
Search for Synergistic Drug Combinations to Treat Chronic Lymphocytic Leukemia
 , , , , ,
, , , , ,  and
and 
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Reagents
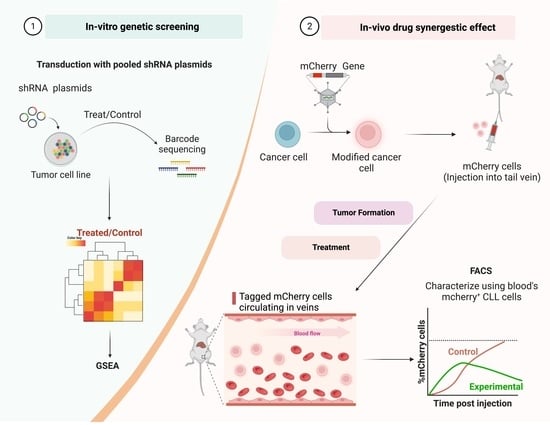
2.2. Decipher Pooled shRNA Library
2.3. shRNA Library Lentivirus Preparation
2.4. Infection of HL-60 Cells with shRNA Lentivirus
2.5. Collection of Viable HL-60 Cells
2.6. Genomic DNA Extraction
2.7. Amplification of the Barcodes
2.8. Bioinformatic Pathway Analysis
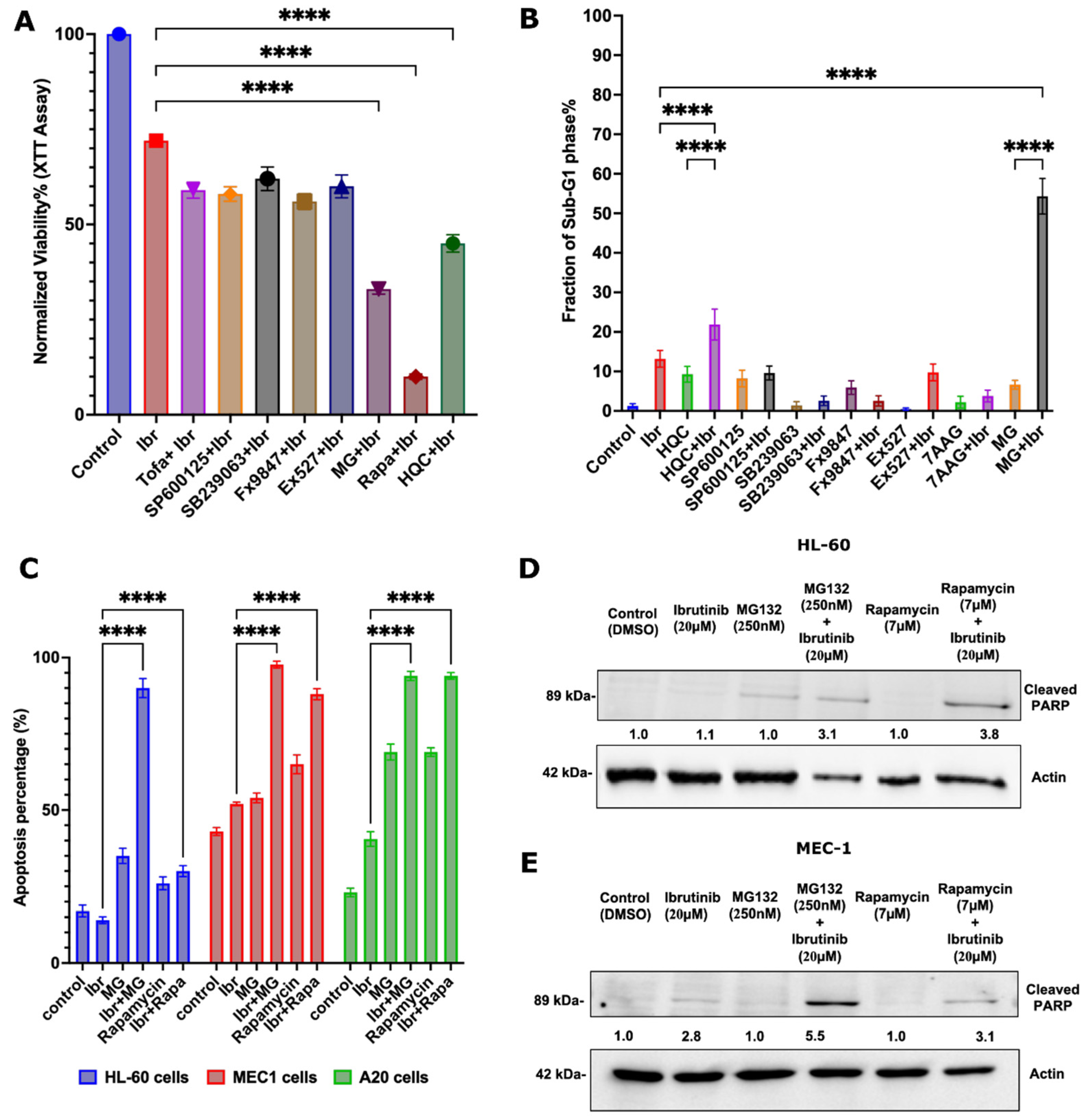
2.9. Synergy Tests
2.10. Lentivirus Preparation
2.11. A20 Cell Infection with Luc-mCherry Lentivirus
2.12. Real-Time PCR Analysis
2.13. Immunoblotting
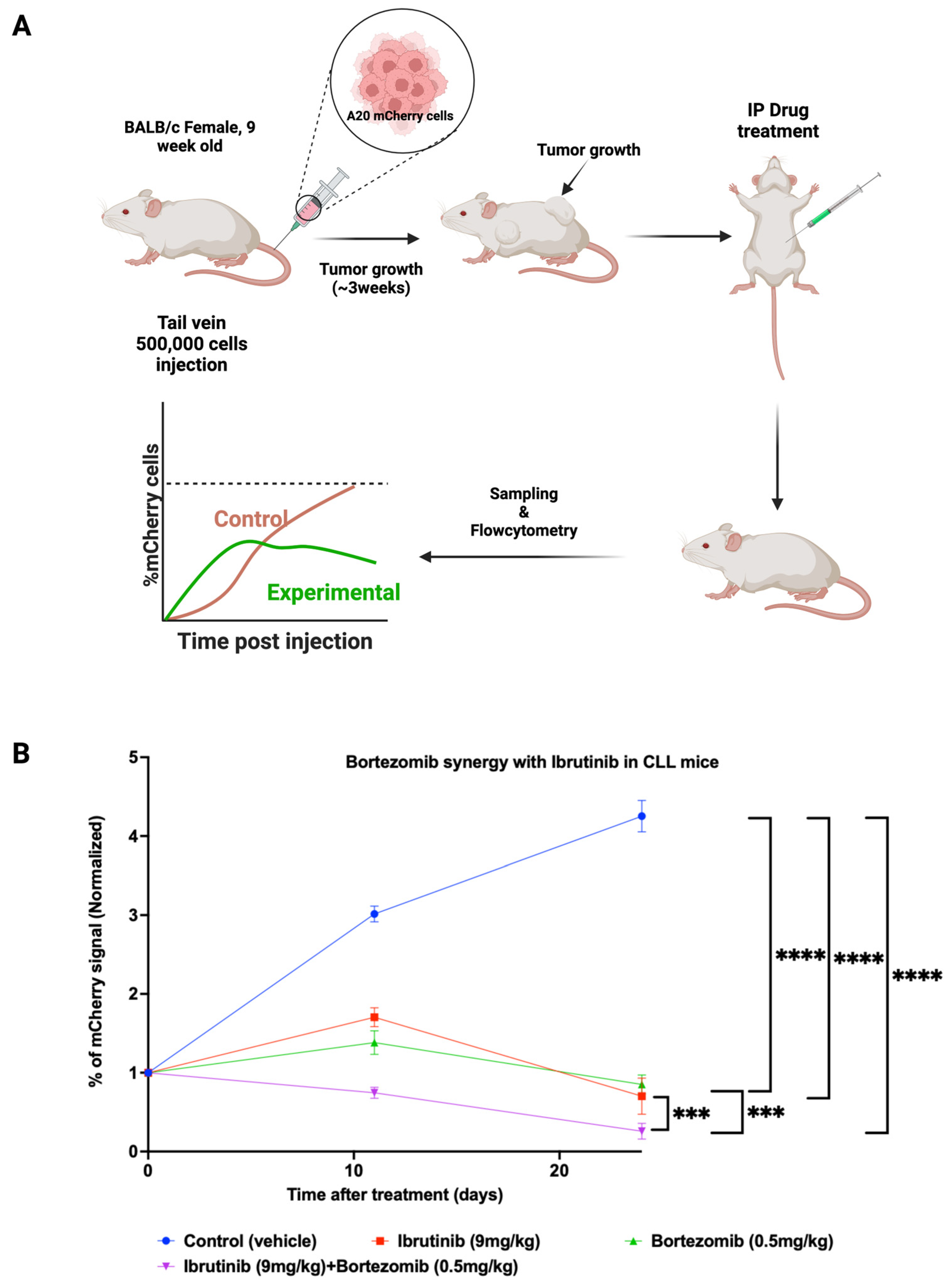
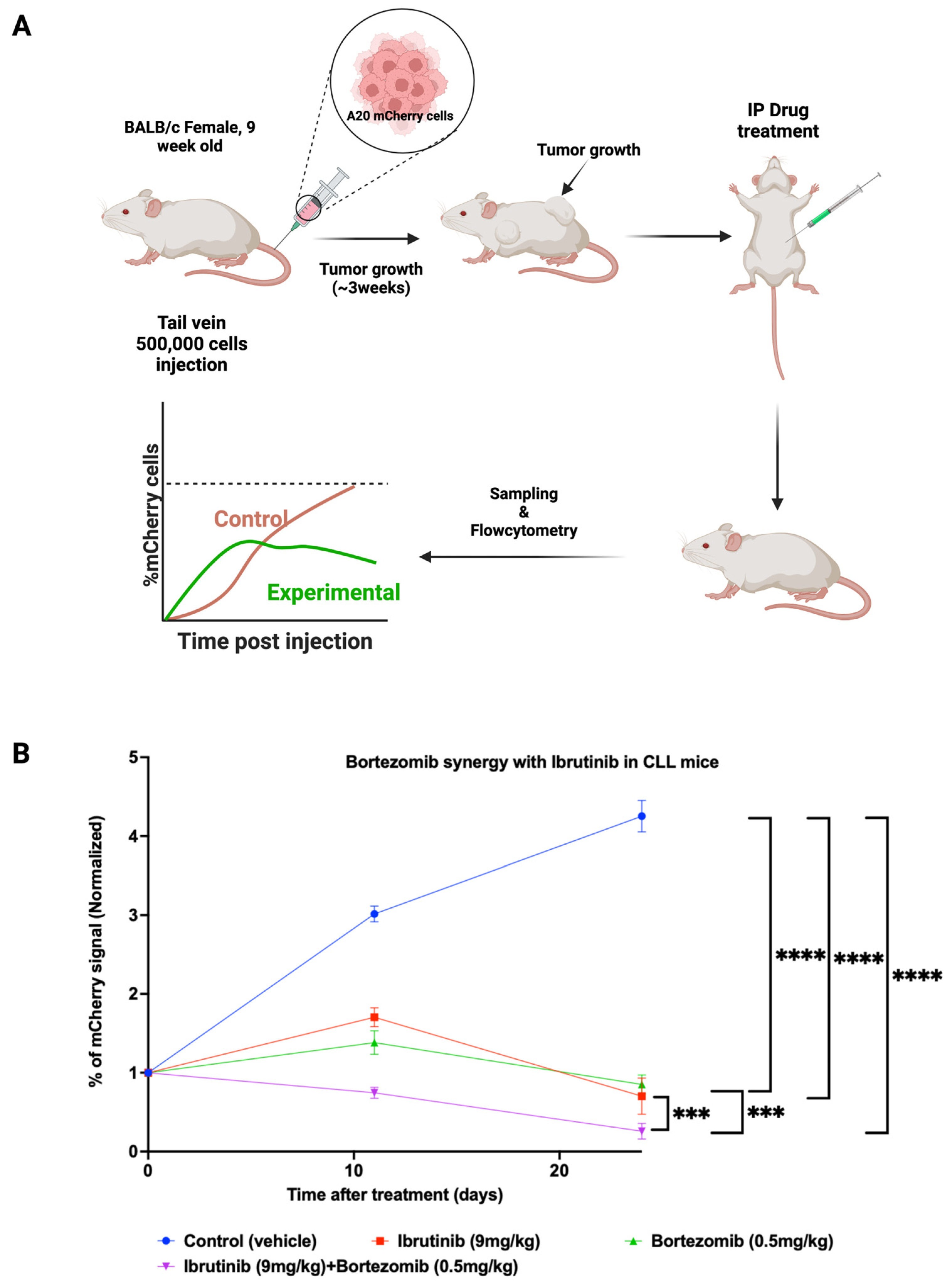
2.14. In Vivo Assessment of Drug Combination in the Animal Model
3. Results
3.1. Identifying Genes Involved in the Enhancement of the Response to Ibrutinib
3.2. Pathways Involved in the Ibrutinib Response
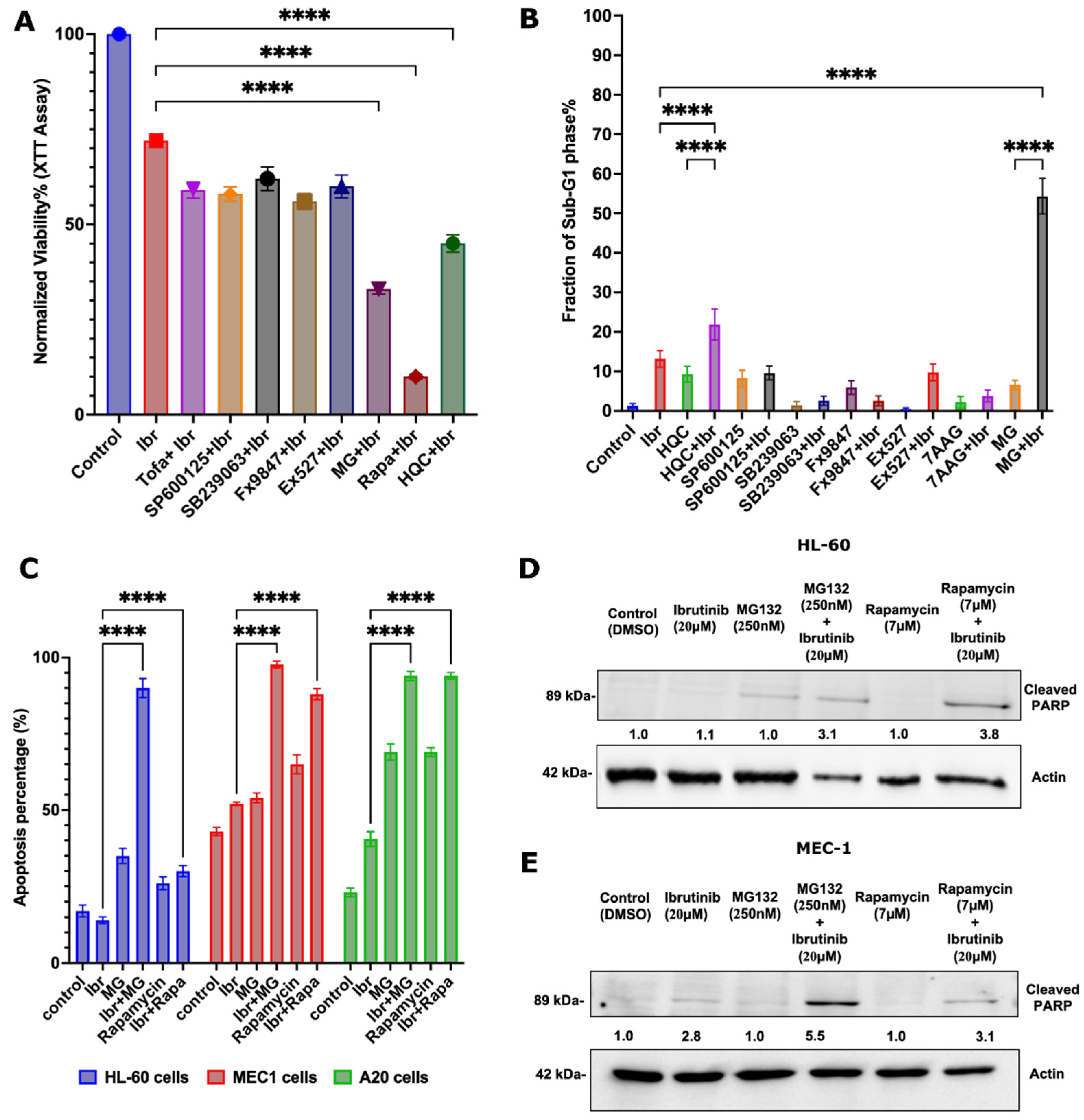
3.3. Identifying FDA-Approved Drugs That Synergize with Ibrutinib
3.4. Synergistic Effects of the Proteasome Inhibitor Bortezomib and Ibrutinib in the Mouse CLL Model
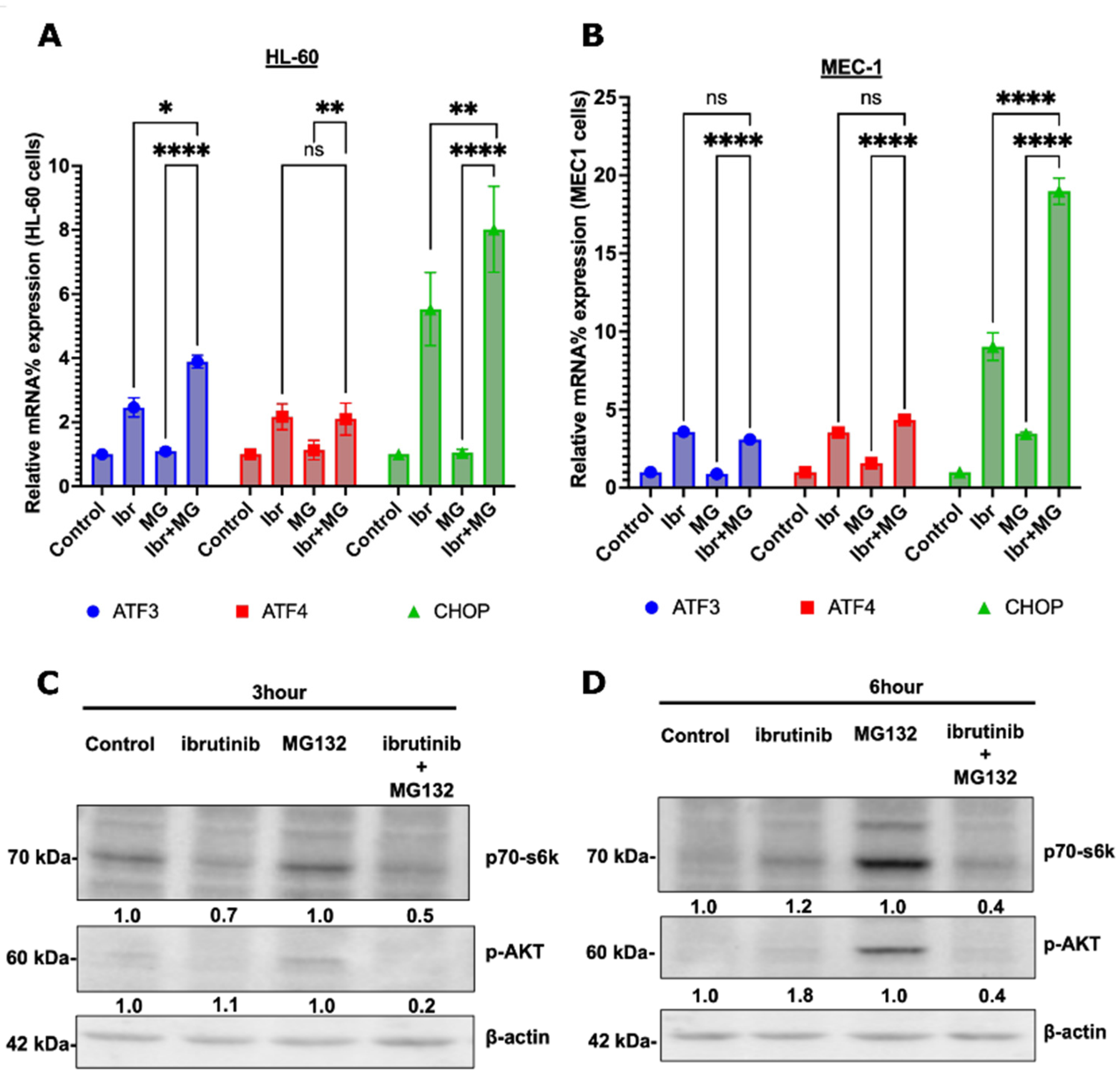
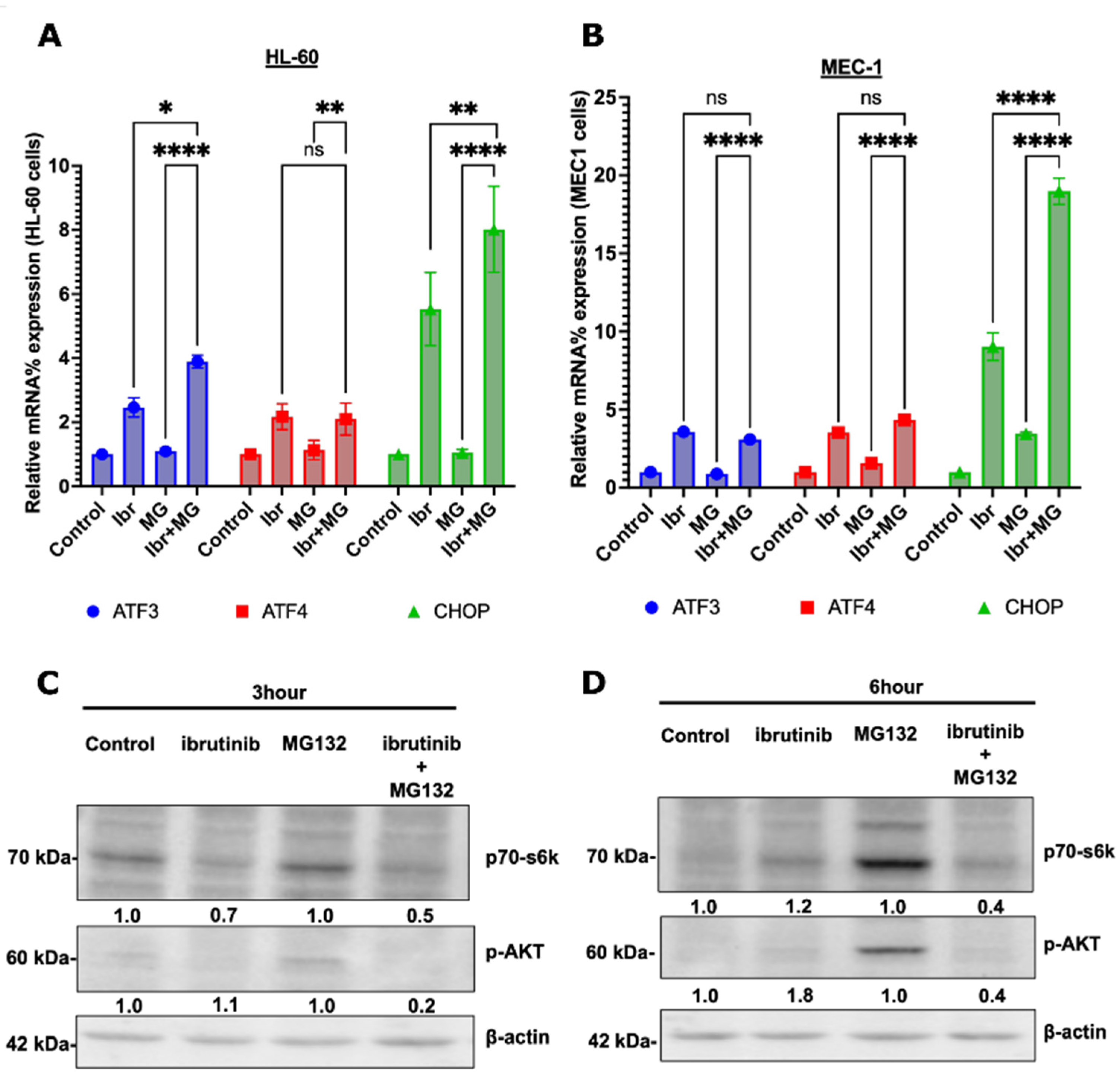
