
Replicative Senescence-Associated LINE1 Methylation and LINE1-Alu Expression Levels in Human Endothelial Cells

 , ,
, ,  , ,
, , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines and Cell Culture
2.2. Biomarkers of the HUVEC and NHDF Replicative Senescence
2.3. Genome-Wide DNA Methylation Analysis
2.4. Primer Design
2.5. RNA Extraction
2.6. Quantitative RT-PCR of Mature microRNAs
2.7. mRNA Expression Level
2.8. Nucleus/cytoplasm Fractionation and DNA Extraction and Quantification
2.9. Western Blot
2.10. Statistical Analysis
3. Results
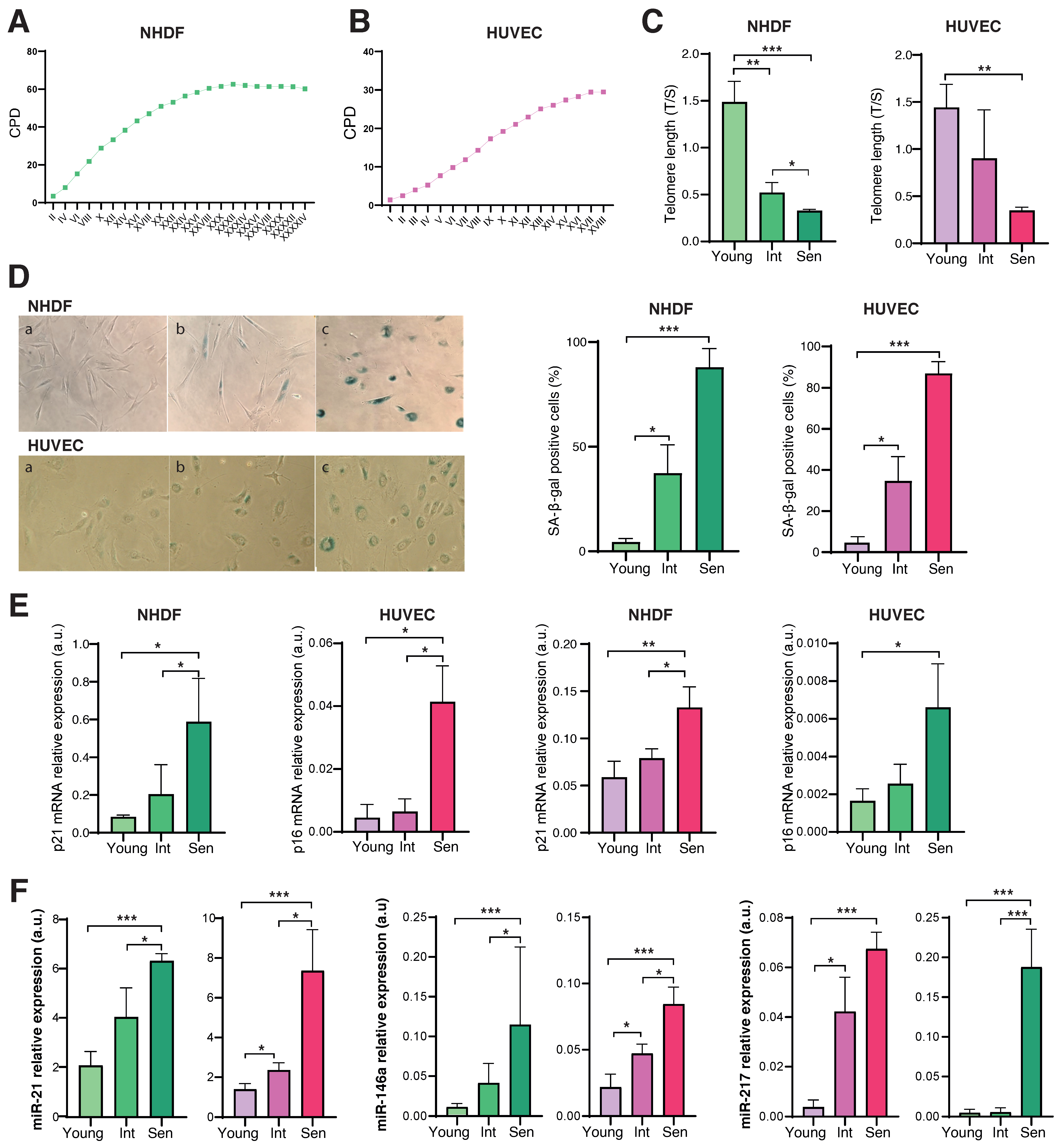
3.1. Characterization of the Senescence Status of HUVECs and NHDFs
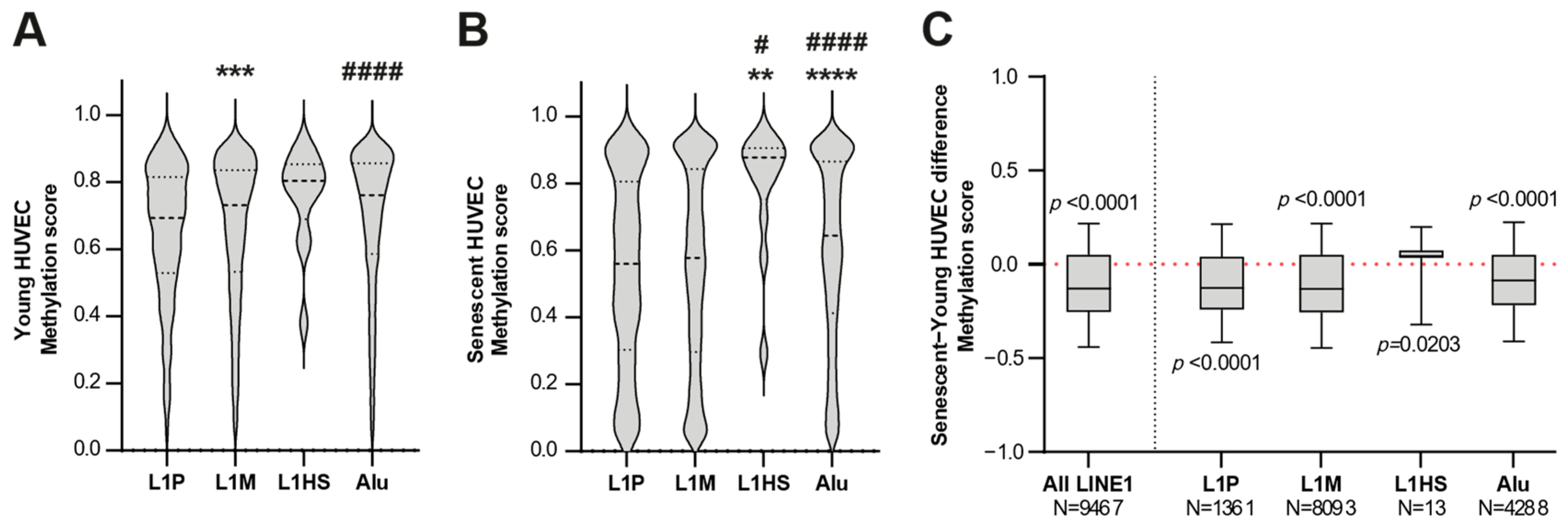
3.2. The Genome-Wide Methylation Analysis of Young and Senescent HUVECs
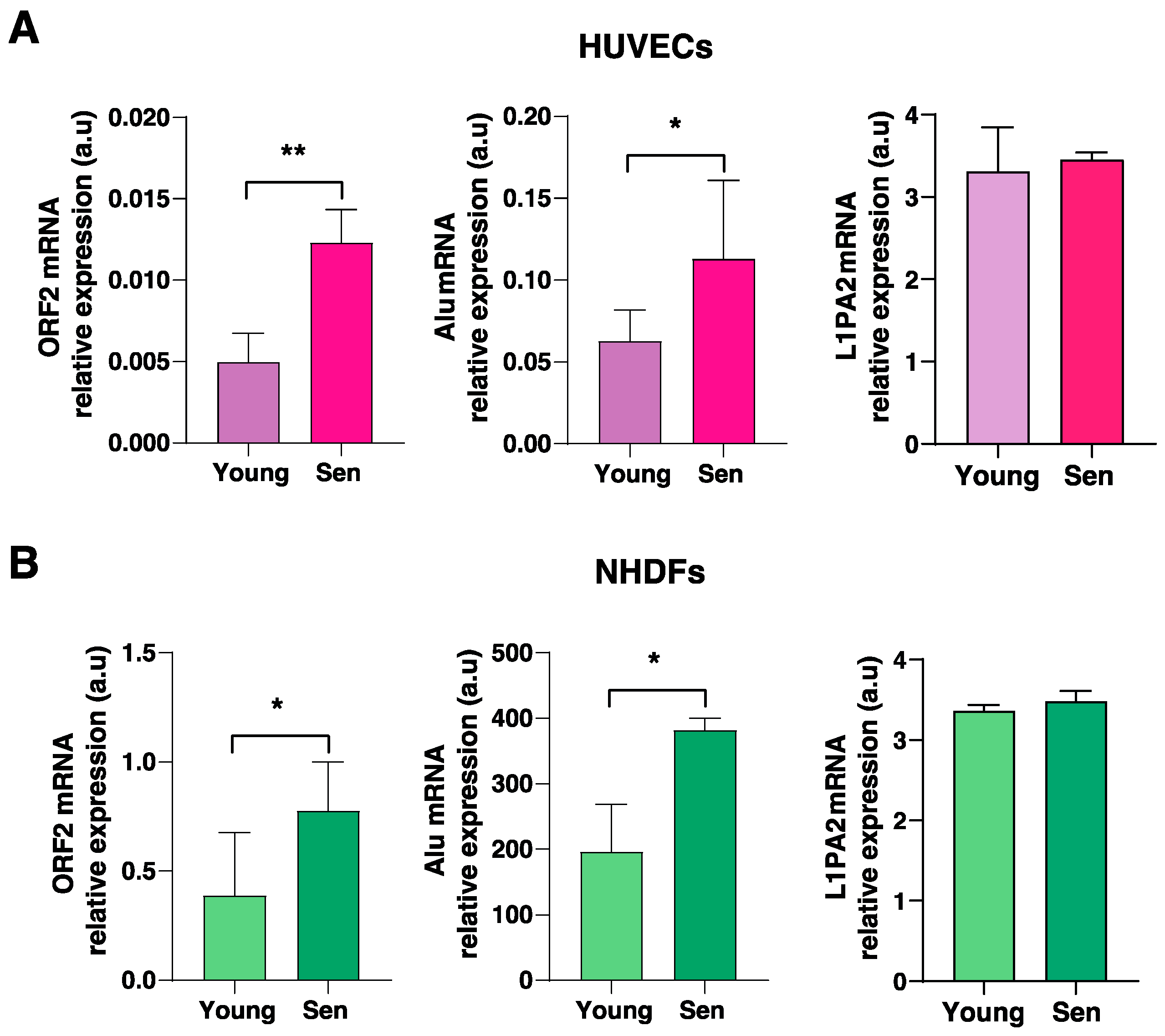
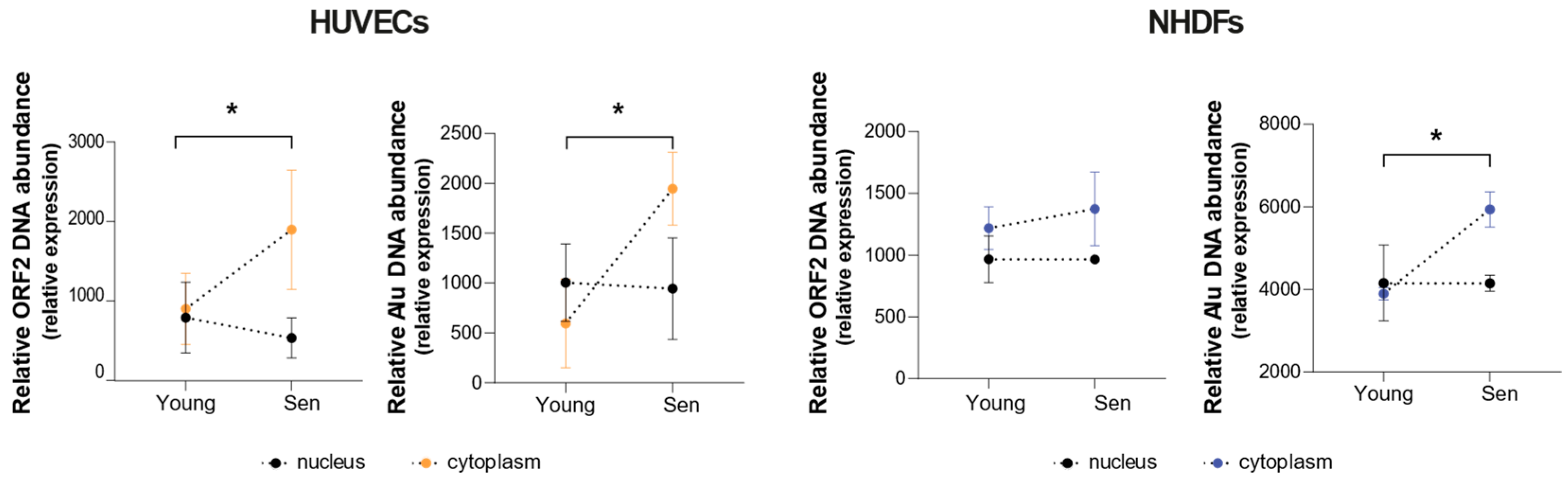
3.3. Cellular Expression and Compartmentation of Alu and LINE1 Transcripts
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Paluvai, H.; Di Giorgio, E.; Brancolini, C. The Histone Code of Senescence. Cells 2020, 9, 466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gorbunova, V.; Seluanov, A.; Mita, P.; McKerrow, W.; Fenyö, D.; Boeke, J.D.; Linker, S.B.; Gage, F.H.; Kreiling, J.A.; Petrashen, A.P.; et al. The role of retrotransposable elements in ageing and age-associated diseases. Nature 2021, 596, 43–53. [Google Scholar] [CrossRef] [PubMed]
- Bure, I.V.; Nemtsova, M.V.; Kuznetsova, E.B. Histone Modifications and Non-Coding RNAs: Mutual Epigenetic Regulation and Role in Pathogenesis. Int. J. Mol. Sci. 2022, 23, 5801. [Google Scholar] [CrossRef] [PubMed]
- Lettieri-Barbato, D.; Aquilano, K.; Punziano, C.; Minopoli, G.; Faraonio, R. MicroRNAs, Long Non-Coding RNAs, and Circular RNAs in the Redox Control of Cell Senescence. Antioxidants 2022, 11, 480. [Google Scholar] [CrossRef]
- Dan, J.; Chen, T. Genetic Studies on Mammalian DNA Methyltransferases. Adv. Exp. Med. Biol. 2016, 945, 123–150. [Google Scholar] [CrossRef]
- Horvath, S. DNA methylation age of human tissues and cell types. Genome Biol. 2013, 14, R115. [Google Scholar] [CrossRef] [Green Version]
