
WWOX Controls Cell Survival, Immune Response and Disease Progression by pY33 to pS14 Transition to Alternate Signaling Partners


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
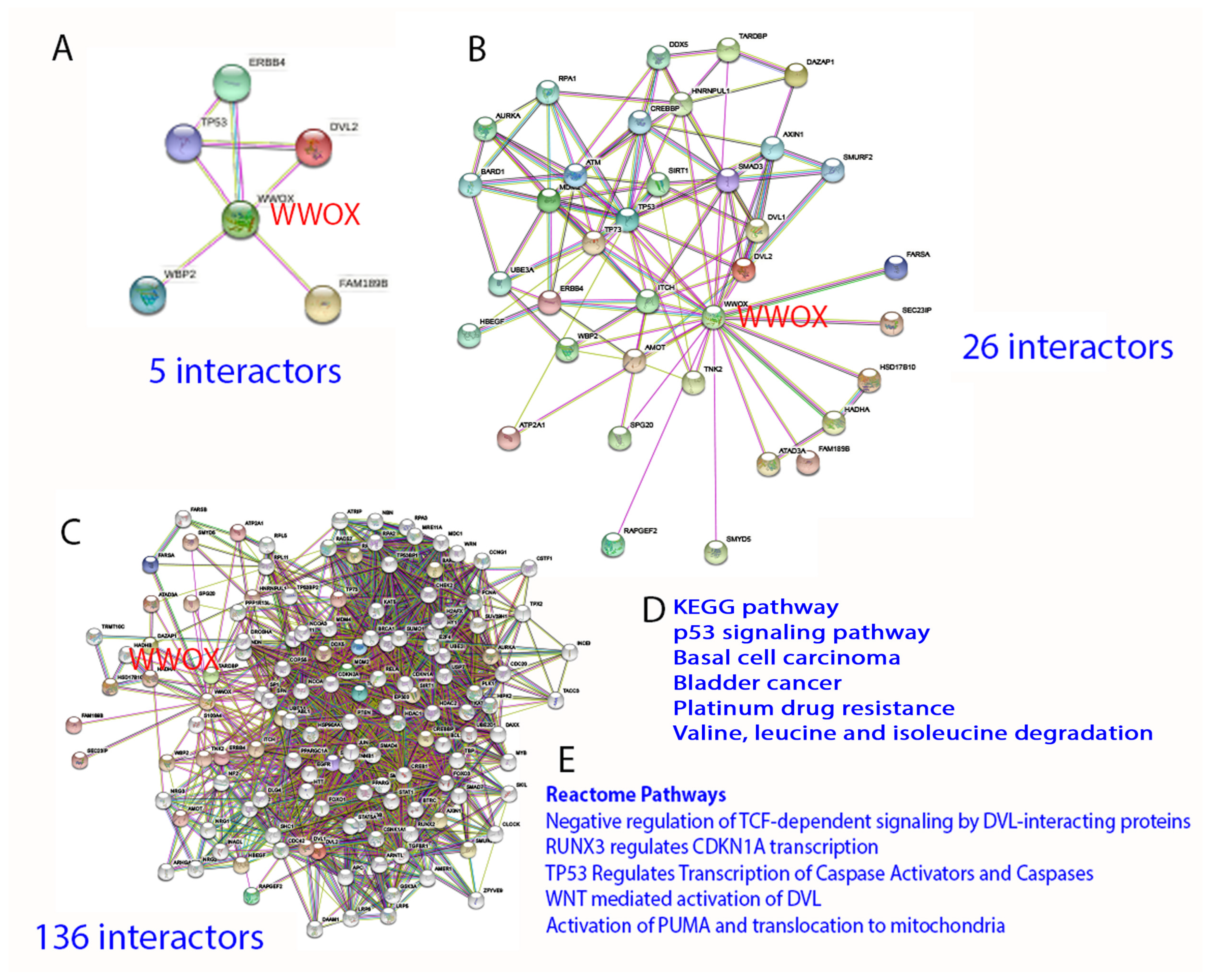
2. Protein Interaction Network in Normal Signaling and Diseases
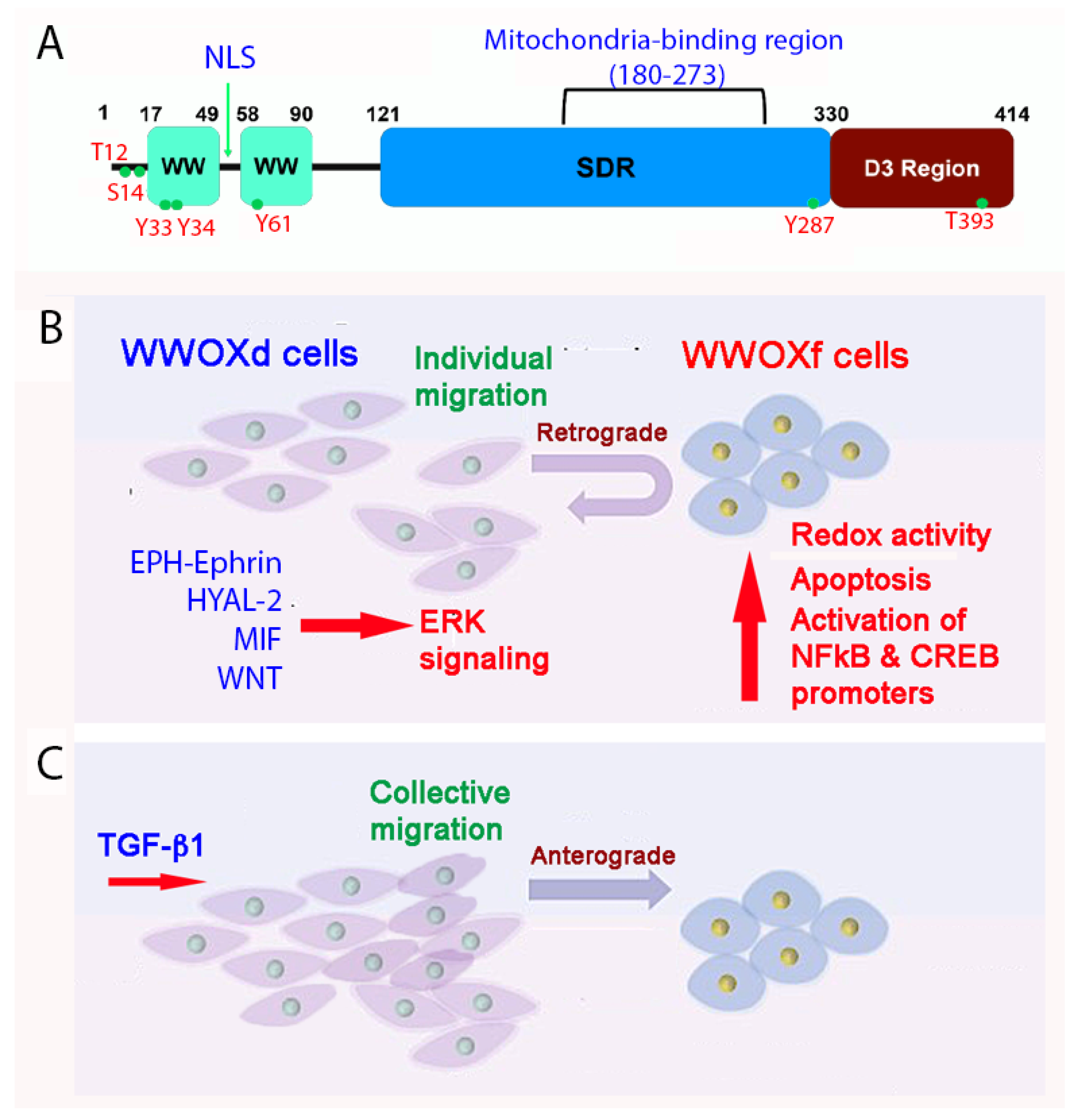
3. WW Domain-Containing Oxidoreductase (WWOX)
4. WWOX Controls Cell Migration, Cell-Cell Recognition, and Neuronal Heterotopia
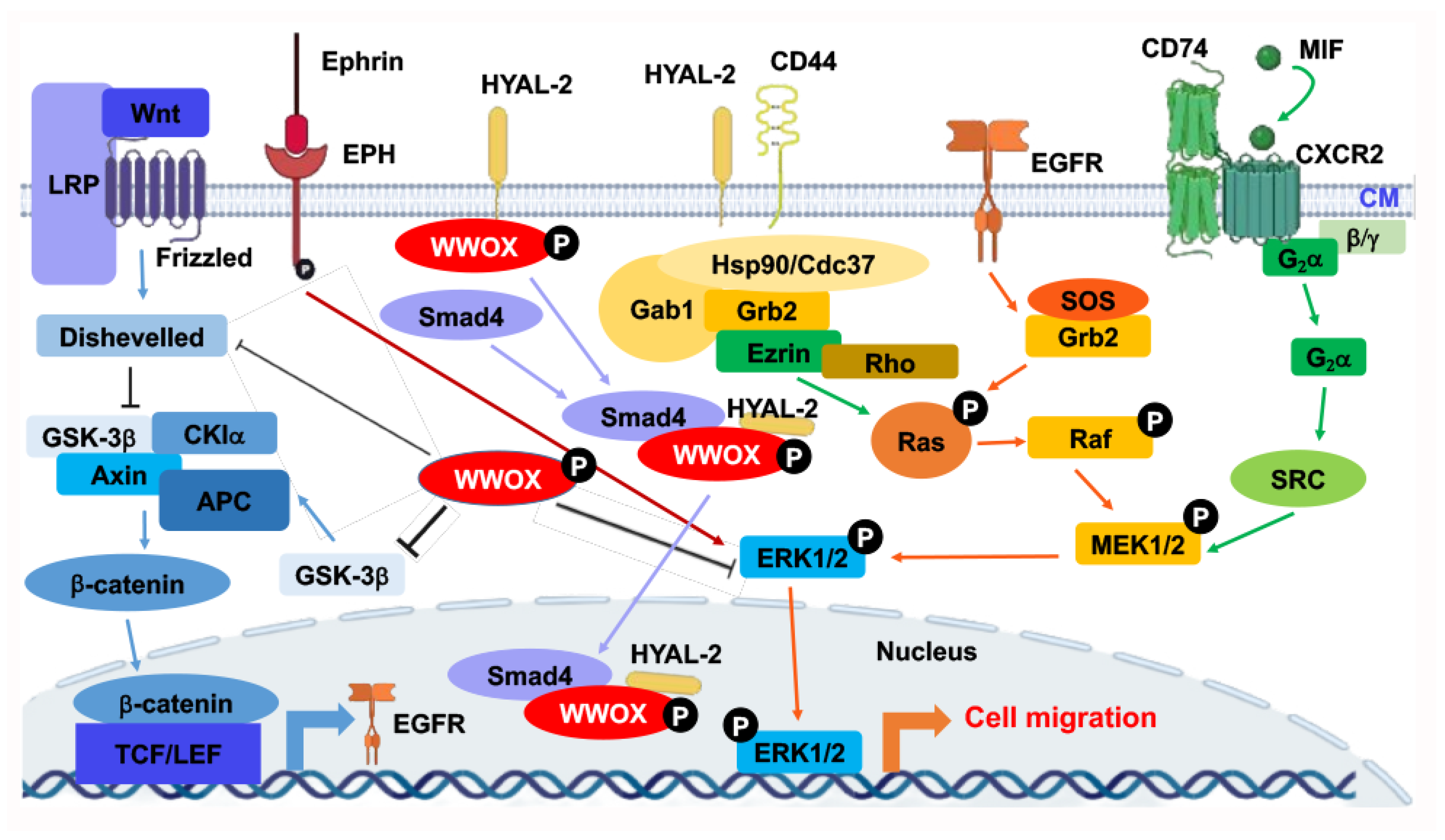
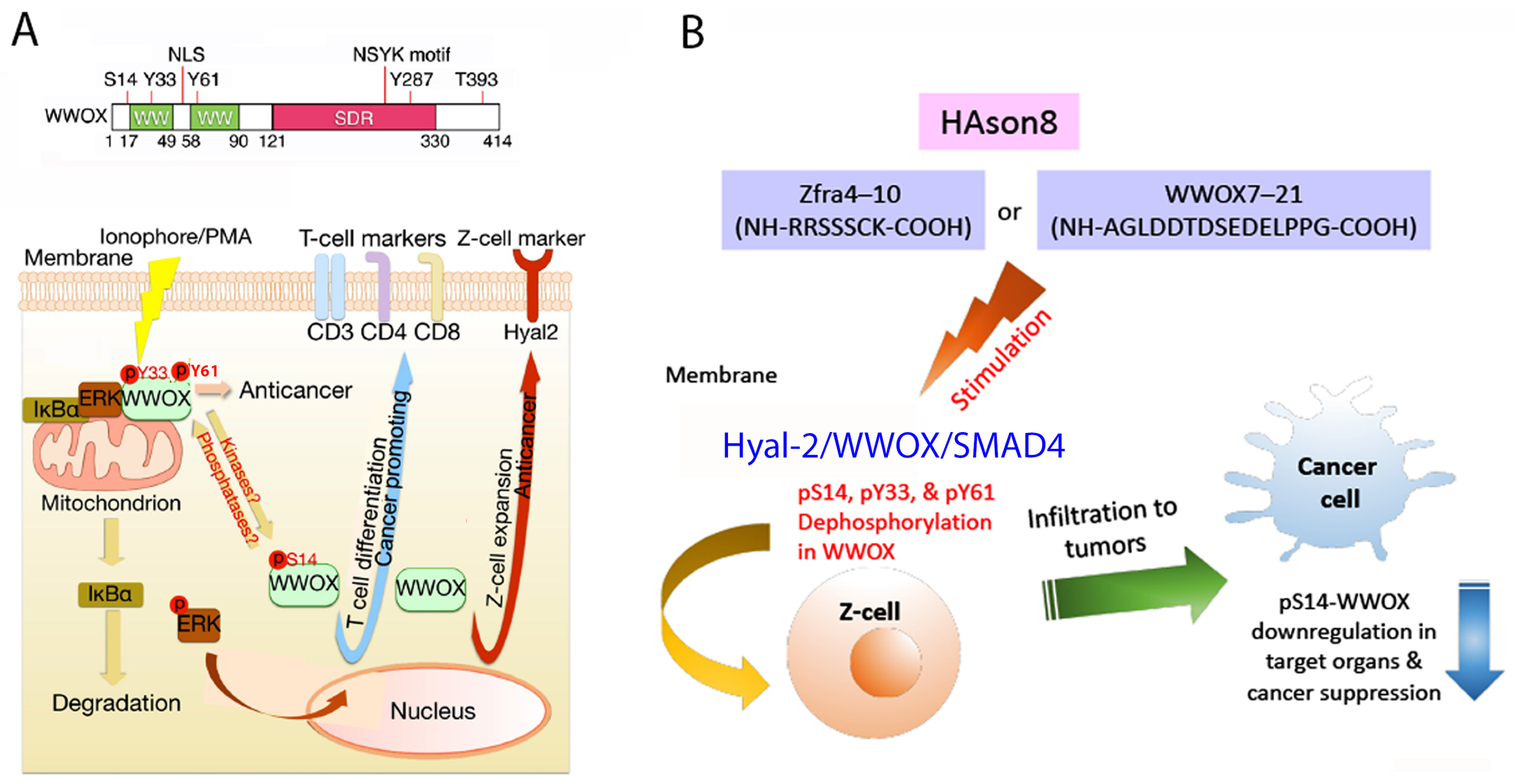
5. WWOX Signaling Network
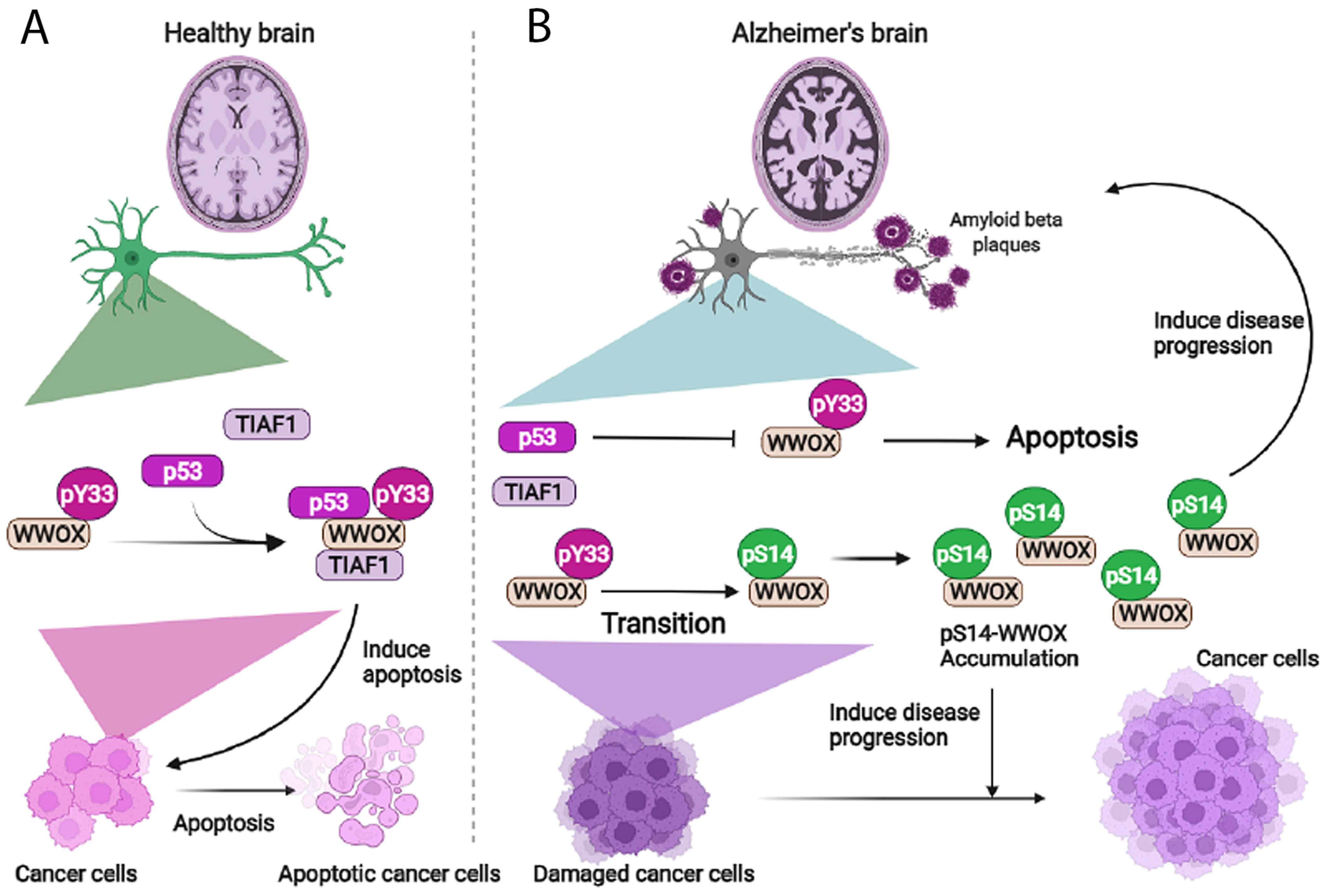
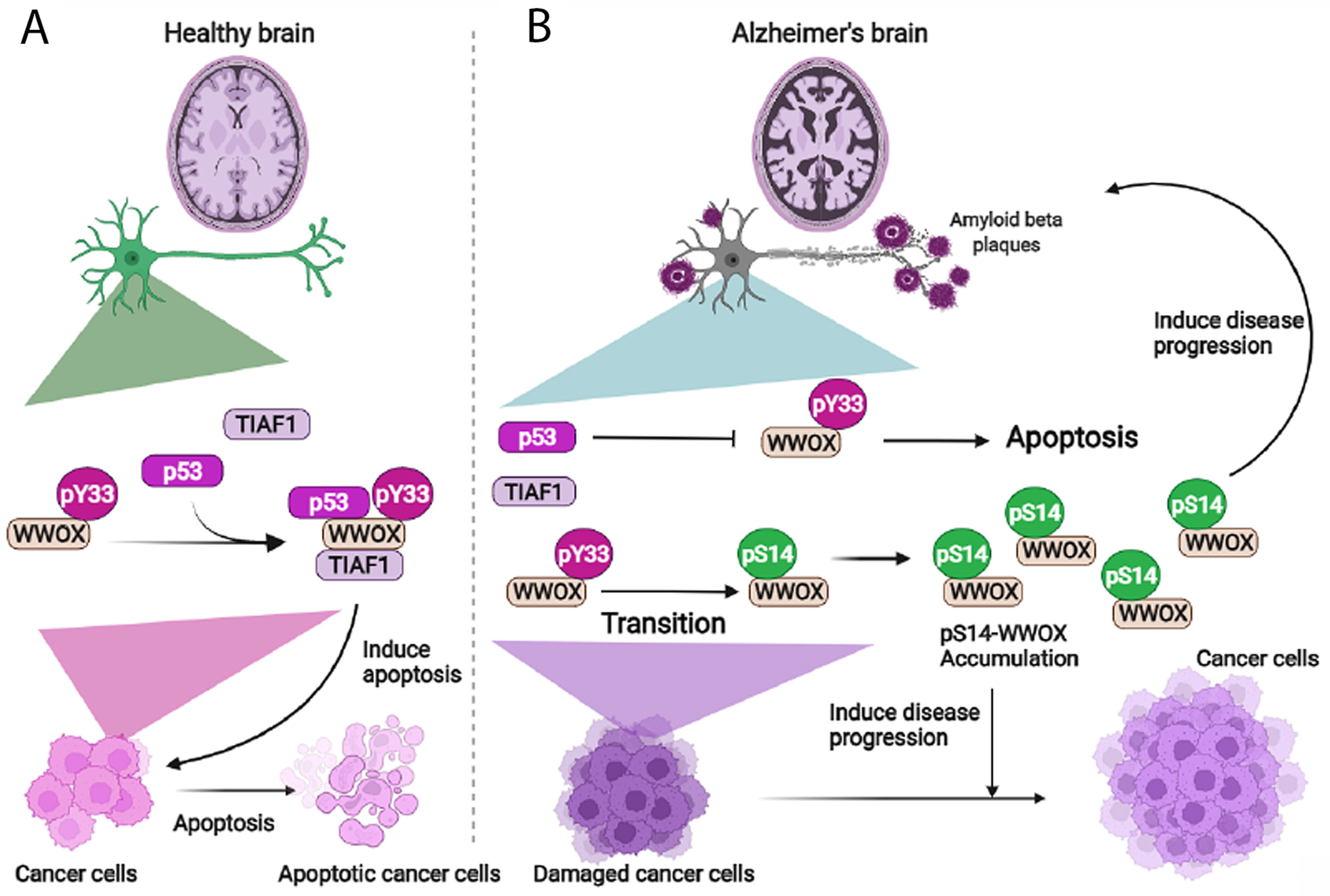
6. pY33 to pS14 Transition in WWOX during Disease Progression
7. WWOX Functional Measurement by Time-Lapse FRET Microscopy
8. TGF-β1 Induction of Initial Driving Force and Then Execution Force for Protein-Protein Binding and Cell Death: TIAF1 Is a Blocker of TGF-β1/SMAD Signaling
9. The Dynamics of WWOX/TIAF1/p53 Triad Formation and Functional Antagonism between p53 and WWOX for Enhancing the Progression of Cancer and Alzheimer’s Disease
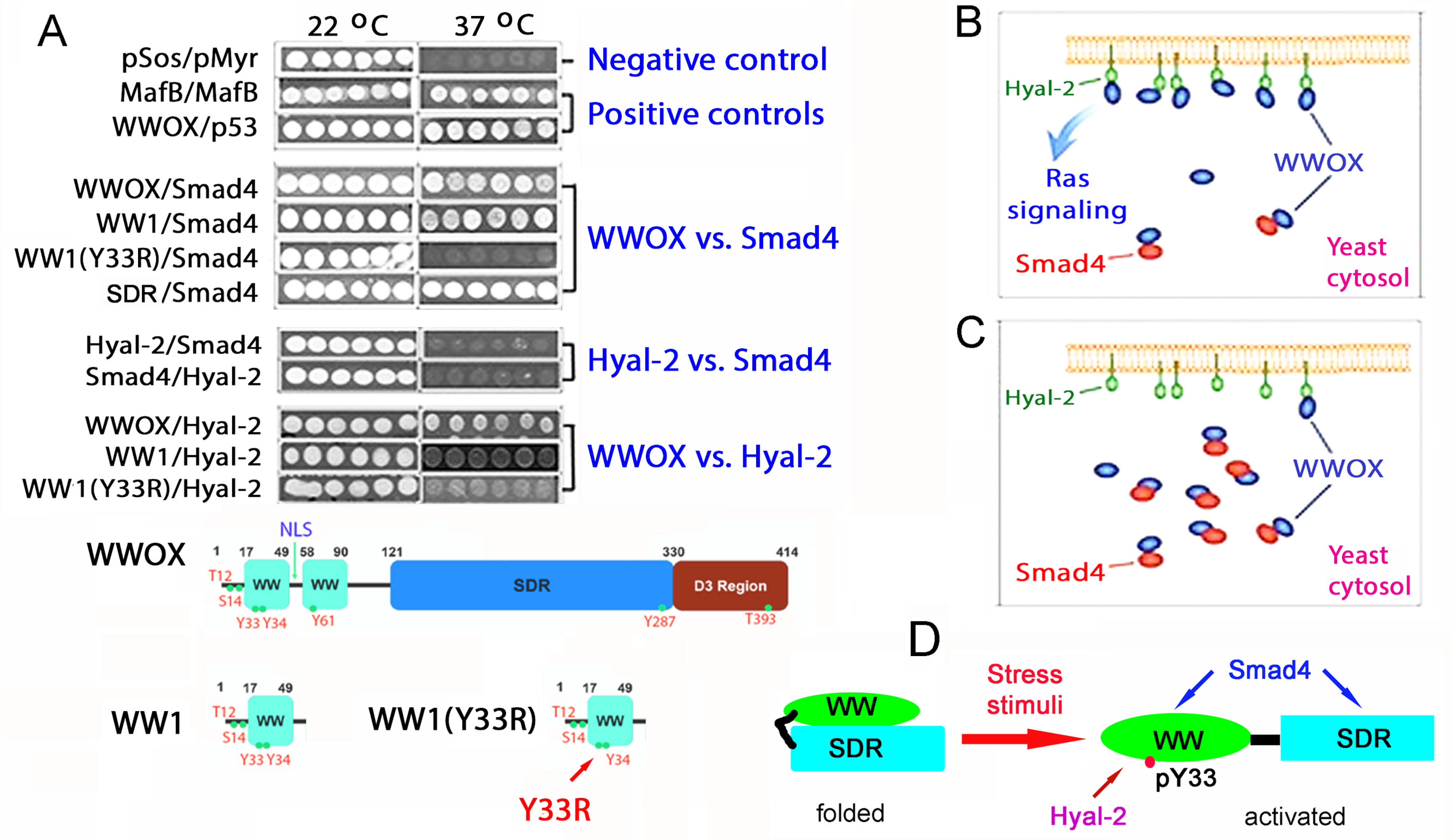
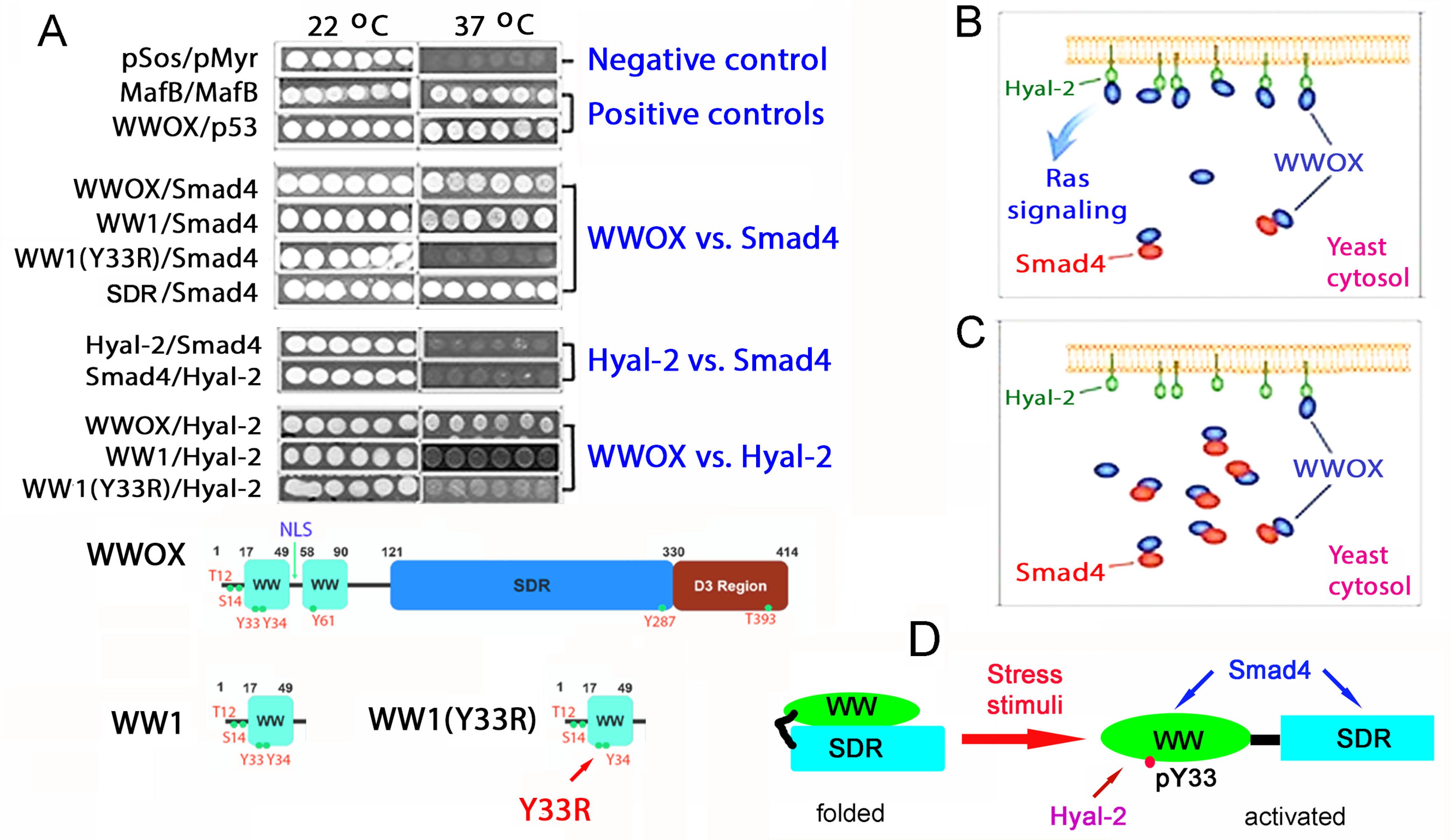
10. Identification of HYAL-2/WWOX/SMAD4 Signaling in Regulating Physiological and Pathological Events
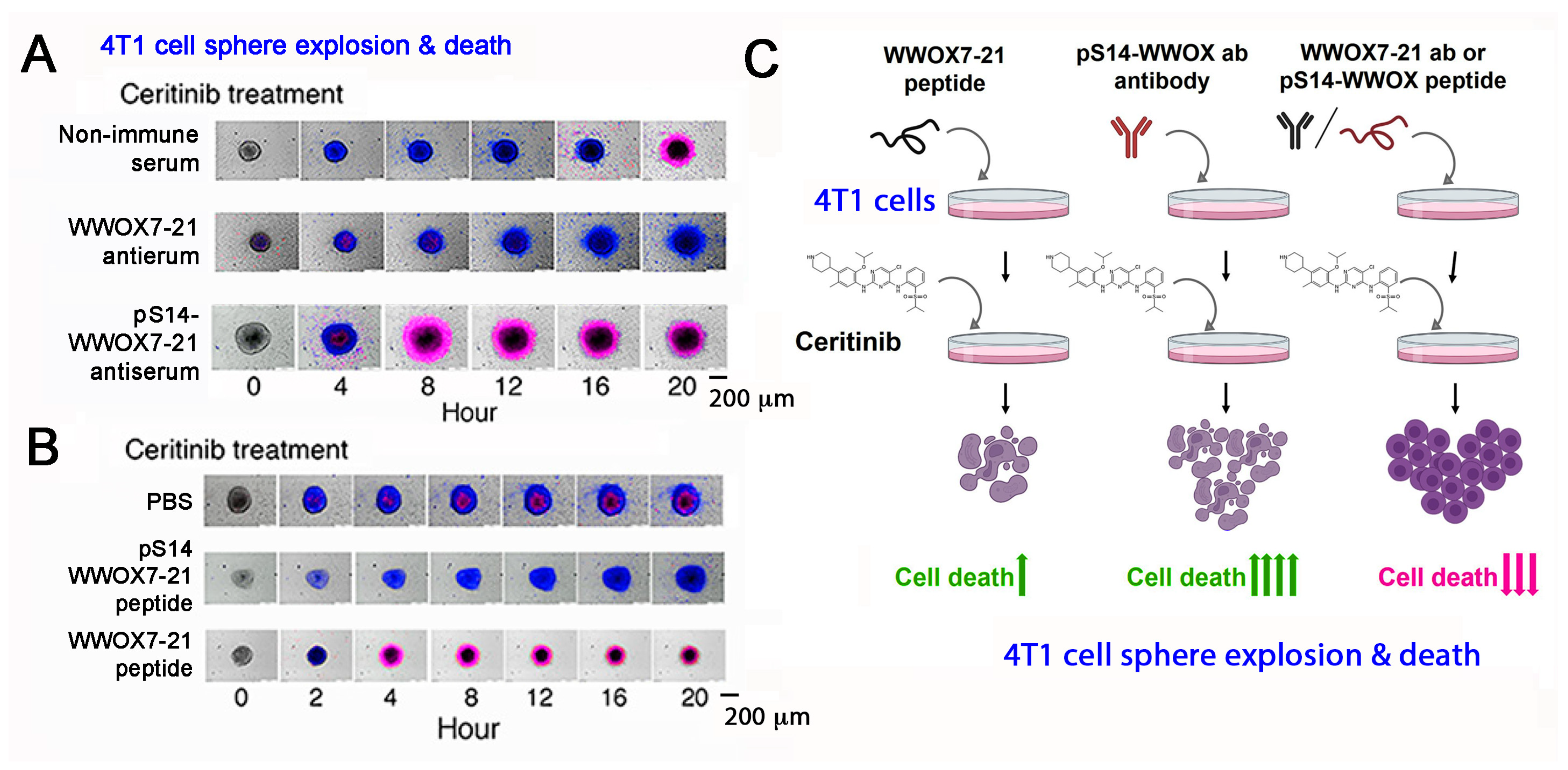
11. A WWOX7-21 Epitope Peptide Drives the HYAL-2/WWOX/SMAD4 Signaling
12. Phosphorylation Status of WWOX in the HYAL-2/WWOX/SMAD4 Complex and Disease Progression
13. Zfra4-10 or WWOX7-21 Activates the HYAL-2/WWOX/SMAD4 Signaling for Z Cell Activation and Suppression of Disease Progression In Vivo
14. Zfra-Induced Spleen Z Cell Activation Requires De-Phosphorylation at S14, Y33 and Y61 in WWOX In Vivo
15. Zfra4-10 or WWOX7-21 Increases the Binding of Endogenous WWOX with Intracellular Protein Partners, Which Contributes to Cancer Growth Suppression In Vivo
16. Switching the HYAL-2/WWOX/SMAD4 Signaling from Bubbling Cell Death to Membrane Blebbing by Replacing HYAL-2 with p53
17. Discussion and Perspectives
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gulfidan, G.; Turanli, B.; Beklen, H.; Sinha, R.; Arga, K.Y. Pan-cancer mapping of differential protein-protein interactions. Sci. Rep. 2020, 10, 3272. [Google Scholar] [CrossRef] [PubMed]
- Guda, P.; Chittur, S.V.; Guda, C. Comparative analysis of protein-protein interactions in cancer-associated genes. Genom. Proteom. Bioinform. 2009, 7, 25–36. [Google Scholar] [CrossRef] [Green Version]
- Guda, C.; King, B.R.; Pal, L.R.; Guda, P. A top-down approach to infer and compare domain-domain interactions across eight model organisms. PLoS ONE 2009, 4, e5096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pal, S.; Goswami, S.; Das, D. Cross β amyloid assemblies as complex catalytic machinery. Chem. Commun. 2021, 57, 7597–7609. [Google Scholar] [CrossRef]
- Capitanio, G.; Papa, F.; Papa, S. The allosteric protein interactions in the proton-motive function of mammalian redox enzymes of the respiratory chain. Biochimie 2021, 189, 1–12. [Google Scholar] [CrossRef]