3.5. Possible Synergy Pathways
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Vaisitti, T.; Arruga, F.; Ferrajoli, A. Chronic Lymphocytic Leukemia. Cancers 2020, 12, 2504. [Google Scholar] [CrossRef]
- Chiorazzi, N.; Rai, K.R.; Ferrarini, M. Chronic Lymphocytic Leukemia. N. Engl. J. Med. 2005, 352, 804–815. [Google Scholar] [CrossRef] [Green Version]
- Puła, B.; Gołos, A.; Górniak, P.; Jamroziak, K. Overcoming Ibrutinib Resistance in Chronic Lymphocytic Leukemia. Cancers 2019, 11, 1834. [Google Scholar] [CrossRef] [Green Version]
- Timofeeva, N.; Gandhi, V. Ibrutinib Combinations in CLL Therapy: Scientific Rationale and Clinical Results. Blood Cancer J 2021, 11, 79. [Google Scholar] [CrossRef]
- Morris, D.V.M.S. Guidelines Provide Strategies for Managing Common IrAEs. Target. Ther. Oncol. 2022, 10, 81. [Google Scholar]
- Lisa, A.; Raedler, P. Imbruvica (Ibrutinib) Now FDA Approved as First-Line Treatment for Chronic Lymphocytic Leukemia and Small Lymphocytic Lymphoma; The Oncology Pharmacist: Cranbury, NJ, USA, 2017. [Google Scholar]
- Sharma, S.; Rai, K.R. Chronic Lymphocytic Leukemia (CLL) Treatment: So Many Choices, Such Great Options. Cancer 2019, 125, 1432–1440. [Google Scholar] [CrossRef]