- Bell, C.G.; Lowe, R.; Adams, P.D.; Baccarelli, A.A.; Beck, S.; Bell, J.T.; Christensen, B.C.; Gladyshev, V.N.; Heijmans, B.T.; Horvath, S.; et al. DNA methylation aging clocks: Challenges and recommendations. Genome Biol. 2019, 20, 249. [Google Scholar] [CrossRef] [Green Version]
- Hannum, G.; Guinney, J.; Zhao, L.; Zhang, L.; Hughes, G.; Sadda, S.; Klotzle, B.; Bibikova, M.; Fan, J.B.; Gao, Y.; et al. Genome-wide methylation profiles reveal quantitative views of human aging rates. Mol. Cell 2013, 49, 359–367. [Google Scholar] [CrossRef] [Green Version]
- Liu, R.; Xu, F.; Bi, S.; Zhao, X.; Jia, B.; Cen, Y. Mitochondrial DNA-Induced Inflammatory Responses and Lung Injury in Thermal Injury Murine Model: Protective Effect of Cyclosporine-A. J. Burn Care Res. 2019, 40, 355–360. [Google Scholar] [CrossRef] [Green Version]
- Jones, M.J.; Goodman, S.J.; Kobor, M.S. DNA methylation and healthy human aging. Aging. Cell 2015, 14, 924–932. [Google Scholar] [CrossRef]
- D’Aquila, P.; Montesanto, A.; Mandala, M.; Garasto, S.; Mari, V.; Corsonello, A.; Bellizzi, D.; Passarino, G. Methylation of the ribosomal RNA gene promoter is associated with aging and age-related decline. Aging Cell 2017, 16, 966–975. [Google Scholar] [CrossRef] [PubMed]
- Bacalini, M.G.; Deelen, J.; Pirazzini, C.; De Cecco, M.; Giuliani, C.; Lanzarini, C.; Ravaioli, F.; Marasco, E.; van Heemst, D.; Suchiman, H.E.D.; et al. Systemic Age-Associated DNA Hypermethylation of ELOVL2 Gene: In Vivo and In Vitro Evidences of a Cell Replication Process. J. Gerontol. A Biol. Sci. Med. Sci. 2017, 72, 1015–1023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marioni, R.E.; Suderman, M.; Chen, B.H.; Horvath, S.; Bandinelli, S.; Morris, T.; Beck, S.; Ferrucci, L.; Pedersen, N.L.; Relton, C.L.; et al. Tracking the Epigenetic Clock Across the Human Life Course: A Meta-analysis of Longitudinal Cohort Data. J. Gerontol. A Biol. Sci. Med. Sci. 2019, 74, 57–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Min, B.; Jeon, K.; Park, J.S.; Kang, Y.-K. Demethylation and derepression of genomic retroelements in the skeletal muscles of aged mice. Aging Cell 2019, 18, e13042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.; Ji, W.; Yang, J.; Yang, L.; Chen, W.; Zhuang, Z. Comparison of global DNA methylation profiles in replicative versus premature senescence. Life Sci. 2008, 83, 475–480. [Google Scholar] [CrossRef]