- Moracci, L.; Crotti, S.; Traldi, P.; Agostini, M.; Cosma, C.; Lapolla, A. Role of mass spectrometry in the study of interactions between amylin and metal ions. Mass Spectrom. Rev. 2021. [Google Scholar] [CrossRef]
- Yang, J.; Perrett, S.; Wu, S. Single Molecule Characterization of Amyloid Oligomers. Molecules 2021, 26, 948. [Google Scholar] [CrossRef]
- Barik, S. The Uniqueness of Tryptophan in Biology: Properties, Metabolism, Interactions and Localization in Proteins. Int. J. Mol. Sci. 2020, 21, 8776. [Google Scholar] [CrossRef]
- Bailly, C.; Vergoten, G. Flurbiprofen as a biphenyl scaffold for the design of small molecules binding to PD-L1 protein dimer. Biochem. Pharmacol. 2020, 178, 114042. [Google Scholar] [CrossRef]
- Meiser, N.; Fuks, C.; Hengesbach, M. Cooperative Analysis of Structural Dynamics in RNA-Protein Complexes by Single-Molecule Förster Resonance Energy Transfer Spectroscopy. Molecules 2020, 25, 2057. [Google Scholar] [CrossRef]
- Chang, N.S.; Pratt, N.; Heath, J.; Schultz, L.; Sleve, D.; Carey, G.B.; Zevotek, N. Hyaluronidase induction of a WW domain-containing oxidoreductase that enhances tumor necrosis factor cytotoxicity. J. Biol. Chem. 2001, 276, 3361–3370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, N.S.; Hsu, L.J.; Lin, Y.S.; Lai, F.J.; Sheu, H.M. WW domain-containing oxidoreductase: A candidate tumor suppressor. Trends Mol. Med. 2007, 13, 12–22. [Google Scholar] [CrossRef]
- Abu-Remaileh, M.; Joy-Dodson, E.; Schueler-Furman, O.; Aqeilan, R.I. Pleiotropic Functions of Tumor Suppressor WWOX in Normal and Cancer Cells. J. Biol. Chem. 2015, 290, 30728–30735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hussain, T.; Lee, J.; Abba, M.C.; Chen, J.; Aldaz, C.M. Delineating WWOX Protein Interactome by Tandem Affinity Purification-Mass Spectrometry: Identification of Top Interactors and Key Metabolic Pathways Involved. Front. Oncol. 2018, 8, 591. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.T.; Liu, C.C.; Chen, S.T.; Yap, Y.V.; Chang, N.S.; Sze, C.I. WW domain-containing oxidoreductase in neuronal injury and neurological diseases. Oncotarget 2014, 5, 11792–11799. [Google Scholar] [CrossRef]
- Chang, N.S. Bubbling cell death: A hot air balloon released from the nucleus in the cold. Exp. Biol. Med. 2016, 241, 1306–1315. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.J.; Lin, P.W.; Lin, H.P.; Huang, S.S.; Lai, F.J.; Sheu, H.M.; Hsu, L.J.; Chang, N.S. UV irradiation/cold shock-mediated apoptosis is switched to bubbling cell death at low temperatures. Oncotarget 2015, 6, 8007–8018. [Google Scholar] [CrossRef] [Green Version]
- Hsu, L.J.; Chiang, M.F.; Sze, C.I.; Su, W.P.; Yap, Y.V.; Lee, I.T.; Kuo, H.L.; Chang, N.S. HYAL-2-WWOX-SMAD4 Signaling in Cell Death and Anticancer Response. Front. Cell Dev. Biol. 2016, 4, 141. [Google Scholar] [CrossRef] [Green Version]
- Hsu, C.Y.; Lee, K.T.; Sun, T.Y.; Sze, C.I.; Huang, S.S.; Hsu, L.J.; Chang, N.S. WWOX and Its Binding Proteins in Neurodegeneration. Cells 2021, 10, 1781. [Google Scholar] [CrossRef]
