
Hippocampal Endoplasmic Reticulum Stress Hastens Motor and Cognitive Decline in Adult Male Rats Sustainedly Exposed to High-Sucrose Diet

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals and Experimental Design
2.2. Assessment of Liver Fat Content
2.3. Assessment of Glucose-Insulin Axis Function
2.4. Assessment of Serum Biochemical and Redox Profiles
2.5. Assessment of Behavioral, Cognitive, and Motor Functions
2.6. Gene Expression by Real-Time PCR (qPCR)
2.7. Protein Levels by Immunoblotting
2.8. Statistical Analysis
3. Results
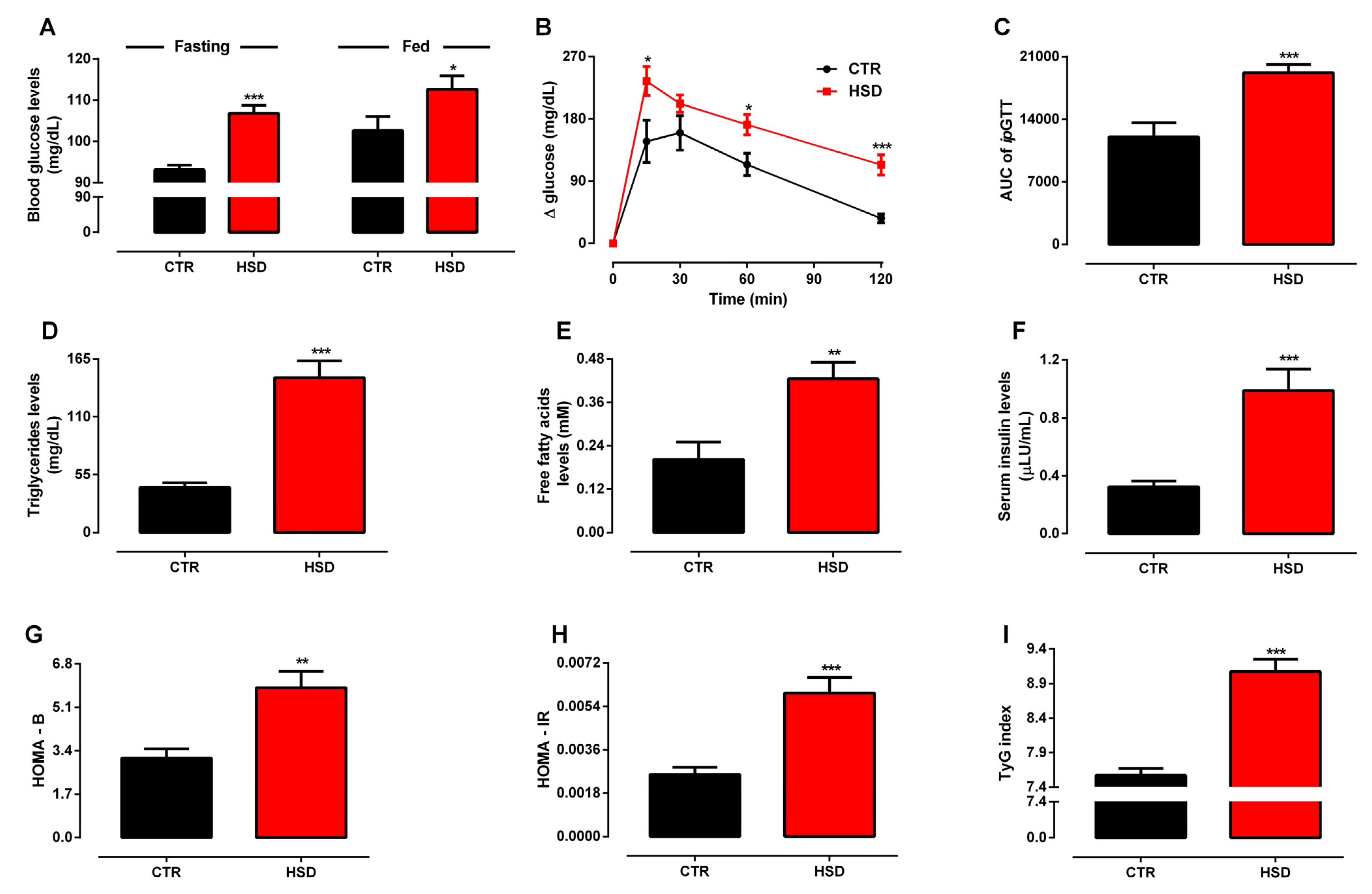
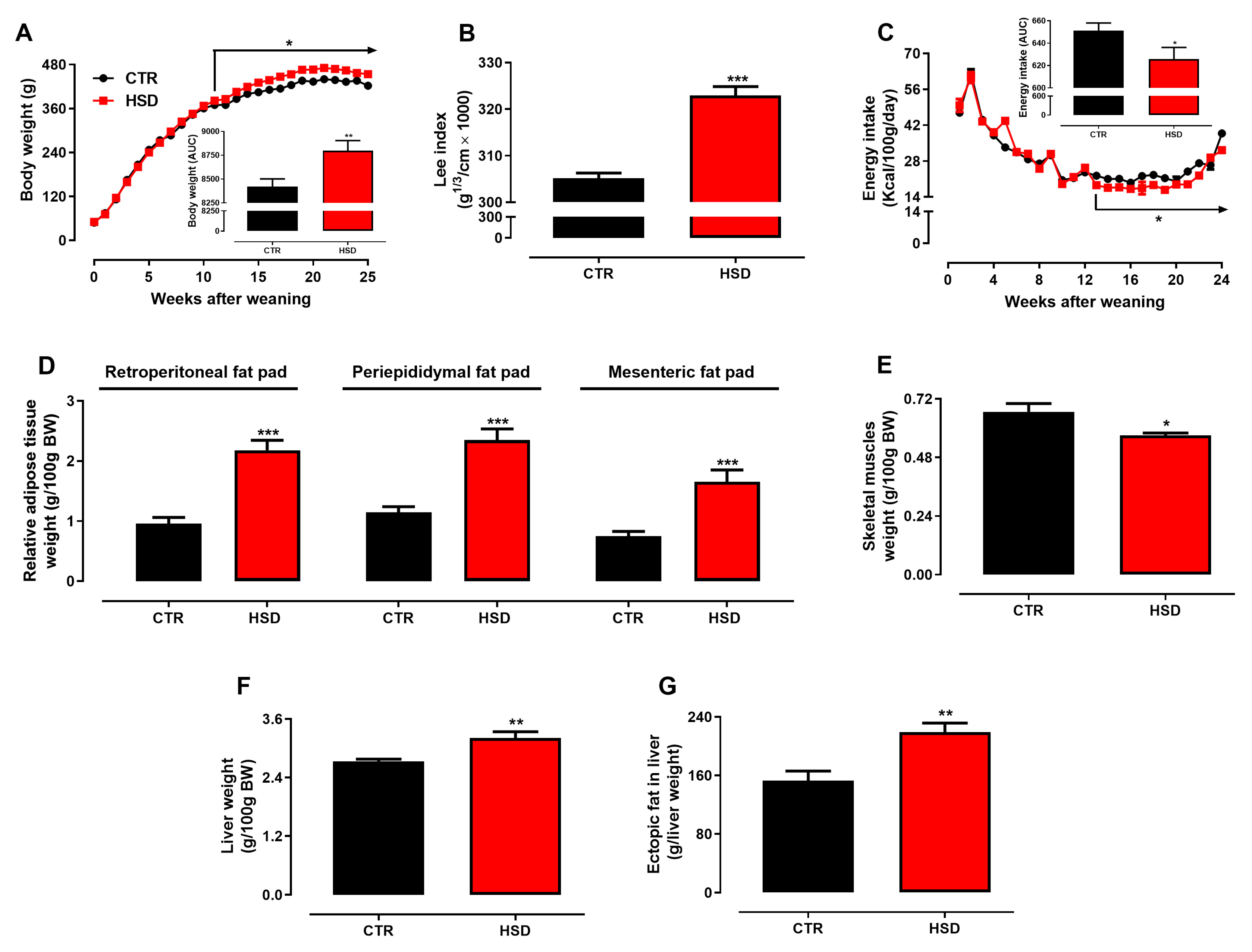
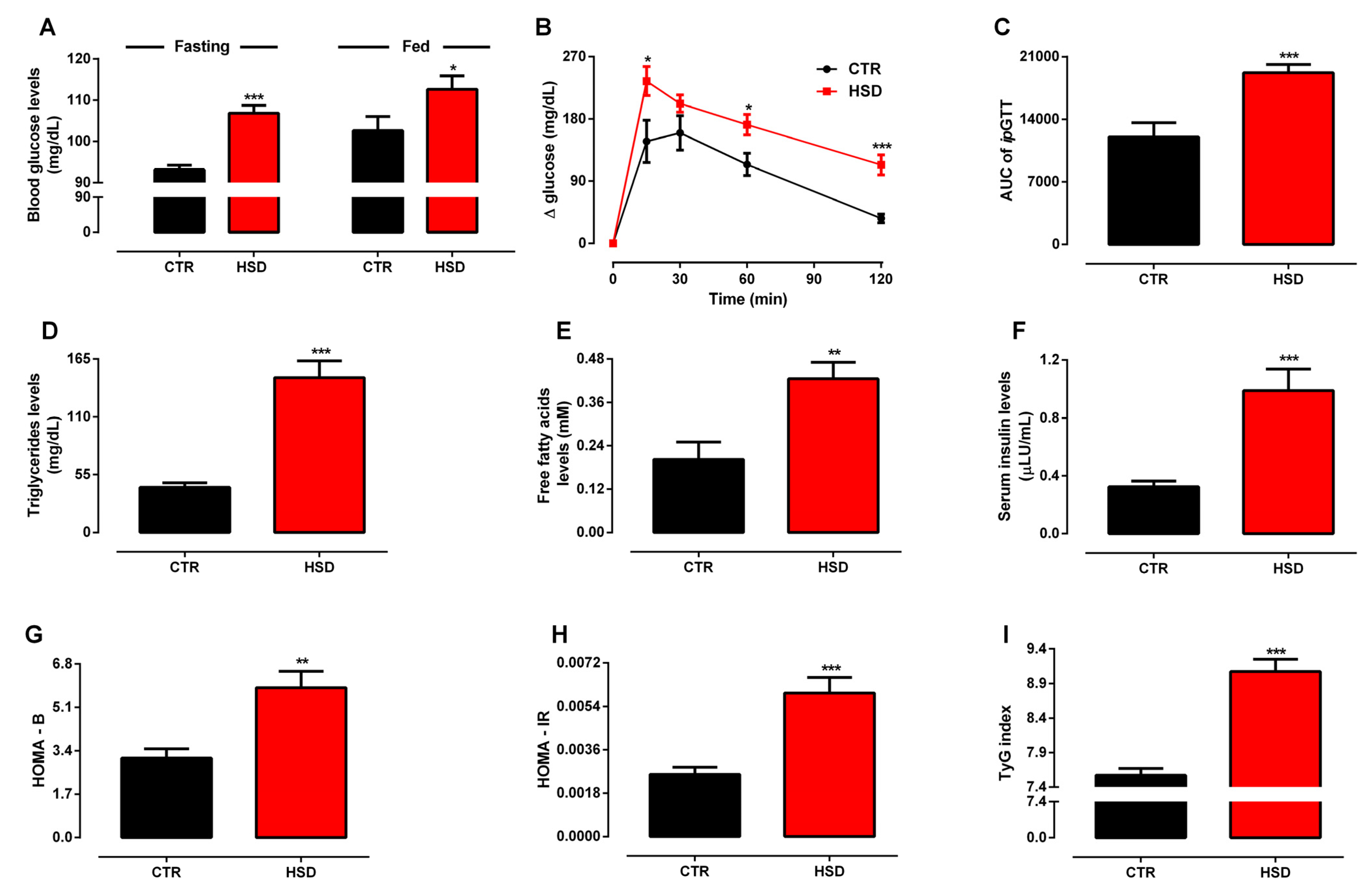
3.1. Long-Term Exposure to a High-Sucrose Diet Induces the Metabolic Syndrome Phenotype in Adult Rats
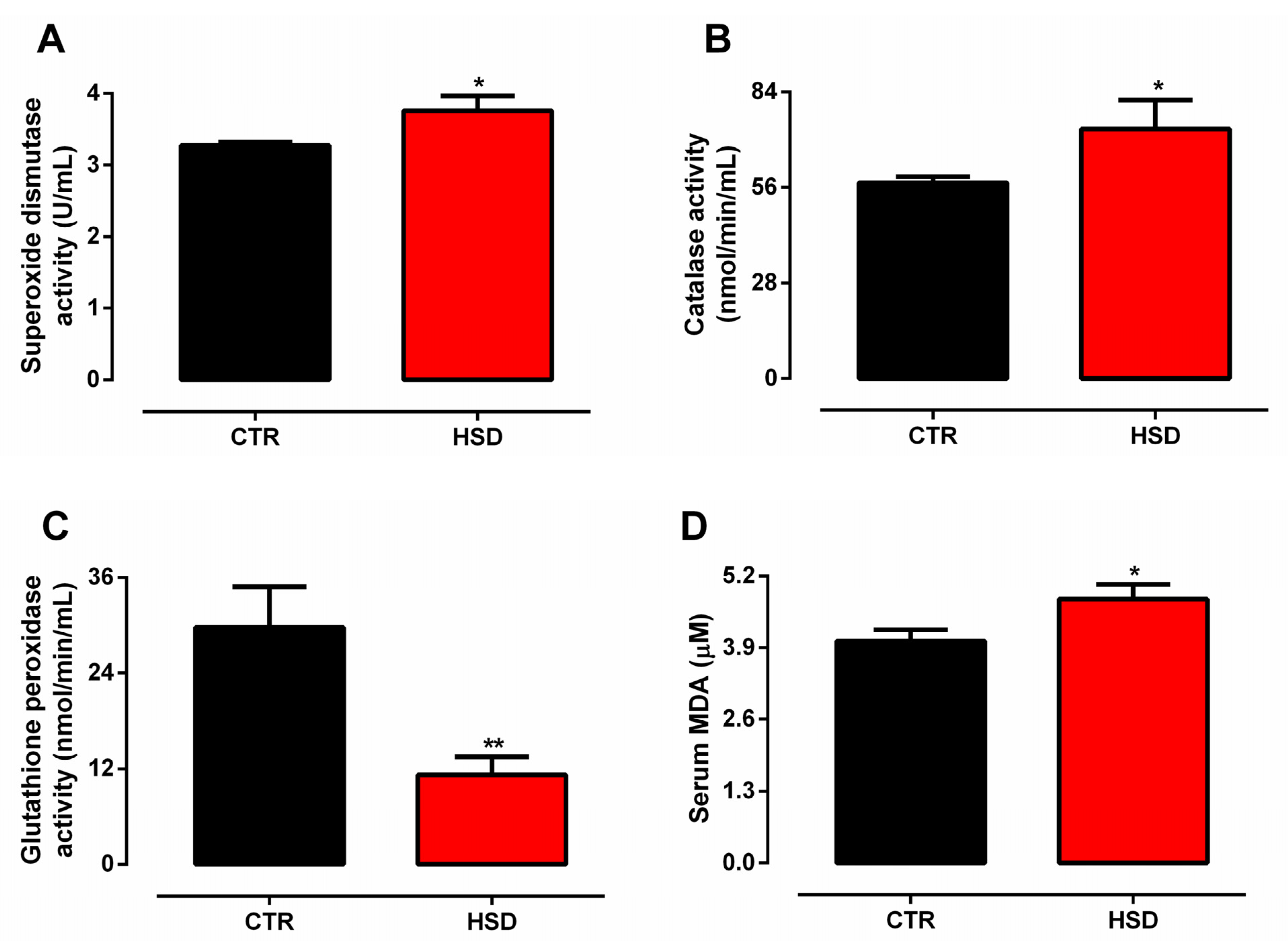
3.2. Metabolic Syndrome Induced by High-Sucrose Consumption Promotes Serum Redox Unbalance and Oxidative Damage
3.3. Metabolic Syndrome Induced by High-Sucrose Consumption Impairs Motor, Behavioral, and Cognitive Functions
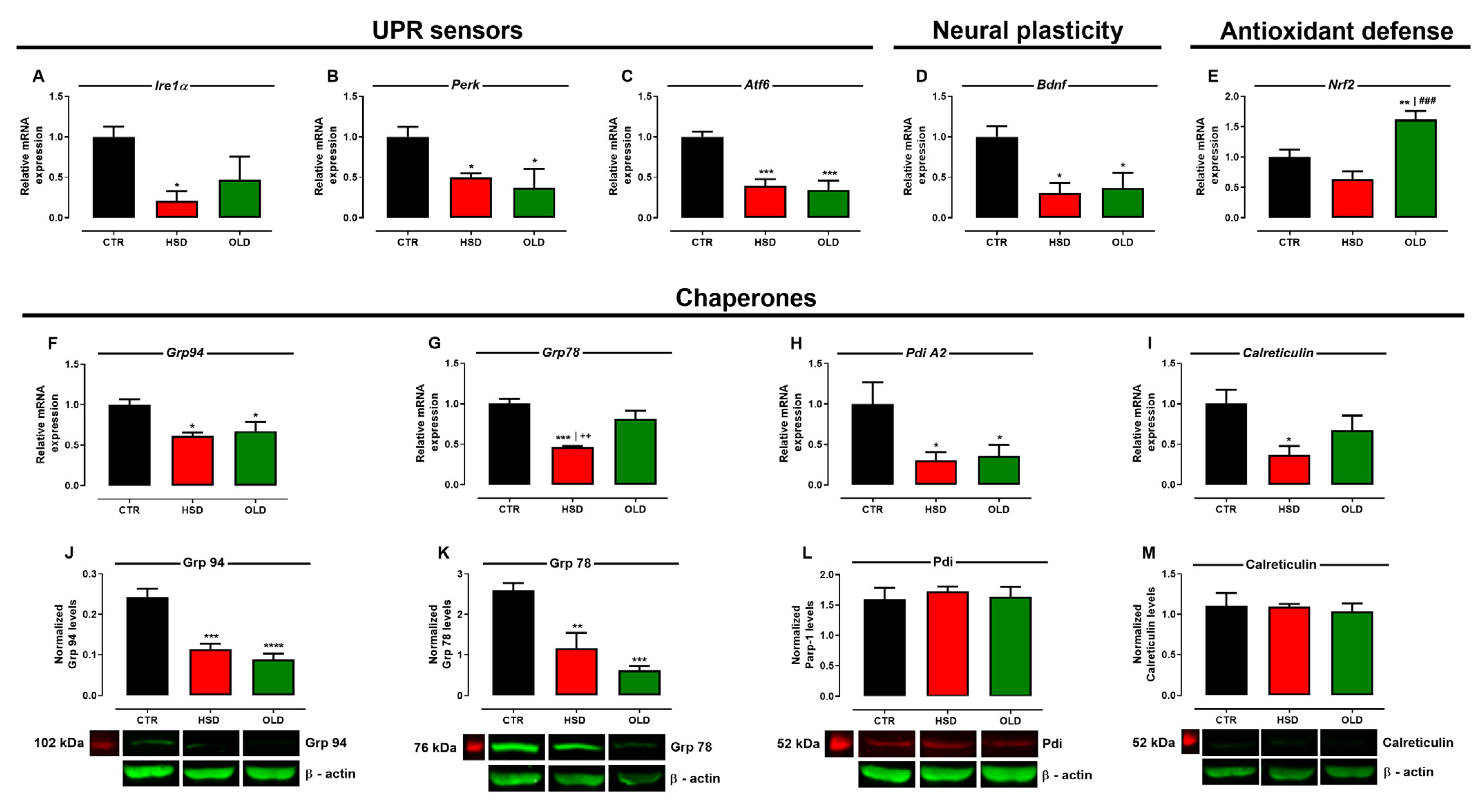
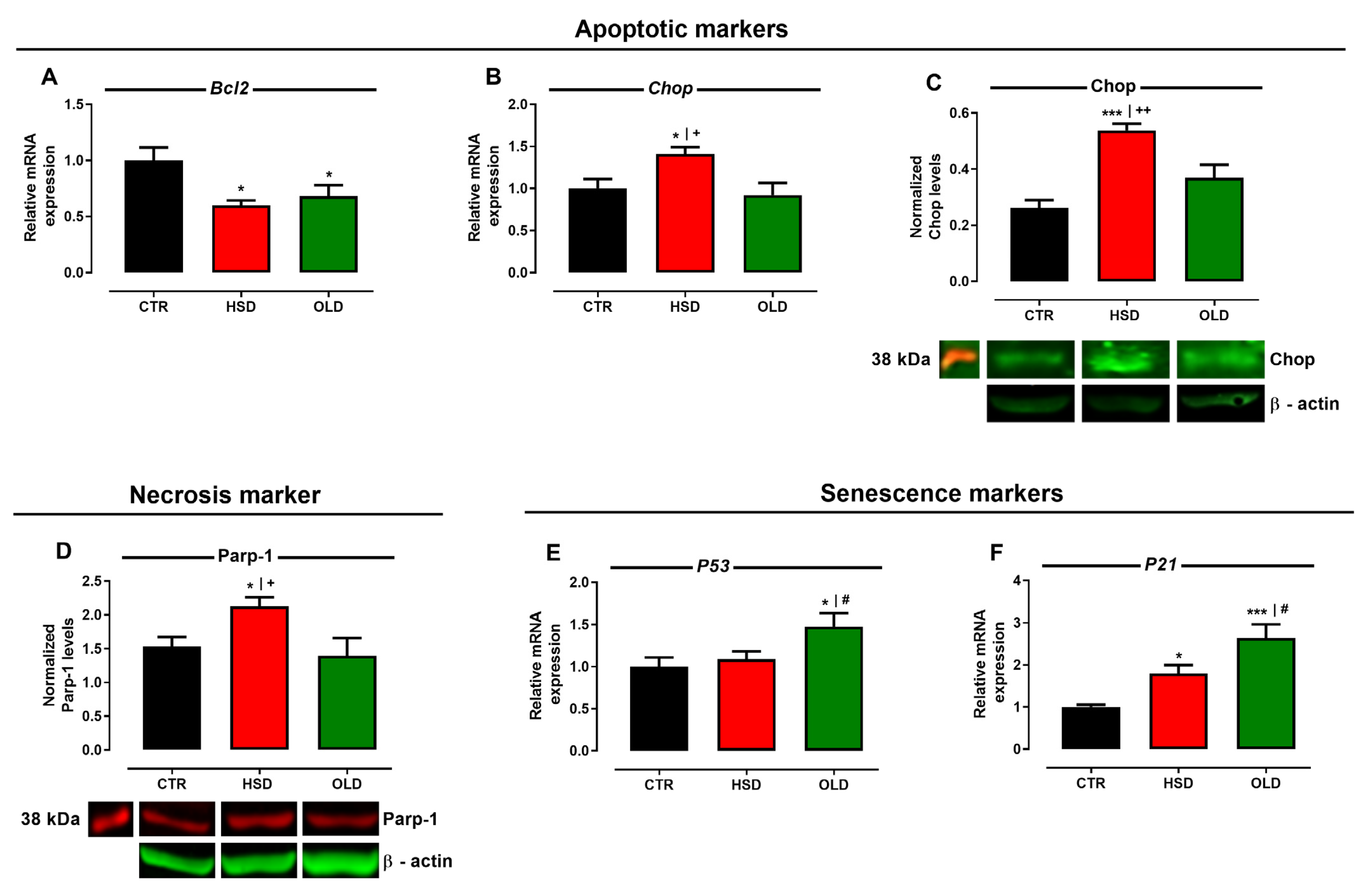
3.4. Failure of Pro-Adaptive and Activation of Pro-Apoptotic Pathways Anticipate Hippocampal Senescence in Cognitively Deficient HSD Rats
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Alberti, K.G.M.; Zimmet, P.; Shaw, J. The metabolic syndrome-a new worldwide definition. Lancet 2005, 366, 1059. [Google Scholar] [CrossRef]
- Saklayen, M.G. The Global Epidemic of the Metabolic Syndrome. Curr. Hypertens Rep. 2018, 20, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alberti, K.G.; Eckel, R.H.; Grundy, S.M.; Zimmet, P.Z.; Cleeman, J.I.; Donato, K.A.; Fruchart, J.C.; James, W.P.; Loria, C.M.; Smith, S.C., Jr.; et al. Harmonizing the metabolic syndrome: A joint interim statement of the International Diabetes Federation Task Force on Epidemiology and Prevention; National Heart, Lung, and Blood Institute; American Heart Association; World Heart Federation; International Atherosclerosis Society; and International Association for the Study of Obesity. Circulation 2009, 120, 1640–1645. [Google Scholar] [CrossRef] [Green Version]
- Haase Alasantro, L.; Hicks, T.H.; Green-Krogmann, E.; Murphy, C. Metabolic syndrome and cognitive performance across the adult lifespan. PLoS ONE 2021, 16, e0249348. [Google Scholar] [CrossRef] [PubMed]
- Pal, K.; Mukadam, N.; Petersen, I.; Cooper, C. Mild cognitive impairment and progression to dementia in people with diabetes, prediabetes and metabolic syndrome: A systematic review and meta-analysis. Soc. Psychiatry Psychiatr. Epidemiol. 2018, 53, 1149–1160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fakih, W.; Zeitoun, R.; AlZaim, I.; Eid, A.H.; Kobeissy, F.; Abd-Elrahman, K.S.; El-Yazbi, A.F. Early metabolic impairment as a contributor to neurodegenerative disease: Mechanisms and potential pharmacological intervention. Obesity 2022, 30, 982–993. [Google Scholar] [CrossRef]