- Singh, S.P.; Dammeijer, F.; Hendriks, R.W. Role of Bruton’s Tyrosine Kinase in B Cells and Malignancies. Mol. Cancer 2018, 17, 57. [Google Scholar] [CrossRef]
- Lu, D.-Y.; Chen, E.-H.; Wu, H.-Y.; Lu, T.-R.; Xu, B.; Ding, J. Anticancer Drug Combinations, How Far We Can Go Through? Anti-Cancer Agents Med. Chem. 2017, 17, 21–28. [Google Scholar] [CrossRef]
- George, B.; Chowdhury, S.M.; Hart, A.; Sircar, A.; Singh, S.K.; Nath, U.K.; Mamgain, M.; Singhal, N.K.; Sehgal, L.; Jain, N. Ibrutinib Resistance Mechanisms and Treatment Strategies for B-Cell Lymphomas. Cancers 2020, 12, 1328. [Google Scholar] [CrossRef]
- Lama, T.G.; Kyung, D.; O’Brien, S. Mechanisms of Ibrutinib Resistance in Chronic Lymphocytic Leukemia and Alternative Treatment Strategies. Expert Rev. Hematol. 2020, 13, 871–883. [Google Scholar] [CrossRef]
- Kumar, S.; Gahramanov, V.; Yaglom, J.; Patel, S.; Kaczmarczyk, L.; Alexandrov, I.; Gerlitz, G.; Salmon-Divon, M.; Sherman, M.Y. Homologous Recombination Repair Creates Mutations in the Non-Coding Genome That Alter Topoisomerase-1 Cleavage Sites & Orchestrates Irinotecan Resistance. bioRxiv 2021. [Google Scholar] [CrossRef]
