes were prelithiated by assembling half cells with Li metal as a counter electrode, with 1 M LiPF6 in EC:DEC:FEC (v/v ratio of 5:70:25) electrolyte, and a polypropylene separator. The cells were fully discharged to 0.01 V at 200 mA g−1. To prepare the cathode electrode, 90% commercial LiCoO2 (LCO, purity 99.5%, Thermo Fisher Scientific Inc., Waltham, MA, USA) powder was added to 5% Super-P carbon as a conductive additive and 5% PVDF in NMP as a binder. All components were thoroughly mixed using a mortar and pestle for 30 min to form a uniform slurry. The slurry was then coated on Al foil using the traditional doctor blade method (wet film thickness: 0–3000 microns, controllable accuracy: 5 μm) with a thickness of 45 μm and dried in an oven at 80 °C for 2 h. Later, the electrode was heat-treated in a vacuum oven at 100 °C for 1 h. Then, 14 mm diameter disks were punched and used as cathode electrodes. The specific capacity of the working electrode was calculated based on the weight of the active material.
4. Electrochemical Evaluation in LIBs
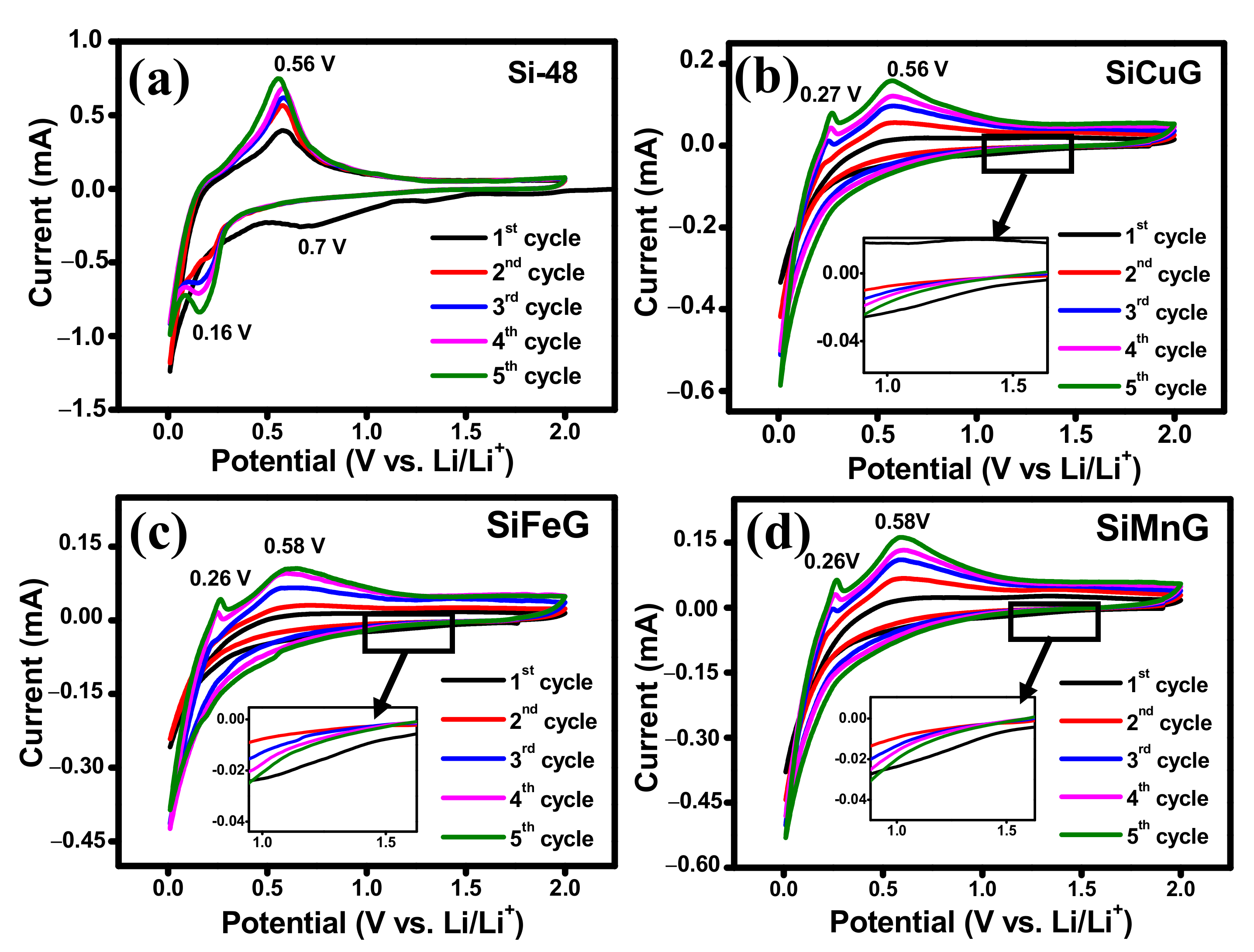
Different electrochemical tests were carried out to investigate the electrochemical performance of the prepared electrode materials. To compare the performance of electrodes, all tests were conducted under the same conditions. The CV results for the first five cycles of Si-48, SiCuG, SiFeG, and SiMnG are shown in
Figure 8a, 8b, 8c, and 8d, respectively, and SiCu, SiFe, and SiMn are shown in
Figure S3a,
Figure S3b, and
Figure S3c, respectively. In the first lithiation of the cathodic scan, Si-48 shows a typical broad reduction peak around ~0.7 V, resulting from the SEI formation layer due to the electrolyte decomposition on the surface of the electrode material. the SiCuG, SiFeG, and SiMnG materials show a smaller extent of SEI formation, indicated by a small reduction peak around ~1.2 V (shown in the insets of