- Moser, M.B.; Moser, E.I. Functional differentiation in the hippocampus. Hippocampus 1998, 8, 608–619. [Google Scholar] [CrossRef]
- Strange, B.A.; Witter, M.P.; Lein, E.S.; Moser, E.I. Functional organization of the hippocampal longitudinal axis. Nat. Rev. Neurosci. 2014, 15, 655–669. [Google Scholar] [CrossRef] [Green Version]
- Lakhan, S.E.; Kirchgessner, A. The emerging role of dietary fructose in obesity and cognitive decline. Nutr. J. 2013, 12, 114. [Google Scholar] [CrossRef] [Green Version]
- Guillemot-Legris, O.; Muccioli, G.G. Obesity-Induced Neuroinflammation: Beyond the Hypothalamus. Trends Neurosci. 2017, 40, 237–253. [Google Scholar] [CrossRef]
- Wiley, C.D.; Campisi, J. The metabolic roots of senescence: Mechanisms and opportunities for intervention. Nat. Metab. 2021, 3, 1290–1301. [Google Scholar] [CrossRef] [PubMed]
- Ahima, R.S. Connecting obesity, aging and diabetes. Nat. Med. 2009, 15, 996–997. [Google Scholar] [CrossRef] [PubMed]
- Hossain, M.M.; Toltin, A.C.; Gamba, L.M.; Molina, M.A. Deltamethrin-Evoked ER Stress Promotes Neuroinflammation in the Adult Mouse Hippocampus. Cells 2022, 11, 1961. [Google Scholar] [CrossRef] [PubMed]
- Shen, F.Y.; Song, Y.C.; Guo, F.; Xu, Z.D.; Li, Q.; Zhang, B.; Ma, Y.Q.; Zhang, Y.Q.; Lin, R.; Li, Y.; et al. Cognitive Impairment and Endoplasmic Reticulum Stress Induced by Repeated Short-Term Sevoflurane Exposure in Early Life of Rats. Front. Psychiatry 2018, 9, 332. [Google Scholar] [CrossRef] [Green Version]
- Hafycz, J.M.; Strus, E.; Naidoo, N. Reducing ER stress with chaperone therapy reverses sleep fragmentation and cognitive decline in aged mice. Aging Cell 2022, 21, e13598. [Google Scholar] [CrossRef]
- Hetz, C. The unfolded protein response: Controlling cell fate decisions under ER stress and beyond. Nat. Rev. Mol. Cell Biol. 2012, 13, 89–102. [Google Scholar] [CrossRef]
- Malhotra, J.D.; Kaufman, R.J. The endoplasmic reticulum and the unfolded protein response. Semin. Cell Dev. Biol. 2007, 18, 716–731. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Kaufman, R.J. The impact of the unfolded protein response on human disease. J. Cell Biol. 2012, 197, 857–867. [Google Scholar] [CrossRef] [Green Version]
- Lemmer, I.L.; Willemsen, N.; Hilal, N.; Bartelt, A. A guide to understanding endoplasmic reticulum stress in metabolic disorders. Mol. Metab. 2021, 47, 101169. [Google Scholar] [CrossRef]
- Brown, M.K.; Naidoo, N. The endoplasmic reticulum stress response in aging and age-related diseases. Front. Physiol. 2012, 3, 263. [Google Scholar] [CrossRef] [Green Version]
- Taylor, R.C. Aging and the UPR(ER). Brain Res. 2016, 1648, 588–593. [Google Scholar] [CrossRef]