- Kreamer, B.L.; Staecker, J.L.; Sawada, N.; Sattler, G.L.; Hsia, M.T.; Pitot, H.C. Use of a low-speed, iso-density percoll centrifugation method to increase the viability of isolated rat hepatocyte preparations. In Vitro Cell Dev Biol. 1986, 4, 201–211. [Google Scholar] [CrossRef]
- Tamura, A.; Urakami, H.; Tsuruhara, T. Purification of Rickettsia Tsutsugamushi by Percoll Density Gradient Centrifugation. Microbiol. Immunol. 1982, 26, 321–328. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene Set Enrichment Analysis: A Knowledge-Based Approach for Interpreting Genome-Wide Expression Profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [Green Version]
- Patel, S.; Kumar, S.; Baldan, S.; Hesin, A.; Yaglom, J.; Sherman, M.Y. Cytoplasmic Proteotoxicity Regulates HRI-Dependent Phosphorylation of EIF2α via the Hsp70-Bag3 Module. iScience 2022, 25, 104282. [Google Scholar] [CrossRef]
- Yang, X.; Feng, Y.; Liu, Y.; Zhang, N.; Lin, W.; Sa, Y.; Hu, X.-H. A Quantitative Method for Measurement of HL-60 Cell Apoptosis Based on Diffraction Imaging Flow Cytometry Technique. Biomed. Opt. Express BOE 2014, 5, 2172–2183. [Google Scholar] [CrossRef] [Green Version]