- Aldaz, C.M.; Hussain, T. WWOX Loss of Function in Neurodevelopmental and Neurodegenerative Disorders. Int. J. Mol. Sci. 2020, 21, 8922. [Google Scholar] [CrossRef]
- Liu, C.C.; Ho, P.C.; Lee, I.T.; Chen, Y.A.; Chu, C.H.; Teng, C.C.; Wu, S.N.; Sze, C.I.; Chiang, M.F.; Chang, N.S. WWOX Phosphorylation, Signaling, and Role in Neurodegeneration. Front. Neurosci. 2018, 12, 563. [Google Scholar] [CrossRef] [PubMed]
- Kunkle, B.W.; Grenier-Boley, B.; Sims, R.; Bis, J.C.; Damotte, V.; Naj, A.C.; Boland, A.; Vronskaya, M.; van der Lee, S.J.; Amlie-Wolf, A.; et al. Genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates Aβ, tau, immunity and lipid processing. Nat. Genet. 2019, 51, 414–430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abu-Odeh, M.; Bar-Mag, T.; Huang, H.; Kim, T.; Salah, Z.; Abdeen, S.K.; Sudol, M.; Reichmann, D.; Sidhu, S.; Kim, P.M.; et al. Characterizing WW domain interactions of tumor suppressor WWOX reveals its association with multiprotein networks. J. Biol. Chem. 2014, 289, 8865–8880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouteille, N.; Driouch, K.; Hage, P.E.; Sin, S.; Formstecher, E.; Camonis, J.; Lidereau, R.; Lallemand, F. Inhibition of the Wnt/beta-catenin pathway by the WWOX tumor suppressor protein. Oncogene 2009, 28, 2569–2580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, N.S.; Doherty, J.; Ensign, A. JNK1 physically interacts with WW domain-containing oxidoreductase (WOX1) and inhibits WOX1-mediated apoptosis. J. Biol. Chem. 2003, 278, 9195–9202. [Google Scholar] [CrossRef] [Green Version]
- Hsu, L.J.; Schultz, L.; Hong, Q.; Van Moer, K.; Heath, J.; Li, M.Y.; Lai, F.J.; Lin, S.R.; Lee, M.H.; Lo, C.P.; et al. Transforming growth factor beta1 signaling via interaction with cell surface Hyal-2 and recruitment of WWOX/WOX1. J. Biol. Chem. 2009, 284, 16049–16059. [Google Scholar] [CrossRef] [Green Version]
- Aldaz, C.M.; Ferguson, B.W.; Abba, M.C. WWOX at the crossroads of cancer, metabolic syndrome related traits and CNS pathologies. Biochim. Biophys. Acta 2014, 1846, 188–200. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.A.; Lu, C.Y.; Cheng, T.Y.; Pan, S.H.; Chen, H.F.; Chang, N.S. WW Domain-Containing Proteins YAP and TAZ in the Hippo Pathway as Key Regulators in Stemness Maintenance, Tissue Homeostasis, and Tumorigenesis. Front. Oncol. 2019, 9, 60. [Google Scholar] [CrossRef] [Green Version]
- Bendinelli, P.; Maroni, P.; Matteucci, E.; Desiderio, M.A. HGF and TGFβ1 differently influenced Wwox regulatory function on Twist program for mesenchymal-epithelial transition in bone metastatic versus parental breast carcinoma cells. Mol. Cancer 2015, 14, 112. [Google Scholar] [CrossRef] [Green Version]
- Chang, R.; Song, L.; Xu, Y.; Wu, Y.; Dai, C.; Wang, X.; Sun, X.; Hou, Y.; Li, W.; Zhan, X.; et al. Loss of Wwox drives metastasis in triple-negative breast cancer by JAK2/STAT3 axis. Nat. Commun. 2018, 9, 3486. [Google Scholar] [CrossRef]
- Taouis, K.; Driouch, K.; Lidereau, R.; Lallemand, F. Molecular Functions of WWOX Potentially Involved in Cancer Development. Cells 2021, 10, 1051. [Google Scholar] [CrossRef] [PubMed]