- Hetz; Mollereau, B. Disturbance of endoplasmic reticulum proteostasis in neurodegenerative diseases. Nat. Rev. Neurosci. 2014, 15, 233–249. [Google Scholar] [CrossRef]
- Pinto, B.A.S.; Melo, T.M.; Flister, K.F.T.; França, L.M.; Kajihara, D.; Tanaka, L.Y.; Laurindo, F.R.M.; Paes, A.M.A. Early and sustained exposure to high-sucrose diet triggers hippocampal ER stress in young rats. Metab. Brain Dis. 2016, 31, 917–927. [Google Scholar] [CrossRef] [PubMed]
- Bernardis, L.; Patterson, B. Correlation between ’Lee index’ and carcass fat content in weanling and adult female rats with hypothalamic lesions. J. Endocrinol. 1968, 40, 527–528. [Google Scholar] [CrossRef] [PubMed]
- França, L.M.; Freitas, L.N.C.; Chagas, V.T.; Coêlho, C.F.F.; Barroso, W.A.; Costa, G.C.; Silva, L.A.; Debbas, V.; Laurindo, F.R.M.; Paes, A.M.A. Mechanisms underlying hypertriglyceridemia in rats with monosodium L-glutamate-induced obesity: Evidence of XBP-1/PDI/MTP axis activation. Biochem. Biophys. Res. Commun. 2014, 443, 725–730. [Google Scholar] [CrossRef] [PubMed]
- Rafacho, A.; Roma, L.; Taboga, S.; Boschero, A.; Bosqueiro, J. Dexamethasone-induced insulin resistance is associated with increased connexin 36 mRNA and protein expression in pancreatic rat islets. Can. J. Physiol. Pharmacol. 2007, 85, 536–545. [Google Scholar] [CrossRef] [PubMed]
- Matthews, D.; Hosker, J.; Rudenski, A.; Naylor, B.; Treacher, D.; Turner, R. Homeostasis model assessment: Insulin resistance and β-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia 1985, 28, 412–419. [Google Scholar] [CrossRef] [Green Version]
- Guerrero-Romero, F.; Simental-Mendía, L.E.; González-Ortiz, M.; Martínez-Abundis, E.; Ramos-Zavala, M.G.; Hernández-González, S.O.; Jacques-Camarena, O.; Rodríguez-Morán, M. The product of triglycerides and glucose, a simple measure of insulin sensitivity. Comparison with the euglycemic-hyperinsulinemic clamp. J. Clin. Endocrinol. Metab. 2010, 95, 3347–3351. [Google Scholar] [CrossRef] [Green Version]
- Marcelino, T.; Longoni, A.; Kudo, K.; Stone, V.; Rech, A.; De Assis, A.; Scherer, E.; Da Cunha, M.; Wyse, A.; Pettenuzzo, L. Evidences that maternal swimming exercise improves antioxidant defenses and induces mitochondrial biogenesis in the brain of young Wistar rats. Neuroscience 2013, 246, 28–39. [Google Scholar] [CrossRef]
- Srodulski, S.; Sharma, S.; Bachstetter, A.B.; Brelsfoard, J.M.; Pascual, C.; Xie, X.S.; Saatman, K.E.; Van Eldik, L.J.; Despa, F. Neuroinflammation and neurologic deficits in diabetes linked to brain accumulation of amylin. Mol. Neurodegener. 2014, 9, 30. [Google Scholar] [CrossRef] [Green Version]
- Hall, C.; Ballachey, E.L. A study of the rat’s behavior in a field. A contribution to method in comparative psychology. Univ. Calif. Publ. Psychol. 1932, 6, 1–12. [Google Scholar]