- Frenk, S.; Houseley, J. Gene expression hallmarks of cellular ageing. Biogerontology 2018, 19, 547–566. [Google Scholar] [CrossRef] [Green Version]
- Mensà, E.; Guescini, M.; Giuliani, A.; Bacalini, M.G.; Ramini, D.; Corleone, G.; Ferracin, M.; Fulgenzi, G.; Graciotti, L.; Prattichizzo, F.; et al. Small extracellular vesicles deliver miR-21 and miR-217 as pro-senescence effectors to endothelial cells. J. Extracell. Vesicles 2020, 9, 1725285. [Google Scholar] [CrossRef]
- Kane, A.E.; Sinclair, D.A. Epigenetic changes during aging and their reprogramming potential. Crit. Rev. Biochem. Mol. Biol. 2019, 54, 61–83. [Google Scholar] [CrossRef]
- Pal, S.; Tyler, J.K. Epigenetics and aging. Sci. Adv. 2016, 2, e1600584. [Google Scholar] [CrossRef] [Green Version]
- Yang, A.S.; Estecio, M.R.H.; Doshi, K.; Kondo, Y.; Tajara, E.H.; Issa, J.P. A simple method for estimating global DNA methylation using bisulfite PCR of repetitive DNA elements. Nucleic. Acids Res. 2004, 32, e38. [Google Scholar] [CrossRef]
- Puca, A.A.; Spinelli, C.; Accardi, G.; Villa, F.; Caruso, C. Centenarians as a model to discover genetic and epigenetic signatures of healthy ageing. Mech. Ageing Dev. 2018, 174, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Brouha, B.; Schustak, J.; Badge, R.M.; Lutz-Prigge, S.; Farley, A.H.; Moran, J.V.; Kazazian, H.H., Jr. Hot L1s account for the bulk of retrotransposition in the human population. Proc. Natl. Acad. Sci. USA 2003, 100, 5280–5285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kramerov, D.A.; Vassetzky, N.S. Origin and evolution of SINEs in eukaryotic genomes. Heredity 2011, 107, 487–495. [Google Scholar] [CrossRef]
- De Cecco, M.; Criscione, S.W.; Peterson, A.L.; Neretti, N.; Sedivy, J.M.; Kreiling, J.A. Transposable elements become active and mobile in the genomes of aging mammalian somatic tissues. Aging 2013, 5, 867–883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cardelli, M. The epigenetic alterations of endogenous retroelements in aging. Mech. Ageing Dev. 2018, 174, 30–46. [Google Scholar] [CrossRef] [PubMed]