- Abu-Odeh, M.; Salah, Z.; Herbel, C.; Hofmann, T.G.; Aqeilan, R.I. WWOX, the common fragile site FRA16D gene product, regulates ATM activation and the DNA damage response. Proc. Natl. Acad. Sci. USA 2014, 111, 4716–4725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aqeilan, R.I.; Pekarsky, Y.; Herrero, J.J.; Palamarchuk, A.; Letofsky, J.; Druck, T.; Trapasso, F.; Han, S.Y.; Melino, G.; Huebner, K.; et al. Functional association between Wwox tumor suppressor protein and p73, a p53 homolog. Proc. Natl. Acad. Sci. USA 2004, 101, 4401–4406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schuchardt, B.J.; Mikles, D.C.; Bhat, V.; McDonald, C.B.; Sudol, M.; Farooq, A. Allostery mediates ligand binding to WWOX tumor suppressor via a conformational switch. J. Mol. Recognit. 2015, 28, 220–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chou, P.Y.; Lin, S.R.; Lee, M.H.; Schultz, L.; Sze, C.I.; Chang, N.S. A p53/TIAF1/WWOX triad exerts cancer suppression but may cause brain protein aggregation due to p53/WWOX functional antagonism. Cell Commun. Signal. 2019, 17, 76. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.A.; Sie, Y.D.; Liu, T.Y.; Kuo, H.L.; Chou, P.Y.; Chen, Y.J.; Lee, K.T.; Chen, P.J.; Chen, S.T.; Chang, N.S. Normal cells repel WWOX-negative or -dysfunctional cancer cells via WWOX cell surface epitope 286–299. Commun. Biol. 2021, 4, 753. [Google Scholar] [CrossRef]
- Chou, P.Y.; Lai, F.J.; Chen, Y.A.; Sie, Y.D.; Kuo, H.L.; Su, W.P.; Wu, C.Y.; Liu, T.Y.; Wen, K.Y.; Hsu, L.J.; et al. Strategies by which WWOX-deficient metastatic cancer cells utilize to survive via dodging, compromising, and causing damage to WWOX-positive normal microenvironment. Cell Death Discov. 2019, 5, 97. [Google Scholar] [CrossRef]
- Sze, C.I.; Su, M.; Pugazhenthi, S.; Jambal, P.; Hsu, L.J.; Heath, J.; Schultz, L.; Chang, N.S. Down-regulation of WW domain-containing oxidoreductase induces Tau phosphorylation in vitro. A potential role in Alzheimer’s disease. J. Biol. Chem. 2004, 279, 30498–30506. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.Y.; Juo, L.I.; Lin, Y.T.; Hsiao, M.; Lin, J.T.; Tsai, C.H.; Tzeng, Y.H.; Chuang, Y.C.; Chang, N.S.; Yang, C.N.; et al. WW domain-containing oxidoreductase promotes neuronal differentiation via negative regulation of glycogen synthase kinase 3β. Cell Death Differ. 2012, 19, 1049–1059. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Y.Y.; Chou, Y.T.; Lai, F.J.; Jan, M.S.; Chang, T.H.; Jou, I.M.; Chen, P.S.; Lo, J.Y.; Huang, S.S.; Chang, N.S.; et al. Wwox deficiency leads to neurodevelopmental and degenerative neuropathies and glycogen synthase kinase 3β-mediated epileptic seizure activity in mice. Acta Neuropathol. Commun. 2020, 8, 6. [Google Scholar] [CrossRef]
- Steinberg, D.J.; Aqeilan, R.I. WWOX-Related Neurodevelopmental Disorders: Models and Future Perspectives. Cells 2021, 10, 3082. [Google Scholar] [CrossRef] [PubMed]