- Morris, R. Developments of a water-maze procedure for studying spatial learning in the rat. J. Neurosci. Methods 1984, 11, 47–60. [Google Scholar] [CrossRef]
- Faul, F.; Erdfelder, E.; Lang, A.G.; Buchner, A. G*Power 3: A flexible statistical power analysis program for the social, behavioral, and biomedical sciences. Behav. Res. Methods 2007, 39, 175–191. [Google Scholar] [CrossRef] [PubMed]
- Cenquizca, L.A.; Swanson, L.W. Spatial organization of direct hippocampal field CA1 axonal projections to the rest of the cerebral cortex. Brain Res. Rev. 2007, 56, 1–26. [Google Scholar] [CrossRef] [Green Version]
- McGinnis, K.M.; Gnegy, M.E.; Park, Y.H.; Mukerjee, N.; Wang, K.K. Procaspase-3 and poly (ADP) ribose polymerase (PARP) are calpain substrates. Biochem. Biophys. Res. Commun. 1999, 263, 94–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bray, G.A.; Nielsen, S.J.; Popkin, B.M. Consumption of high-fructose corn syrup in beverages may play a role in the epidemic of obesity. Am. J. Clin. Nutr. 2004, 79, 537–543. [Google Scholar] [CrossRef]
- Bray, G.A. Fructose: Pure, white, and deadly? Fructose, by any other name, is a health hazard. J. Diabetes Sci. Technol. 2010, 4, 1003–1007. [Google Scholar] [CrossRef] [Green Version]
- Kanarek, R.B.; Orthen-Gambill, N. Differential effects of sucrose, fructose and glucose on carbohydrate-induced obesity in rats. J. Nutr. 1982, 112, 1546–1554. [Google Scholar] [CrossRef] [Green Version]
- Kanazawa, M.; Xue, C.Y.; Kageyama, H.; Suzuki, E.; Ito, R.; Namba, Y.; Osaka, T.; Kimura, S.; Inoue, S. Effects of a high-sucrose diet on body weight, plasma triglycerides, and stress tolerance. Nutr. Rev. 2003, 61, S27–S33. [Google Scholar] [CrossRef]
- Basciano, H.; Federico, L.; Adeli, K. Fructose, insulin resistance, and metabolic dyslipidemia. Nutr. Metab. 2005, 2, 5. [Google Scholar] [CrossRef] [Green Version]
- Botezelli, J.D.; Cambri, L.T.; Ghezzi, A.C.; Dalia, R.A.; Voltarelli, F.A.; de Mello, M.A.R. Fructose-rich diet leads to reduced aerobic capacity and to liver injury in rats. Lipids Health Dis. 2012, 11, 78. [Google Scholar] [CrossRef] [Green Version]
- Newens, K.J.; Walton, J. A review of sugar consumption from nationally representative dietary surveys across the world. J. Hum. Nutr. Diet. 2016, 29, 225–240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sousa, R.M.L.; Ribeiro, N.L.X.; Pinto, B.A.S.; Sanches, J.R.; da Silva, M.U.; Coelho, C.F.F.; Franca, L.M.; de Figueiredo Neto, J.A.; Paes, A.M.A. Long-term high-protein diet intake reverts weight gain and attenuates metabolic dysfunction on high-sucrose-fed adult rats. Nutr. Metab. 2018, 15, 53. [Google Scholar] [CrossRef] [Green Version]