- Sedivy, J.M.; Kreiling, J.A.; Neretti, N.; De Cecco, M.; Criscione, S.W.; Hofmann, J.W.; Zhao, X.; Ito, T.; Peterson, A.L. Death by transposition—The enemy within? Bioessays 2013, 35, 1035–1043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Cecco, M.; Ito, T.; Petrashen, A.P.; Elias, A.E.; Skvir, N.J.; Criscione, S.W.; Caligiana, A.; Brocculi, G.; Adney, E.M.; Boeke, J.D.; et al. L1 drives IFN in senescent cells and promotes age-associated inflammation. Nature 2019, 566, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Cawthon, R.M. Telomere measurement by quantitative PCR. Nucleic Acids Res. 2002, 30, e47. [Google Scholar] [CrossRef] [Green Version]
- Bonfigli, A.R.; Spazzafumo, L.; Prattichizzo, F.; Bonafe, M.; Mensa, E.; Micolucci, L.; Giuliani, A.; Fabbietti, P.; Testa, R.; Boemi, M.; et al. Leukocyte telomere length and mortality risk in patients with type 2 diabetes. Oncotarget 2016, 7, 50835–50844. [Google Scholar] [CrossRef] [Green Version]
- Giuliani, A.; Cirilli, I.; Prattichizzo, F.; Mensà, E.; Fulgenzi, G.; Sabbatinelli, J.; Graciotti, L.; Olivieri, F.; Procopio, A.D.; Tiano, L.; et al. The mitomiR/Bcl-2 axis affects mitochondrial function and autophagic vacuole formation in senescent endothelial cells. Aging 2018, 10, 2855–2873. [Google Scholar] [CrossRef]
- Asada, K.; Ito, K.; Yui, D.; Tagaya, H.; Yokota, T. Cytosolic Genomic DNA functions as a Natural Antisense. Sci. Rep. 2018, 8, 8551. [Google Scholar] [CrossRef] [PubMed]
- Olivieri, F.; Lazzarini, R.; Babini, L.; Prattichizzo, F.; Rippo, M.R.; Tiano, L.; Di Nuzzo, S.; Graciotti, L.; Festa, R.; Brugè, F.; et al. Anti-inflammatory effect of ubiquinol-10 on young and senescent endothelial cells via miR-146a modulation. Free Radic. Biol. Med. 2013, 63, 410–420. [Google Scholar] [CrossRef] [PubMed]
- Pinson, M.-E.; Court, F.; Masson, A.; Renaud, Y.; Fantini, A.; Bacoeur-Ouzillou, O.; Barriere, M.; Pereira, B.; Guichet, P.-O.; Chautard, E.; et al. L1 chimeric transcripts are expressed in healthy brain and their deregulation in glioma follows that of their host locus. Hum. Mol. Genet. 2022, 31, 2606–2622. [Google Scholar] [CrossRef] [PubMed]