Figure 8b–d). A possible reason could be because the formation of the SEI layer may be a sluggish process during the initial cycles, so the characteristic SEI reduction peaks were not obvious in the initial cycles of CV curves [
52]. This SEI reduction peak disappeared in the following cycles, indicating stable SEI layer formation. The sharp peak at 0.05 V vs. Li
+/Li in all electrodes corresponds to the alloying of Li
+ in crystalline Si and the formation of an amorphous Li
xSi phase. In the anodic scan, SiCuG, SiFeG, and SiMnG materials show two peaks at around ~0.26 V and ~0.58 V, which are related to the characteristic two-step dealloying process of Li
xSi phase to metastable amorphous Si. However, in the anodic scan, Si-48 shows one sharp peak around 0.56 V, resulting from the dealloying of Li
+ from the Li
xSi phase and the formation of amorphous Si, indicating that dealloying in Si-48 is a slow and sluggish process. This occurrence is normally observed in many Si-based anodes. The increase in peak intensities in all electrodes increases with cycles due to the ongoing activation process of the active materials [
25,
45]. The CV results of SiCu, SiFe, and SiMn are similar to the CV curves of Si-48, indicating that the reaction path of the lithiation and delithiation process was not changed after metal doping into Si, due to inactivity of metal dopants towards Li-ions [
30].
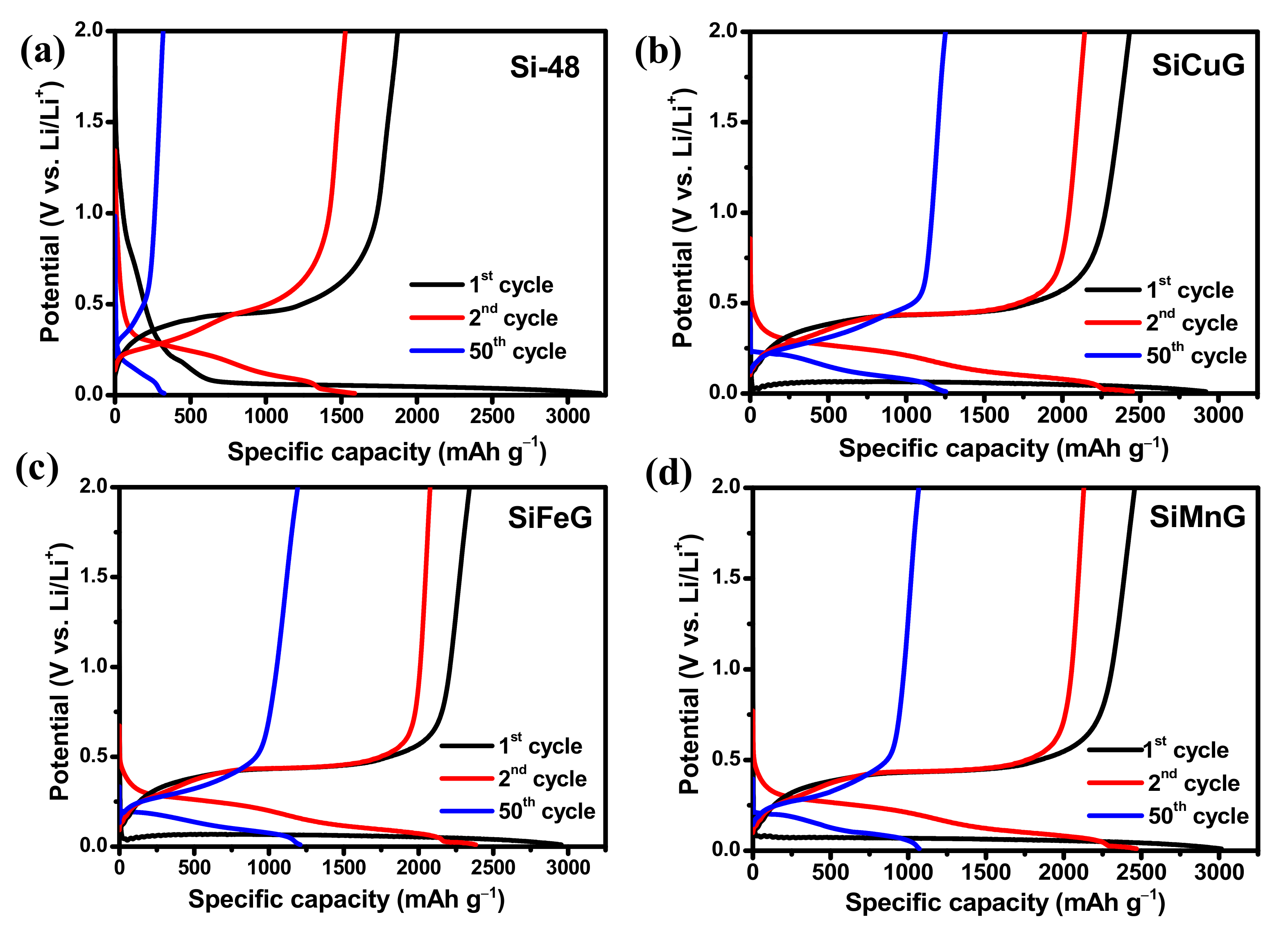
The specific capacity vs. voltage profiles for the 1st, 2nd, and 50th cycles at a current density of 200 mA g
−1 for Si-48, SiCuG, SiFeG, and SiMnG are shown in the
Figure 9a,
Figure 9b,
Figure 9c, and
