


Epigenomes 2026, 10(1), 9; https://doi.org/10.3390/epigenomes10010009 - 5 Feb 2026
Abstract
Background/Objectives: Atrial fibrillation (AF) is currently the most common arrhythmia worldwide, and it is linked to increased mortality and morbidity, hence the need for a better clinical stratification of AF patients. Histone complexes or nucleosomes, released into the blood circulation, are found
[...] Read more.
Background/Objectives: Atrial fibrillation (AF) is currently the most common arrhythmia worldwide, and it is linked to increased mortality and morbidity, hence the need for a better clinical stratification of AF patients. Histone complexes or nucleosomes, released into the blood circulation, are found elevated in acute conditions such as stroke, trauma, and sepsis. The aim of this pilot single-centre study was to assess whether circulating histone levels could be used for diagnostic purposes in patients with AF. Methods: A total of 40 patients, well characterised for their biochemical and clinical characteristics, were recruited from outpatient clinics. Patients were randomly recruited into two groups (n = 20 per group), i.e., persistent AF and hypertensive controls. A multi-channel flow imaging methodology based on ImageStreamX was used with a well-optimised protocol to image and quantify five individual histones (H2A, H2B, H3, H4, and macroH2A1.1) together with the dimers (H2A/H2B, and H3/H4). Results: In the AF groups, plasma levels of histone dimers H2A/H2B and H3/H4 were elevated compared to hypertensive controls, 1.8% vs. 1.06% (p-value = 0.03). H2A/H2B dimer levels were increased in AF patients irrespective of gender, smoking status, diabetes, and pharmacological therapy. In the overall population, an inverse correlation between H2A and BMI was detected. Conclusions: Our pilot study, although limited in sample size, suggests that circulating histone complexes may be epigenetic sentinels for AF, offering mechanistic insights while addressing unmet needs in risk stratification.
Full article
(This article belongs to the Special Issue Epigenetic Signatures in Metabolic Health and Cancer)
►
Show Figures

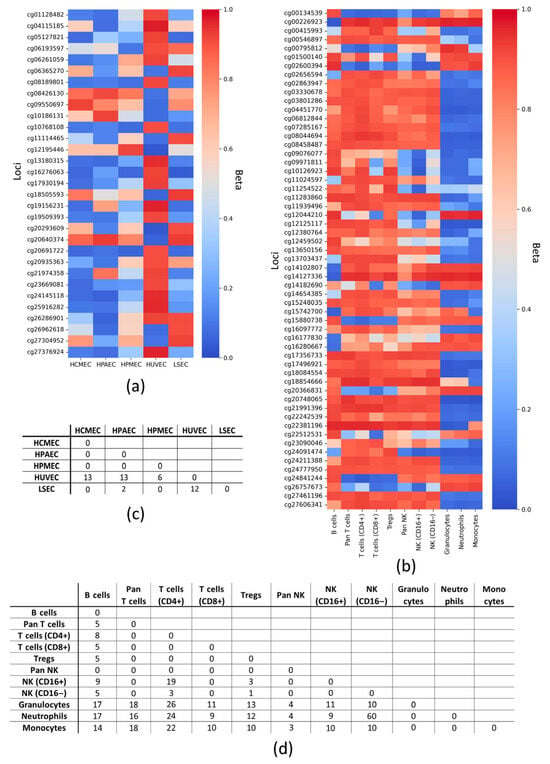
Figure 1




















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}