- Jordà, M.; Diez-Villanueva, A.; Mallona, I.; Martin, B.; Lois, S.; Barrera, V.; Esteller, M.; Vavouri, T.; Peinado, M.A. The epigenetic landscape of Alu repeats delineates the structural and functional genomic architecture of colon cancer cells. Genome Res. 2017, 27, 118–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phokaew, C.; Kowudtitham, S.; Subbalekha, K.; Shuangshoti, S.; Mutirangura, A. LINE-1 methylation patterns of different loci in normal and cancerous cells. Nucleic Acids Res. 2008, 36, 5704–5712. [Google Scholar] [CrossRef] [PubMed]
- Simon, M.; Van Meter, M.; Ablaeva, J.; Ke, Z.; Gonzalez, R.S.; Taguchi, T.; De Cecco, M.; Leonova, K.I.; Kogan, V.; Helfand, S.L.; et al. LINE1 Derepression in Aged Wild-Type and SIRT6-Deficient Mice Drives Inflammation. Cell Metab. 2019, 29, 871–885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maugeri, A.; Barchitta, M.; Lio, R.M.S.; Favara, G.; La Mastra, C.; La Rosa, M.C.; Agodi, A. The Relationship between Body Mass Index, Obesity, and LINE-1 Methylation: A Cross-Sectional Study on Women from Southern Italy. Dis. Mrk. 2021, 2021, 9910878. [Google Scholar] [CrossRef]
- Pearce, M.S.; McConnell, J.C.; Potter, C.; Barrett, L.M.; Parker, L.; Mathers, J.C.; Relton, C.L. Global LINE-1 DNA methylation is associated with blood glycaemic and lipid profiles. Int. J. Epidemiol. 2012, 41, 210–217. [Google Scholar] [CrossRef] [Green Version]
- Martin-Ruiz, C.M.; Baird, D.; Roger, L.; Boukamp, P.; Krunic, D.; Cawthon, R.; Dokter, M.M.; van der Harst, P.; Bekaert, S.; de Meyer, T.; et al. Reproducibility of telomere length assessment: An international collaborative study. Int. J. Epidemiol. 2015, 44, 1673–1683. [Google Scholar] [CrossRef]
- Cardelli, M.; van Doorn, R.; Larcher, L.; Di Donato, M.; Piacenza, F.; Pierpaoli, E.; Giacconi, R.; Malavolta, M.; Rachakonda, S.; Gruis, N.A.; et al. Association of HERV-K and LINE-1 hypomethylation with reduced disease-free survival in melanoma patients. Epigenomics 2020, 12, 1689–1706. [Google Scholar] [CrossRef]