- Repudi, S.; Steinberg, D.J.; Elazar, N.; Breton, V.L.; Aquilino, M.S.; Saleem, A.; Abu-Swai, S.; Vainshtein, A.; Eshed-Eisenbach, Y.; Vijayaragavan, B.; et al. Neuronal deletion of Wwox, associated with WOREE syndrome, causes epilepsy and myelin defects. Brain 2021, 144, 3061–3077. [Google Scholar] [CrossRef] [PubMed]
- Li, M.Y.; Lai, F.J.; Hsu, L.J.; Lo, C.P.; Cheng, C.L.; Lin, S.R.; Lee, M.H.; Chang, J.Y.; Subhan, D.; Tsai, M.S.; et al. Dramatic co-activation of WWOX/WOX1 with CREB and NF-kappaB in delayed loss of small dorsal root ganglion neurons upon sciatic nerve transection in rats. PLoS ONE 2009, 4, e7820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, L.J.; Hong, Q.; Chen, S.T.; Kuo, H.L.; Schultz, L.; Heath, J.; Lin, S.R.; Lee, M.H.; Li, D.Z.; Li, Z.L.; et al. Hyaluronan activates Hyal-2/WWOX/Smad4 signaling and causes bubbling cell death when the signaling complex is overexpressed. Oncotarget 2017, 8, 19137–19155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aqeilan, R.I.; Donati, V.; Palamarchuk, A.; Trapasso, F.; Kaou, M.; Pekarsky, Y.; Sudol, M.; Croce, C.M. WW domain-containing proteins, WWOX and YAP, compete for interaction with ErbB-4 and modulate its transcriptional function. Cancer Res. 2005, 65, 6764–6772. [Google Scholar] [CrossRef] [Green Version]
- Yang, T.; Xu, R.; Huo, J.; Wang, B.; Du, X.; Dai, B.; Zhu, M.; Zhan, Y.; Zhang, D.; Zhang, Y. WWOX activation by toosendanin suppresses hepatocellular carcinoma metastasis through JAK2/Stat3 and Wnt/β-catenin signaling. Cancer Lett. 2021, 513, 50–62. [Google Scholar] [CrossRef]
- Zhang, H.; Huang, C.J.; Tian, Y.; Wang, Y.P.; Han, Z.G.; Li, X.C. Ectopic overexpression of COTE1 promotes cellular invasion of hepatocellular carcinoma. Asian Pac. J. Cancer Prev. 2012, 13, 5799–5804. [Google Scholar] [CrossRef] [Green Version]
- Chang, N.S.; Doherty, J.; Ensign, A.; Schultz, L.; Hsu, L.J.; Hong, Q. WOX1 is essential for tumor necrosis factor-, UV light-, staurosporine-, and p53-mediated cell death, and its tyrosine 33-phosphorylated form binds and stabilizes serine 46-phosphorylated p53. J. Biol. Chem. 2005, 280, 43100–43108. [Google Scholar] [CrossRef] [Green Version]
- Jin, C.; Ge, L.; Ding, X.; Chen, Y.; Zhu, H.; Ward, T.; Wu, F.; Cao, X.; Wang, Q.; Yao, X. PKA-mediated protein phosphorylation regulates ezrin-WWOX interaction. Biochem. Biophys. Res. Commun. 2006, 341, 784–791. [Google Scholar] [CrossRef]
- Lee, M.H.; Shih, Y.H.; Lin, S.R.; Chang, J.Y.; Lin, Y.H.; Sze, C.I.; Kuo, Y.M.; Chang, N.S. Zfra restores memory deficits in Alzheimer’s disease triple-transgenic mice by blocking aggregation of TRAPPC6AΔ, SH3GLB2, tau, and amyloid β, and inflammatory NF-κB activation. Alzheimers Dement. 2017, 3, 189–204. [Google Scholar] [CrossRef]