- Boulares, A.H.; Yakovlev, A.G.; Ivanova, V.; Stoica, B.A.; Wang, G.; Iyer, S.; Smulson, M. Role of Poly(ADP-Ribose) Polymerase (PARP) Cleavage in Apoptosis: CASPASE 3-RESISTANT PARP MUTANT INCREASES RATES OF APOPTOSIS IN TRANSFECTED CELLS*. J. Biol. Chem. 1999, 274, 22932–22940. [Google Scholar] [CrossRef] [Green Version]
- Simbulan-Rosenthal, C.M.; Rosenthal, D.S.; Hilz, H.; Hickey, R.; Malkas, L.; Applegren, N.; Wu, Y.; Bers, G.; Smulson, M.E. The Expression of Poly(ADP-Ribose) Polymerase during Differentiation-Linked DNA Replication Reveals That It Is a Component of the Multiprotein DNA Replication Complex. Biochemistry 1996, 35, 11622–11633. [Google Scholar] [CrossRef] [PubMed]
- Ponader, S.; Chen, S.-S.; Buggy, J.J.; Balakrishnan, K.; Gandhi, V.; Wierda, W.G.; Keating, M.J.; O’Brien, S.; Chiorazzi, N.; Burger, J.A. The Bruton Tyrosine Kinase Inhibitor PCI-32765 Thwarts Chronic Lymphocytic Leukemia Cell Survival and Tissue Homing in Vitro and in Vivo. Blood 2012, 119, 1182–1189. [Google Scholar] [CrossRef] [PubMed]
- Sagiv-Barfi, I.; Kohrt, H.E.; Burckhardt, L.; Czerwinski, D.K.; Levy, R. Ibrutinib Enhances the Antitumor Immune Response Induced by Intratumoral Injection of a TLR9 Ligand in Mouse Lymphoma. Blood 2015, 125, 2079–2086. [Google Scholar] [CrossRef]