- Muse, M.E.; Bergman, D.T.; Salas, L.A.; Tom, L.N.; Tan, J.M.; Laino, A.; Lambie, D.; Sturm, R.A.; Schaider, H.; Soyer, H.P.; et al. Genome-Scale DNA Methylation Analysis Identifies Repeat Element Alterations that Modulate the Genomic Stability of Melanocytic Nevi. J. Investig. Dermatol. 2022, 142, 1893–1902. [Google Scholar] [CrossRef] [PubMed]
- Bravo, J.I.; Nozownik, S.; Danthi, P.S.; Benayoun, B.A. Transposable elements, circular RNAs and mitochondrial transcription in age-related genomic regulation. Development 2020, 147, dev175786. [Google Scholar] [CrossRef] [PubMed]
- Justice, J.N.; Ferrucci, L.; Newman, A.B.; Aroda, V.R.; Bahnson, J.L.; Divers, J.; Espeland, M.A.; Marcovina, S.; Pollak, M.N.; Kritchevsky, S.B.; et al. A framework for selection of blood-based biomarkers for geroscience-guided clinical trials: Report from the TAME Biomarkers Workgroup. Geroscience 2018, 40, 419–436. [Google Scholar] [CrossRef] [PubMed]
- Saito, K.; Kawakami, K.; Matsumoto, I.; Oda, M.; Watanabe, G.; Minamoto, T. Long Interspersed Nuclear Element 1 Hypomethylation Is a Marker of Poor Prognosis in Stage IA Non–Small Cell Lung CancerLINE-1 Methylation and Prognosis in NSCLC. Clin. Cancer Res. 2010, 16, 2418–2426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stenz, L. The L1-dependant and Pol III transcribed Alu retrotransposon, from its discovery to innate immunity. Mol. Biol. Rep. 2021, 48, 2775–2789. [Google Scholar] [CrossRef]
- Colombo, A.R.; Elias, H.K.; Ramsingh, G. Senescence induction universally activates transposable element expression. Cell Cycle 2018, 17, 1846–1857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Capone, S.; Connor, K.M.; Colombo, A.; Li, X.; Triche, T.J., Jr.; Ramsingh, G. Senescent human hematopoietic progenitors show elevated expression of transposable elements and inflammatory genes. Exp. Hematol. 2018, 62, 33–38. [Google Scholar] [CrossRef]
- Ardeljan, D.; Taylor, M.S.; Ting, D.T.; Burns, K.H. The Human Long Interspersed Element-1 Retrotransposon: An Emerging Biomarker of Neoplasia. Clin. Chem. 2017, 63, 816–822. [Google Scholar] [CrossRef] [Green Version]