- Lee, M.H.; Su, W.P.; Wang, W.J.; Lin, S.R.; Lu, C.Y.; Chen, Y.A.; Chang, J.Y.; Huang, S.S.; Chou, P.Y.; Ye, S.R.; et al. Zfra activates memory Hyal-2+ CD3- CD19- spleen cells to block cancer growth, stemness, and metastasis in vivo. Oncotarget 2015, 6, 3737–3751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, W.P.; Wang, W.J.; Sze, C.I.; Chang, N.S. Zfra induction of memory anticancer response via a novel immune cell. Oncoimmunology 2016, 5, e1213935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, W.P.; Wang, W.J.; Chang, J.Y.; Ho, P.C.; Liu, T.Y.; Wen, K.Y.; Kuo, H.L.; Chen, Y.J.; Huang, S.S.; Subhan, D.; et al. Therapeutic Zfra4-10 or WWOX7-21 Peptide Induces Complex Formation of WWOX with Selective Protein Targets in Organs that Leads to Cancer Suppression and Spleen Cytotoxic Memory Z Cell Activation In Vivo. Cancers 2020, 12, 2189. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.J.; Ho, P.C.; Nagarajan, G.; Chen, Y.A.; Kuo, H.L.; Subhan, D.; Su, W.P.; Chang, J.Y.; Lu, C.Y.; Chang, K.T.; et al. WWOX Possesses N-Terminal Cell Surface-Exposed Epitopes WWOX(7-21) and WWOX(7-11) for Signaling Cancer Growth Suppression and Prevention In Vivo. Cancers 2019, 11, 1818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, P.C. The Contrast Formation in Optical Microscopy. In Handbook of Biological Confocal Microscopy; Pawley, J.B., Ed.; Springer: Boston, MA, USA, 2006; pp. 162–206. [Google Scholar]
- Huang, S.S.; Su, W.P.; Lin, H.P.; Kuo, H.L.; Wei, H.L.; Chang, N.S. Role of WW Domain-containing Oxidoreductase WWOX in Driving T Cell Acute Lymphoblastic Leukemia Maturation. J. Biol. Chem. 2016, 291, 17319–17331. [Google Scholar] [CrossRef] [Green Version]
- Che, Y.; Khavari, P.A. Research Techniques Made Simple: Emerging Methods to Elucidate Protein Interactions through Spatial Proximity. J. Investig. Dermatol. 2017, 137, e197–e203. [Google Scholar] [CrossRef] [Green Version]
- Kuo, H.L.; Ho, P.C.; Huang, S.S.; Chang, N.S. Chasing the signaling run by tri-molecular time-lapse FRET microscopy. Cell Death Discov. 2018, 4, 45. [Google Scholar] [CrossRef]
- Liu, Q.; Zheng, J.; Sun, W.; Huo, Y.; Zhang, L.; Hao, P.; Wang, H.; Zhuang, M. A proximity-tagging system to identify membrane protein-protein interactions. Nat. Methods 2018, 15, 715–722. [Google Scholar] [CrossRef]
- Imani, M.; Mohajeri, N.; Rastegar, M.; Zarghami, N. Recent advances in FRET-Based biosensors for biomedical applications. Anal. Biochem. 2021, 630, 114323. [Google Scholar] [CrossRef]
- Feng, X.A.; Poyton, M.F.; Ha, T. Multicolor single-molecule FRET for DNA and RNA processes. Curr. Opin. Struct. Biol. 2021, 70, 26–33. [Google Scholar] [CrossRef]
- Nakamura, A.; Goto, Y.; Kondo, Y.; Aoki, K. Shedding light on developmental ERK signaling with genetically encoded biosensors. Development 2021, 148, dev199767. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.H.; Lin, S.R.; Chang, J.Y.; Schultz, L.; Heath, J.; Hsu, L.J.; Kuo, Y.M.; Hong, Q.; Chiang, M.F.; Gong, C.X.; et al. TGF-β induces TIAF1 self-aggregation via type II receptor-independent signaling that leads to generation of amyloid β plaques in Alzheimer’s disease. Cell Death Dis. 2010, 1, e110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, J.Y.; Chiang, M.F.; Lin, S.R.; Lee, M.H.; He, H.; Chou, P.Y.; Chen, S.J.; Chen, Y.A.; Yang, L.Y.; Lai, F.J.; et al. TIAF1 self-aggregation in peritumor capsule formation, spontaneous activation of SMAD-responsive promoter in p53-deficient environment, and cell death. Cell Death Dis. 2012, 3, e302. [Google Scholar] [CrossRef]