Figure 9d, respectively. The long discharge plateau below 0.1 V in the first cycle in all electrodes is related to the alloying reaction of Li-ions with crystalline Si and the formation of the Li
xSi phase. In the following cycles, the discharge plateaus are shifted to above ~0.22 V due to the formation of amorphous Si. The charge plateaus above ~0.5 V are in all electrodes, representing the dealloying reaction Li
xSi phase to amorphous Si [
12]. These charge/discharge profiles are in agreement with CV profiles. The first discharge/charge capacities of Si-48, SiCuG, SiFeG, and SiMnG are 3219/1874, 2919/2430, 2954/2340, and 2915/2456 mAh g
–1 with Coulombic efficiencies (CEs) of 58.2%, 83.2%, 79.2%, and 84.2%, respectively. The irreversible capacity loss is accompanied by the formed SEI layer on the electrode’s surface. The lower CE% of Si-48 could be due to the formation of a thick SEI layer, and is consistent with CV results (
Figure 8). On the other hand, the prepared anode materials showed better initial CE because of the thin SEI layer formation resulting from the metal dopant, which controls the Si volume expansion, and the graphite carbon, which maintains the overall structural stability. Furthermore, Si-48 capacities declined rapidly and reached 321/312 mAh g
−1 with 97% CE after 50 cycles, but the prepared SiCuG, SiFeG, and SiMnG electrodes delivered 1253/1244, 1208/1197, and 1170/1165 mAh g
−1 specific capacity with 99.2%, 99%, and 99.5% CE, respectively. The discharge/charge profiles for the 1st, 2nd, and 50th cycles for SiCu, SiFe, and SiMn are shown in
Figure S4a,
Figure S4b, and
Figure S4c, respectively. These materials also show similar capacity vs. voltage profiles, indicating the same reaction path as other prepared materials.