- Tappy, L.; Lê, K.-A. Metabolic effects of fructose and the worldwide increase in obesity. Physiol. Rev. 2010, 90, 23–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parks, E.J.; Skokan, L.E.; Timlin, M.T.; Dingfelder, C.S. Dietary sugars stimulate fatty acid synthesis in adults. J. Nutr. 2008, 138, 1039–1046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolderup, A.; Svihus, B. Fructose metabolism and relation to atherosclerosis, type 2 diabetes, and obesity. J. Nutr. Metab. 2015, 2015, 823081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solon-Biet, S.M.; McMahon, A.C.; Ballard, J.W.; Ruohonen, K.; Wu, L.E.; Cogger, V.C.; Warren, A.; Huang, X.; Pichaud, N.; Melvin, R.G.; et al. The ratio of macronutrients, not caloric intake, dictates cardiometabolic health, aging, and longevity in ad libitum-fed mice. Cell Metab. 2014, 19, 418–430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, X.; Hancock, D.P.; Gosby, A.K.; McMahon, A.C.; Solon, S.M.; Le Couteur, D.G.; Conigrave, A.D.; Raubenheimer, D.; Simpson, S.J. Effects of dietary protein to carbohydrate balance on energy intake, fat storage, and heat production in mice. Obesity 2013, 21, 85–92. [Google Scholar] [CrossRef]
- Sorensen, A.; Mayntz, D.; Raubenheimer, D.; Simpson, S.J. Protein-leverage in mice: The geometry of macronutrient balancing and consequences for fat deposition. Obesity 2008, 16, 566–571. [Google Scholar] [CrossRef]
- Lomeli, N.; Bota, D.A.; Davies, K.J. Diminished stress resistance and defective adaptive homeostasis in age-related diseases. Clin. Sci. 2017, 131, 2573–2599. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.S.; Kim, A.Y.; Choi, J.W.; Kim, M.; Yasue, S.; Son, H.J.; Masuzaki, H.; Park, K.S.; Kim, J.B. Dysregulation of adipose glutathione peroxidase 3 in obesity contributes to local and systemic oxidative stress. Mol. Endocrinol. 2008, 22, 2176–2189. [Google Scholar] [CrossRef] [Green Version]
- Sastre, J.; Pallardó, F.V.; Llopis, J.; Furukawa, T.; Vinã, J.R.; Viña, J. Glutathione depletion by hyperphagia-induced obesity. Life Sci. 1989, 45, 183–187. [Google Scholar] [CrossRef]
- Huang, T.-T.; Leu, D.; Zou, Y. Oxidative stress and redox regulation on hippocampal-dependent cognitive functions. Arch. Biochem. Biophys. 2015, 576, 2–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finkel, T.; Holbrook, N.J. Oxidants, oxidative stress and the biology of ageing. Nature 2000, 408, 239–247. [Google Scholar] [CrossRef]
- Brooks, S.P.; Dunnett, S.B. Tests to assess motor phenotype in mice: A user’s guide. Nat. Rev. Neurosci. 2009, 10, 519–529. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Fischer, K.E.; Soto, V.; Liu, Y.; Sosnowska, D.; Richardson, A.; Salmon, A.B. Obesity-induced oxidative stress, accelerated functional decline with age and increased mortality in mice. Arch. Biochem. Biophys. 2015, 576, 39–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jurdak, N.; Lichtenstein, A.H.; Kanarek, R.B. Diet-induced obesity and spatial cognition in young male rats. Nutr. Neurosci. 2008, 11, 48–54. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, R.; Gomez-Pinilla, F. ‘Metabolic syndrome’ in the brain: Deficiency in omega-3 fatty acid exacerbates dysfunctions in insulin receptor signalling and cognition. J. Physiol. 2012, 590, 2485–2499. [Google Scholar] [CrossRef]