- Wallace, N.A.; Belancio, V.P.; Deininger, P.L. L1 mobile element expression causes multiple types of toxicity. Gene 2008, 419, 75–81. [Google Scholar] [CrossRef] [Green Version]
- Chan, C.-P.; Jin, D.-Y. Cytoplasmic RNA sensors and their interplay with RNA-binding partners in innate antiviral response: Theme and variations. RNA 2022, 28, 449–477. [Google Scholar] [CrossRef]
- Hopfner, K.-P.; Hornung, V. Molecular mechanisms and cellular functions of cGAS-STING signalling. Nat. Rev. Mol. Cell Biol. 2020, 21, 501–521. [Google Scholar] [CrossRef] [PubMed]
- Andrenacci, D.; Cavaliere, V.; Lattanzi, G. The role of transposable elements activity in aging and their possible involvement in laminopathic diseases. Ageing Res. Rev. 2020, 57, 100995. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chromosomal Location | CpG IDs | Repeat Masker Annotation | DNAm Young HUVEC | DNAm Senescent HUVEC | DNAm Difference (Senescent − Young) | p-Value | Adjusted BH p-Value |
|---|---|---|---|---|---|---|---|
| Chr3: complement (127793383-4) | cg13228884 | L1ME2z | 0.703 | 0.045 | 0.658 ↓ | 1.85 × 10−8 | 1.04 × 10−4 |
| Chr4: complement 84255411-2 | cg25428494 | L1PB4 | 0.693 | 0.077 | 0.616 ↓ | 1.91 × 10−8 | 1.01 × 10−4 |
| Chr5: complement (180561011-2) | cg15102731 | L1MEg | 0.879 | 0.092 | 0.787 ↓ | 4.13 × 10−9 | 8.82 × 10−5 |
| Chr7: (80642464-5) | cg17270475 | L1MEg1 | 0.192 | 0.936 | 0.744 ↑ | 3.52 × 10−8 | 1.17 × 10−4 |
| Chr7: complement (157706117-8) | cg13117953 | L1ME3E | 0.605 | 0.039 | 0.566 ↓ | 8.52 × 10−10 | 6.65 × 10−5 |
| Chr13: complement (113707783-4) | cg00245856 | L1MC4 | 0.862 | 0.107 | 0.755 ↓ | 4.89 × 10−8 | 1.25 × 10−4 |
| Chr14: complement (46358139-40) | cg24170212 | L1PA2 | 0.891 | 0.422 | 0.469 ↓ | 1.19 × 10−8 | 9.97 × 10−5 |
| Chr15: complement (22562214-5) | cg07397108 | L1MEf | 0.636 | 0.152 | 0.484 ↓ | 4.41 × 10−8 | 1.21 × 10−4 |
| Chr17: complement (33780343-4) | cg06138714 | L1ME1 | 0.736 | 0.036 | 0.700 ↓ | 1.15 × 10−8 | 9.97 × 10−5 |
| Chr17: complement (45946982-3) | cg06085038 | L1ME3A | 0.823 | 0.616 | 0.207 ↓ | 1.15 × 10−8 | 9.97 × 10−5 |
| Chr20: complement (58106111-2) | cg19748181 | L1MC1 | 0.889 | 0.109 | 0.780 ↓ | 4.14 × 10−8 | 1.20 × 10−4 |