- Hong, Q.; Hsu, L.J.; Chou, P.Y.; Chou, Y.T.; Lu, C.Y.; Chen, Y.A.; Chang, N.S. Self-aggregating TIAF1 in lung cancer progression. Transl. Respir. Med. 2013, 1, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, J.Y.; Chang, N.S. WWOX dysfunction induces sequential aggregation of TRAPPC6AΔ, TIAF1, tau and amyloid β, and causes apoptosis. Cell Death Discov. 2015, 1, 15003. [Google Scholar] [CrossRef] [PubMed]
- Chang, N.S. Introduction to a thematic issue for WWOX. Exp. Biol. Med. 2015, 240, 281–284. [Google Scholar] [CrossRef] [Green Version]
- Anbarasan, T.; Bourdon, J.C. The Emerging Landscape of p53 Isoforms in Physiology, Cancer and Degenerative Diseases. Int. J. Mol. Sci. 2019, 20, 6257. [Google Scholar] [CrossRef] [Green Version]
- Joruiz, S.M.; Beck, J.A.; Horikawa, I.; Harris, C.C. The Δ133p53 Isoforms, Tuners of the p53 Pathway. Cancers 2020, 12, 3422. [Google Scholar] [CrossRef]
- Free, R.B.; Hazelwood, L.A.; Sibley, D.R. Identifying novel protein-protein interactions using co-immunoprecipitation and mass spectroscopy. Curr. Protoc. Neurosci. 2009, 46, 5–28. [Google Scholar] [CrossRef] [Green Version]
- Vojtek, A.B.; Hollenberg, S.M. Ras-Raf interaction: Two-hybrid analysis. Methods Enzymol. 1995, 255, 331–342. [Google Scholar]
- Chang, N.S. The non-ankyrin C terminus of Ikappa Balpha physically interacts with p53 in vivo and dissociates in response to apoptotic stress, hypoxia, DNA damage, and transforming growth factor-beta 1-mediated growth suppression. J. Biol. Chem. 2002, 277, 10323–10331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deeks, E.D. Ceritinib: A Review in ALK-Positive Advanced NSCLC. Target. Oncol. 2016, 11, 693–700. [Google Scholar] [CrossRef]
- Hsu, L.J.; Schultz, L.; Mattison, J.; Lin, Y.S.; Chang, N.S. Cloning and characterization of a small-size peptide Zfra that regulates the cytotoxic function of tumor necrosis factor by interacting with JNK1. Biochem. Biophys. Res. Commun. 2005, 327, 415–423. [Google Scholar] [CrossRef] [PubMed]
- Hsu, L.J.; Hong, Q.; Schultz, L.; Kuo, E.; Lin, S.R.; Lee, M.H.; Lin, Y.S.; Chang, N.S. Zfra is an inhibitor of Bcl-2 expression and cytochrome c release from the mitochondria. Cell. Signal. 2008, 20, 1303–1312. [Google Scholar] [CrossRef]
- Huppert, J.; Closhen, D.; Croxford, A.; White, R.; Kulig, P.; Pietrowski, E.; Bechmann, I.; Becher, B.; Luhmann, H.J.; Waisman, A.; et al. Cellular mechanisms of IL-17-induced blood-brain barrier disruption. FASEB J. 2010, 24, 1023–1034. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.T.; Chuang, J.I.; Wang, J.P.; Tsai, M.S.; Li, H.; Chang, N.S. Expression of WW domain-containing oxidoreductase WOX1 in the developing murine nervous system. Neuroscience 2004, 124, 831–839. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.T.; Chuang, J.I.; Cheng, C.L.; Hsu, L.J.; Chang, N.S. Light-induced retinal damage involves tyrosine 33 phosphorylation, mitochondrial and nuclear translocation of WW domain-containing oxidoreductase in vivo. Neuroscience 2005, 130, 397–407. [Google Scholar] [CrossRef] [PubMed]