- Hsu, T.M.; Konanur, V.R.; Taing, L.; Usui, R.; Kayser, B.D.; Goran, M.I.; Kanoski, S.E. Effects of sucrose and high fructose corn syrup consumption on spatial memory function and hippocampal neuroinflammation in adolescent rats. Hippocampus 2015, 25, 227–239. [Google Scholar] [CrossRef]
- Wong, A.; Dogra, V.R.; Reichelt, A.C. High-sucrose diets in male rats disrupt aspects of decision making tasks, motivation and spatial memory, but not impulsivity measured by operant delay-discounting. Behav. Brain Res. 2017, 327, 144–154. [Google Scholar] [CrossRef]
- Beilharz, J.; Maniam, J.; Morris, M. Short-term exposure to a diet high in fat and sugar, or liquid sugar, selectively impairs hippocampal-dependent memory, with differential impacts on inflammation. Behav. Brain Res. 2016, 306, 1–7. [Google Scholar] [CrossRef]
- Lemos, C.; Rial, D.; Gonçalves, F.; Pires, J.; Silva, H.; Matheus, F.; Da Silva, A.; Marques, J.; Rodrigues, R.; Jarak, I. High sucrose consumption induces memory impairment in rats associated with electrophysiological modifications but not with metabolic changes in the hippocampus. Neuroscience 2016, 315, 196–205. [Google Scholar] [CrossRef] [PubMed]
- Beilharz, J.E.; Kaakoush, N.O.; Maniam, J.; Morris, M.J. The effect of short-term exposure to energy-matched diets enriched in fat or sugar on memory, gut microbiota and markers of brain inflammation and plasticity. Brain Behav. Immun. 2016, 57, 304–313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stranahan, A.M.; Norman, E.D.; Lee, K.; Cutler, R.G.; Telljohann, R.S.; Egan, J.M.; Mattson, M.P. Diet-induced insulin resistance impairs hippocampal synaptic plasticity and cognition in middle-aged rats. Hippocampus 2008, 18, 1085–1088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitchell, C.S.; Goodman, E.K.; Tedesco, C.R.; Nguyen, K.; Zhang, L.; Herzog, H.; Begg, D.P. The Effect of Dietary Fat and Sucrose on Cognitive Functioning in Mice Lacking Insulin Signaling in Neuropeptide Y Neurons. Front. Physiol. 2022, 13, 841935. [Google Scholar] [CrossRef]
- Maniam, J.; Antoniadis, C.P.; Youngson, N.A.; Sinha, J.K.; Morris, M.J. Sugar consumption produces effects similar to early life stress exposure on hippocampal markers of neurogenesis and stress response. Front. Mol. Neurosci. 2016, 8, 86. [Google Scholar] [CrossRef] [Green Version]
- Hotamisligil, G.S. Inflammation and metabolic disorders. Nature 2006, 444, 860–867. [Google Scholar] [CrossRef]