The long-term cyclic stability and CEs were evaluated for Si-48, SiG, SiCuG, SiFeG, SiMnG, SiCu, SiFe, and SiMn materials at applied currents of 200 mA g
−1 within the voltage of 0.01–2.0 V. The cyclability and CE results for Si-48, SiCuG, SiFeG, and SiMnG are shown in
Figure 10a,b, and those for SiG, SiCu, SiFe, and SiMn are shown in
Figure S5a,b. From
Figure 10a, Si-48 shows a higher initial capacity compared with the other three electrodes, but these capacities declined rapidly with the number of cycles. After 75 cycles, Si-48 reached 226/222 mAh g
−1 with 98.2% CE and 23.4% capacity retention compared with the 10th cycle. The less stability and poor capacities could be due to the massive volume changes during lithium reactions, leading to structural destruction and peeling of active materials from the copper current collector. On the other hand, SiCuG, SiFeG, and SiMnG material showed better cyclic stabilities compared to Si-48. After 75 cycles, SiCuG, SiFeG, and SiMnG delivered 1075/1072, 1015/1009, and 1025/1022 mAh g
−1 capacity with 99.7%, 99.4%, and 99.7% CE with 64.2%, 64.4%, and 63.3% capacity retention (compared with 10th cycle), respectively. From
Figure S5a, Si-48, SiCu, SiFe, and SiMn materials show similar cyclability results. However, compared with Si-48, the prepared SiCu, SiFe, and SiMn materials show slight stable cyclability. After 75 cycles, SiCu, SiFe, and SiMn materials delivered 355/342, 417/412, and 437/431 mAh g
−1 capacity with 61.3%, 67.6%, and 52.5% capacity retention (compared with the 10th cycle), respectively. From the cycling results of all prepared materials, it is evident that SiCu, SiFe, and SiMn show better cyclability results, and this could be due to the enhanced intrinsic conductivity and structural stability given by the metal dopants, which are helpful for faster Li
+ reactions. The SiG material also showed better cyclability compared to Si-48, which could be due to the adequate conductivity and structural stability provided by the graphite. However, after 10 cycles, SiG also showed declined capacities. After 30 cycles, the SiG electrode managed to deliver 935/916 mAh g
−1 specific capacities, which is less compared to SiCuG, SiFeG, and SiMnG electrodes. This result suggests that only the addition of graphite into Si-48 is not sufficient to attain stable capacities. On the other hand, SiCuG, SiFeG, and SiMnG materials showed much better cyclability results compared with SiCu, SiFe, and SiMn materials. These results suggest that metal doping into nano/micro Si is insufficient to afford the required structural stability and conductivity to nano/micro Si particles and the need for graphite carbon. The graphite in the composite successfully afforded the required conductivity and structural and mechanical stability to nano/micro Si and encouraged stable SEI formation on the electrode’s surface during harsh Li
+ reactions. The synergetic properties of metal dopants and graphite together promote faster Li
+ diffusion into the active materials and encourage enhanced electrochemical properties. The rate capability results were evaluated for Si-48, SiCuG, SiFeG, and SiMnG electrodes at wide applied current densities from 200 to 3200 mA g
−1 in the voltage window of 0.01 V to 2.0 V, and the resultant specific capacity vs. cycle number plots and CEs are depicted in
Figure 10c and
Figure 10d, respectively. For all prepared materials, the specific capacities are decreased with increasing applied current densities. All prepared SiCuG, SiFeG, and SiMnG showed stable rate capability results compared to Si-48, which failed at higher current densities. Si-48 delivered 3418, 996, and 301 mAh g
−1 at applied currents of 200, 400, and 800 mA g
−1, respectively. After that, the rate capacities dropped quickly and reached single-digit capacities at 1600 mA g
−1 and failed to sustain at higher current densities. On the contrary, the prepared anodes SiCuG/SiFeG/SiMnG delivered 2910/2924/2893, 1366/1440/1575, 955/943/1106, 615/729/761, and 543/642/695 mAh g
−1 capacities at 200, 400, 800, 1600, and 3200 mA g
−1 applied currents, respectively. The rate capability results suggest that the metal dopants and graphite in the composite material play a critical role in enhancing the conductivity and affording the structural flexibility of nano/micro Si particles. The specific capacities of all prepared anode materials at different cycles are summarized in
Table 1.