- Aalipour, A.; Advani, R.H. Bruton’s Tyrosine Kinase Inhibitors and Their Clinical Potential in the Treatment of B-Cell Malignancies: Focus on Ibrutinib. Ther. Adv. Hematol. 2014, 5, 121–133. [Google Scholar] [CrossRef] [PubMed]
- Bannerman, B.; Xu, L.; Jones, M.; Tsu, C.; Yu, J.; Hales, P.; Monbaliu, J.; Fleming, P.; Dick, L.; Manfredi, M.; et al. Preclinical Evaluation of the Antitumor Activity of Bortezomib in Combination with Vitamin C or with Epigallocatechin Gallate, a Component of Green Tea. Cancer Chemother. Pharmacol. 2011, 68, 1145–1154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guzev, E.; Jadhav, S.S.; Hezkiy, E.E.; Sherman, M.Y.; Firer, M.A.; Bunimovich-Mendrazitsky, S. Validation of a Mathematical Model Describing the Dynamics of Chemotherapy for Chronic Lymphocytic Leukemia In Vivo. Cells 2022, 11, 2325. [Google Scholar] [CrossRef] [PubMed]
- Liberzon, A.; Birger, C.; Thorvaldsdóttir, H.; Ghandi, M.; Mesirov, J.P.; Tamayo, P. The Molecular Signatures Database (MSigDB) Hallmark Gene Set Collection. Cell Syst. 2015, 1, 417–425. [Google Scholar] [CrossRef] [Green Version]
- Yoo, M.; Shin, J.; Kim, J.; Ryall, K.A.; Lee, K.; Lee, S.; Jeon, M.; Kang, J.; Tan, A.C. DSigDB: Drug Signatures Database for Gene Set Analysis. Bioinformatics 2015, 31, 3069–3071. [Google Scholar] [CrossRef] [Green Version]