- Sims-Robinson, C.; Zhao, S.; Hur, J.; Feldman, E. Central nervous system endoplasmic reticulum stress in a murine model of type 2 diabetes. Diabetologia 2012, 55, 2276–2284. [Google Scholar] [CrossRef] [Green Version]
- Gavilan, M.P.; Vela, J.; Castaño, A.; Ramos, B.; Juan, C.; Vitorica, J.; Ruano, D. Cellular environment facilitates protein accumulation in aged rat hippocampus. Neurobiol. Aging 2006, 27, 973–982. [Google Scholar] [CrossRef]
- Hussain, S.G.; Ramaiah, K.V. Reduced eIF2α phosphorylation and increased proapoptotic proteins in aging. Biochem. Biophys. Res. Commun. 2007, 355, 365–370. [Google Scholar] [CrossRef]
- Naidoo, N.; Ferber, M.; Master, M.; Zhu, Y.; Pack, A.I. Aging impairs the unfolded protein response to sleep deprivation and leads to proapoptotic signaling. J. Neurosci. 2008, 28, 6539–6548. [Google Scholar] [CrossRef]
- Chaitanya, G.V.; Alexander, J.S.; Babu, P.P. PARP-1 cleavage fragments: Signatures of cell-death proteases in neurodegeneration. Cell Commun. Signal. 2010, 8, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fonseca, A.C.R.; Ferreiro, E.; Oliveira, C.R.; Cardoso, S.M.; Pereira, C.F. Activation of the endoplasmic reticulum stress response by the amyloid-beta 1–40 peptide in brain endothelial cells. Biochim. Et Biophys. Acta (BBA)-Mol. Basis Dis. 2013, 1832, 2191–2203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayashi, A.; Kasahara, T.; Iwamoto, K.; Ishiwata, M.; Kametani, M.; Kakiuchi, C.; Furuichi, T.; Kato, T. The role of brain-derived neurotrophic factor (BDNF)-induced XBP1 splicing during brain development. J. Biol. Chem. 2007, 282, 34525–34534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayashi, A.; Kasahara, T.; Kametani, M.; Kato, T. Attenuated BDNF-induced upregulation of GABAergic markers in neurons lacking Xbp1. Biochem. Biophys. Res. Commun. 2008, 376, 758–763. [Google Scholar] [CrossRef] [PubMed]
- Martínez, G.; Vidal, R.L.; Mardones, P.; Serrano, F.G.; Ardiles, A.O.; Wirth, C.; Valdes, P.; Thielen, P.; Schneider, B.L.; Kerr, B. Regulation of memory formation by the transcription factor XBP1. Cell Rep. 2016, 14, 1382–1394. [Google Scholar] [CrossRef] [Green Version]
- Minamino, T.; Orimo, M.; Shimizu, I.; Kunieda, T.; Yokoyama, M.; Ito, T.; Nojima, A.; Nabetani, A.; Oike, Y.; Matsubara, H. A crucial role for adipose tissue p53 in the regulation of insulin resistance. Nat. Med. 2009, 15, 1082–1087. [Google Scholar] [CrossRef]
- Bruns, D.R.; Drake, J.C.; Biela, L.M.; Peelor, F.F.; Miller, B.F.; Hamilton, K.L. Nrf2 signaling and the slowed aging phenotype: Evidence from long-lived models. Oxidative Med. Cell. Longev. 2015, 2015, 732596. [Google Scholar] [CrossRef] [Green Version]
- Gounder, S.S.; Kannan, S.; Devadoss, D.; Miller, C.J.; Whitehead, K.S.; Odelberg, S.J.; Firpo, M.A.; Paine III, R.; Hoidal, J.R.; Abel, E.D. Impaired transcriptional activity of Nrf2 in age-related myocardial oxidative stress is reversible by moderate exercise training. PLoS ONE 2012, 7, e45697. [Google Scholar] [CrossRef]