The long cyclability and CEs of prepared materials were evaluated by cycling at high currents of 1000 mA g
−1, and the results are shown in
Figure 11a and
Figure 11b, respectively. The three electrodes showed declined capacities in the initial cycles, but after 20 cycles, the capacities are stabilized. In the initial cycle, SiCuG, SiFeG, and SiMnG delivered 2514/1609, 2810/1740, and 3105/1600 mAh g
−1 capacities with 64%, 61%, and 51% CE. After 100 cycles, the materials managed to deliver 704/696, 779/758, and 754/740 mAh g
−1 specific capacities with 98.8%, 97.3%, and 98.1% CE, respectively. The capacity retention values compared with the 10th cycles are 73.6%, 72%, and 72.4%, respectively, and suggest the highly stable microstructures of metal-doped Si/G composite even at higher applied currents.
The Li
+ kinetics between electrode and electrolyte were analyzed by employing EIS. The resultant Nyquist plots for Si-48, SiCuG, SiFeG, and SiMnG electrodes before and after 25 charge/discharge cycles at 500 mA g
−1 are displayed in
Figure 12a and
Figure 12b, respectively. All three electrodes exhibited a depressed semicircle in the mid- and high-frequency regions related to the charge transfer resistance between the electrode and the electrolyte interface. The inclined line in the low-frequency region corresponds to the lithium diffusion process [
53]. The equivalent circuit model in
Figure 12c consists of solution resistance (R
s), SEI layer resistance (R
SEI), charge transfer resistance (R
ct), constant phase elements (CPE
1, CPE
2), and Warburg impedance (W
b). From
Figure 12a, all electrodes exhibited R
ct of more than 500 Ω (shown in the inset
Figure 10a). After 25 charge/discharge cycles at 500 mA g
−1, the R
ct values are greatly decreased due to the wetting process of electrolyte into the electrodes and electrode kinetics with Li-ions. From
Figure 12b, Si-48 showed 106.5 Ω, which is quite high compared to the other three prepared electrodes, as SiCuG, SiFeG, and SiMnG showed much lower R
ct values of 40.3, 38.2, and 40.7 Ω, respectively, and could be attributed to faster Li
+ diffusion and enhanced electrical conductivity afforded by the metal dopants and graphite in the composite materials. The values of R
s, R
SEI, and R
ct values after 25 cycles are summarized in
Table 2. The evaluation of Li
+ diffusion properties, Z
Re vs. ω
−1/2, is plotted and depicted in
Figure 12d. From the plot, Z
Re linearly depends on ω
−1/2 in the low-frequency region. The following equations were used to calculate the diffusion coefficient (D) of Li
+ into the electrode materials.
Here, R is the gas constant, T is the absolute temperature, C is the molar concentration of Li
+ ions, A is the electrode area, and F is the Faraday constant. The slope of Z
Re vs. ω
−1/2 plots gives the Warburg impedance coefficients σ
w. The lower slope values yield higher diffusion coefficients and suggest better diffusion properties. The calculated Warburg factors for Si-48, SiCuG, SiFeG, and SiMnG are 65.1, 32.5, 19.1, and 37.3 Ω s
−1/2, respectively. The calculated D values from Equation (2) for each electrode material are summarized in
Table 2. All prepared materials show higher D values compared to Si-48, demonstrating the faster Li
+ kinetics owing to enhanced conductivity, more ionic channels due to metal dopants, structural stability, and flexibility afforded by the graphite.