| Chr21: complement (19195209-10) | cg25387466 | L1MEc | 0.729 | 0.121 | 0.608 ↓ | 1.10 × 10−8 | 9.97 × 10−5 |
| Chr1: complement (158904784-5) | cg25464787 | AluSq2 | 0.822 | 0.088 | 0.734 ↓ | 9.41 × 10−9 | 9.97 × 10−5 |
| Chr7: complement (1253999-4000) | cg15877936 | AluSg | 0.767 | 0.195 | 0.572 ↓ | 2.97 × 10−8 | 1.14 × 10−4 |
| Chr16: complement (8974513-4) | cg06611212 | AluSp | 0.810 | 0.588 | 0.222 ↓ | 4.42 × 10−8 | 1.21 × 10−4 |
| Low Gene Density | Intermediate Gene Density | High Gene Density | Total | Z-Test (Low Gene Density) | |
|---|---|---|---|---|---|
| Demethylated LINE 1 CpGs | 1046 (16.6%) | 4528 (72.0%) | 717 (11.4%) | 6291 (100%) | p < 0.00001 |
| All LINE1 CpGs | 3457 (12.1%) | 22,076 (77.5%) | 2940 (10.3%) | 28,473 (100%) | Ref. |
| Demethylated L1M CpGs | 775 (14.5%) | 3936 (73.6%) | 636 (11.9%) | 5347 (100%) | p < 0.00001 |
| All L1M CpGs | 2330 (9.6%) | 19,246 (79.4%) | 2668 (11.0%) | 24,244 (100%) | Ref. |
| Demethylated L1P CpGs | 271 (28.7%) | 591 (62.7%) | 81 (8.6%) | 943 (100%) | p = 0.136 |
| All L1P CpGs | 1099 (26.4%) | 2798 (67.1%) | 272 (6.5%) | 4169 (100%) | Ref. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ramini, D.; Latini, S.; Giuliani, A.; Matacchione, G.; Sabbatinelli, J.; Mensà, E.; Bacalini, M.G.; Garagnani, P.; Rippo, M.R.; Bronte, G.; et al. Replicative Senescence-Associated LINE1 Methylation and LINE1-Alu Expression Levels in Human Endothelial Cells. Cells 2022, 11, 3799. https://doi.org/10.3390/cells11233799
Ramini D, Latini S, Giuliani A, Matacchione G, Sabbatinelli J, Mensà E, Bacalini MG, Garagnani P, Rippo MR, Bronte G, et al. Replicative Senescence-Associated LINE1 Methylation and LINE1-Alu Expression Levels in Human Endothelial Cells. Cells. 2022; 11(23):3799. https://doi.org/10.3390/cells11233799
Chicago/Turabian StyleRamini, Deborah, Silvia Latini, Angelica Giuliani, Giulia Matacchione, Jacopo Sabbatinelli, Emanuela Mensà, Maria Giulia Bacalini, Paolo Garagnani, Maria Rita Rippo, Giuseppe Bronte, and et al. 2022. "Replicative Senescence-Associated LINE1 Methylation and LINE1-Alu Expression Levels in Human Endothelial Cells" Cells 11, no. 23: 3799. https://doi.org/10.3390/cells11233799