- Suh, J.H.; Shenvi, S.V.; Dixon, B.M.; Liu, H.; Jaiswal, A.K.; Liu, R.-M.; Hagen, T.M. Decline in transcriptional activity of Nrf2 causes age-related loss of glutathione synthesis, which is reversible with lipoic acid. Proc. Natl. Acad. Sci. USA 2004, 101, 3381–3386. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.; Sun, Z.; Wang, X.-J.; Jiang, T.; Huang, Z.; Fang, D.; Zhang, D.D. Direct interaction between Nrf2 and p21 Cip1/WAF1 upregulates the Nrf2-mediated antioxidant response. Mol. Cell 2009, 34, 663–673. [Google Scholar] [CrossRef] [Green Version]
- Cullinan, S.B.; Diehl, J.A. PERK-dependent activation of Nrf2 contributes to redox homeostasis and cell survival following endoplasmic reticulum stress. J. Biol. Chem. 2004, 279, 20108–20117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Birkenfeld, A.L.; Lee, H.-Y.; Majumdar, S.; Jurczak, M.J.; Camporez, J.P.; Jornayvaz, F.R.; Frederick, D.W.; Guigni, B.; Kahn, M.; Zhang, D. Influence of the hepatic eukaryotic initiation factor 2α (eIF2α) endoplasmic reticulum (ER) stress response pathway on insulin-mediated ER stress and hepatic and peripheral glucose metabolism. J. Biol. Chem. 2011, 286, 36163–36170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghosh, A.; Abdo, S.; Zhao, S.; Wu, C.-H.; Shi, Y.; Lo, C.-S.; Chenier, I.; Alquier, T.; Filep, J.G.; Ingelfinger, J.R. Insulin Inhibits Nrf2 Gene Expression via Heterogeneous Nuclear Ribonucleoprotein F/K in Diabetic Mice. Endocrinology 2017, 158, 903–919. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pinto, B.A.S.; Melo, T.M.; Flister, K.F.T.; França, L.M.; Moreira, V.R.; Kajihara, D.; Mendes, N.O.; Pereira, S.R.; Laurindo, F.R.M.; Paes, A.M.A. Hippocampal Endoplasmic Reticulum Stress Hastens Motor and Cognitive Decline in Adult Male Rats Sustainedly Exposed to High-Sucrose Diet. Antioxidants 2022, 11, 1395. https://doi.org/10.3390/antiox11071395
Pinto BAS, Melo TM, Flister KFT, França LM, Moreira VR, Kajihara D, Mendes NO, Pereira SR, Laurindo FRM, Paes AMA. Hippocampal Endoplasmic Reticulum Stress Hastens Motor and Cognitive Decline in Adult Male Rats Sustainedly Exposed to High-Sucrose Diet. Antioxidants. 2022; 11(7):1395. https://doi.org/10.3390/antiox11071395
Chicago/Turabian StylePinto, Bruno Araújo Serra, Thamys Marinho Melo, Karla Frida Torres Flister, Lucas Martins França, Vanessa Ribeiro Moreira, Daniela Kajihara, Nelmar Oliveira Mendes, Silma Regina Pereira, Francisco Rafael Martins Laurindo, and Antonio Marcus Andrade Paes. 2022. "Hippocampal Endoplasmic Reticulum Stress Hastens Motor and Cognitive Decline in Adult Male Rats Sustainedly Exposed to High-Sucrose Diet" Antioxidants 11, no. 7: 1395. https://doi.org/10.3390/antiox11071395