To investigate the structure and morphology changes of the Si-48, SiCuG, SiFeG, and SiMnG materials, the cells were carefully disassembled after 20 charge/discharge cycles, and the anode electrodes were collected. To remove the SEI layer, the electrodes were washed with anhydrous dimethyl carbonate solution and dried in an oven. Later, the electrode materials were carefully collected for SEM analysis.
Figure S6 shows the SEM images of Si-48 and
Figures S7–S9 show the SEM images, SEM-EDX elemental mapping, and spectra of SiCuG, SiFeG, and SiMnG, respectively. From
Figure S6, the Si-48 particles are aggregated to a larger extent and no individual particles have appeared (as shown in
Figure S1b), which results in structural damage and leads to declined specific capacities with cycle number (showed in
Figure 10a). From the SEM images of SiCuG, SiFeG, and SiMnG, Si particles and graphite are agglomerated to a small extent. The SEM-EDX mapping and EDX spectra results also specify the presence of Si, metal (Cu/Fe/Mn) dopants, and C in their respective materials. These results suggest the favorable structural and mechanical stability of metal-doped Si/graphite composite materials even after 20 charge/discharge cycles.
The practical applicability of the prepared anodes SiCuG, SiFeG, and SiMnG are evaluated by fabricating full cells. The full cells were assembled with a commercial LiCoO
2 (LCO) cathode. The fundamental electrochemical properties such as voltage profiles, cyclability, and rate capability results of the LCO cathode are shown in
Figure S10a,
Figure S10b, and
Figure S10c, respectively. The cyclability tests were carried out at 0.5 C (1 C = 140 mA g
−1, based on the cathode active material), and the rate capability test was carried out from 0.1 C to 5 C. After 100 cycles, the LCO cathode delivered 110.6/109.1 mAh g
−1 capacity with 98.6% CE, and at the high rate of 5 C, the LCO cathode delivered 51.8/48.9 mAh g
−1 capacity, indicating suitable cyclic stability and rate capability, suggesting LCO as a strong cathode material for LIBs. For full cell fabrication, the N/P ratio was fixed at 1.102, 1.142, and 1.083 for SiCuG, SiFeG, and SiMnG materials, respectively, to match the capacities of cathode and anode active materials. Before fabricating full cells, the prepared anodes (SiCuG, SiFeG, and SiMnG) were prelithiated up to two complete cycles, followed by complete discharge to 0.01 V at a current density of 200 mA g
−1. After prelithiation, the cells were disassembled carefully in a glove box, and prelithiated anodes were used in full cell fabrication with the LCO cathode. The cyclability tests for full cells were carried out at 0.5 C (1 C =140 mA g
−1, based on the cathode active material), and the results are shown in
Figure 13. The 1st and 2nd voltage vs. specific capacity plots, cyclability, and CE results of SiCuG, SiFeG, and SiMnG are depicted in
Figure 13a,
Figure 13b, and
Figure 13c, respectively. After 100 cycles, the full cells delivered 79.5, 73.5, and 85.5 mAh g
−1 capacities with more than 99.5% CE with 79.5%, 72%, and 75% of capacity retention (vs. 2nd cycle charge capacity), respectively. Even though the electrode SiFeG shows a better diffusion coefficient (
Table 2), it shows little lesser capacities compared to SiCuG and SiMnG. This could be due to the N/P ratio being a little higher than the other two electrodes, which significantly affects the full cell performance due to the availability of lower Li
+ ions compared to the other two electrodes. Detailed observations revealed that three prepared composites show little differences in half cell and full cell performances. These small differences could be attributed to the individual metal dopants’ physical and chemical properties such as electrical conductivity, resistivity, how effectively they doped, the internal bonding of the metal matrix with host Si and/or with surface oxygen, etc. These factors could cause differences in the overall performance of the composite material [
54]. The calculated energy densities for SiCuG // LCO, SiFeG // LCO, and SiMnG // LCO are 302, 297.5, and 314 Wh.·kg
−1, respectively, with an average voltage of 3.5 V. The energy densities are reasonable for materials prepared from bulk Si particles by a low-cost and affordable approach and are most suitable and acceptable for Li-ion energy storage applications.

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}