- Willey, C.D.; Bonner, J.A. Chapter 4—Interaction of Chemotherapy and Radiation. In Clinical Radiation Oncology, 3rd ed.; Gunderson, L.L., Tepper, J.E., Eds.; W.B. Saunders: Philadelphia, PA, USA, 2012; pp. 65–82. ISBN 978-1-4377-1637-5. [Google Scholar]
- Wang, M.; Law, M.E.; Castellano, R.K.; Law, B.K. The Unfolded Protein Response as a Target for Anticancer Therapeutics. Crit. Rev. Oncol. Hematol. 2018, 127, 66–79. [Google Scholar] [CrossRef]
- Tarjányi, O.; Haerer, J.; Vecsernyés, M.; Berta, G.; Stayer-Harci, A.; Balogh, B.; Farkas, K.; Boldizsár, F.; Szeberényi, J.; Sétáló, G. Prolonged Treatment with the Proteasome Inhibitor MG-132 Induces Apoptosis in PC12 Rat Pheochromocytoma Cells. Sci. Rep. 2022, 12, 5808. [Google Scholar] [CrossRef]
- Paradzik, T.; Bandini, C.; Mereu, E.; Labrador, M.; Taiana, E.; Amodio, N.; Neri, A.; Piva, R. The Landscape of Signaling Pathways and Proteasome Inhibitors Combinations in Multiple Myeloma. Cancers 2021, 13, 1235. [Google Scholar] [CrossRef]
- Bürkel, F.; Jost, T.; Hecht, M.; Heinzerling, L.; Fietkau, R.; Distel, L. Dual MTOR/DNA-PK Inhibitor CC-115 Induces Cell Death in Melanoma Cells and Has Radiosensitizing Potential. Int. J. Mol. Sci. 2020, 21, 9321. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hezkiy, E.E.; Kumar, S.; Gahramanov, V.; Yaglom, J.; Hesin, A.; Jadhav, S.S.; Guzev, E.; Patel, S.; Avinery, E.; Firer, M.A.; et al. Search for Synergistic Drug Combinations to Treat Chronic Lymphocytic Leukemia. Cells 2022, 11, 3671. https://doi.org/10.3390/cells11223671
Hezkiy EE, Kumar S, Gahramanov V, Yaglom J, Hesin A, Jadhav SS, Guzev E, Patel S, Avinery E, Firer MA, et al. Search for Synergistic Drug Combinations to Treat Chronic Lymphocytic Leukemia. Cells. 2022; 11(22):3671. https://doi.org/10.3390/cells11223671
Chicago/Turabian StyleHezkiy, Eleonora Ela, Santosh Kumar, Valid Gahramanov, Julia Yaglom, Arkadi Hesin, Suchita Suryakant Jadhav, Ekaterina Guzev, Shivani Patel, Elena Avinery, Michael A. Firer, and et al. 2022. "Search for Synergistic Drug Combinations to Treat Chronic Lymphocytic Leukemia" Cells 11, no. 22: 3671. https://doi.org/10.3390/cells11223671
APA StyleHezkiy, E. E., Kumar, S., Gahramanov, V., Yaglom, J., Hesin, A., Jadhav, S. S., Guzev, E., Patel, S., Avinery, E., Firer, M. A., & Sherman, M. Y. (2022). Search for Synergistic Drug Combinations to Treat Chronic Lymphocytic Leukemia. Cells, 11(22), 3671. https://doi.org/10.3390/cells11223671