- Chiang, M.F.; Chen, S.T.; Lo, C.P.; Sze, C.I.; Chang, N.S.; Chen, Y.J. Expression of WW domain-containing oxidoreductase WOX1 in human nervous system tumors. Anal. Cell. Pathol. 2013, 36, 133–147. [Google Scholar] [CrossRef]
- Lo, C.P.; Hsu, L.J.; Li, M.Y.; Hsu, S.Y.; Chuang, J.I.; Tsai, M.S.; Lin, S.R.; Chang, N.S.; Chen, S.T. MPP+-induced neuronal death in rats involves tyrosine 33 phosphorylation of WW domain-containing oxidoreductase WOX1. Eur. J. Neurosci. 2008, 27, 1634–1646. [Google Scholar] [CrossRef]
- Ludes-Meyers, J.H.; Kil, H.; Bednarek, A.K.; Drake, J.; Bedford, M.T.; Aldaz, C.M. WWOX binds the specific proline-rich ligand PPXY: Identification of candidate interacting proteins. Oncogene 2004, 23, 5049–5055. [Google Scholar] [CrossRef] [Green Version]
- McDonald, C.B.; Buffa, L.; Bar-Mag, T.; Salah, Z.; Bhat, V.; Mikles, D.C.; Deegan, B.J.; Seldeen, K.L.; Malhotra, A.; Sudol, M.; et al. Biophysical basis of the binding of WWOX tumor suppressor to WBP1 and WBP2 adaptors. J. Mol. Biol. 2012, 422, 58–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reuven, N.; Shanzer, M.; Shaul, Y. Tyrosine phosphorylation of WW proteins. Exp. Biol. Med. 2015, 240, 375–382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.; Wang, H.; Huang, Y.F.; Li, M.L.; Cheng, J.H.; Hu, P.; Lu, C.H.; Zhang, Y.; Liu, N.; Tzeng, C.M.; et al. WW domain-binding protein 2: An adaptor protein closely linked to the development of breast cancer. Mol. Cancer 2017, 16, 128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saigo, C.; Kito, Y.; Takeuchi, T. Cancerous Protein Network That Inhibits the Tumor Suppressor Function of WW Domain-Containing Oxidoreductase (WWOX) by Aberrantly Expressed Molecules. Front. Oncol. 2018, 8, 350. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Ruthel, G.; Freedman, B.D.; Harty, R.N. WWOX-Mediated Degradation of AMOTp130 Negatively Affects Egress of Filovirus VP40 Virus-Like Particles. J. Virol. 2022, 96, e0202621. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, T.-Y.; Nagarajan, G.; Chiang, M.-F.; Huang, S.-S.; Lin, T.-C.; Chen, Y.-A.; Sze, C.-I.; Chang, N.-S. WWOX Controls Cell Survival, Immune Response and Disease Progression by pY33 to pS14 Transition to Alternate Signaling Partners. Cells 2022, 11, 2137. https://doi.org/10.3390/cells11142137
Liu T-Y, Nagarajan G, Chiang M-F, Huang S-S, Lin T-C, Chen Y-A, Sze C-I, Chang N-S. WWOX Controls Cell Survival, Immune Response and Disease Progression by pY33 to pS14 Transition to Alternate Signaling Partners. Cells. 2022; 11(14):2137. https://doi.org/10.3390/cells11142137
Chicago/Turabian StyleLiu, Tsung-Yun, Ganesan Nagarajan, Ming-Fu Chiang, Shenq-Shyang Huang, Tzu-Chia Lin, Yu-An Chen, Chun-I Sze, and Nan-Shan Chang. 2022. "WWOX Controls Cell Survival, Immune Response and Disease Progression by pY33 to pS14 Transition to Alternate Signaling Partners" Cells 11, no. 14: 2137. https://doi.org/10.3390/cells11142137