
Pathophysiology of Disseminated Intravascular Coagulation in Sepsis: A Clinically Focused Overview

 , , and
, , and 
Abstract
1. Introduction
2. Understanding the Role of Bacterial Virulence in the Pathogenesis of Sepsis and Associated Conditions
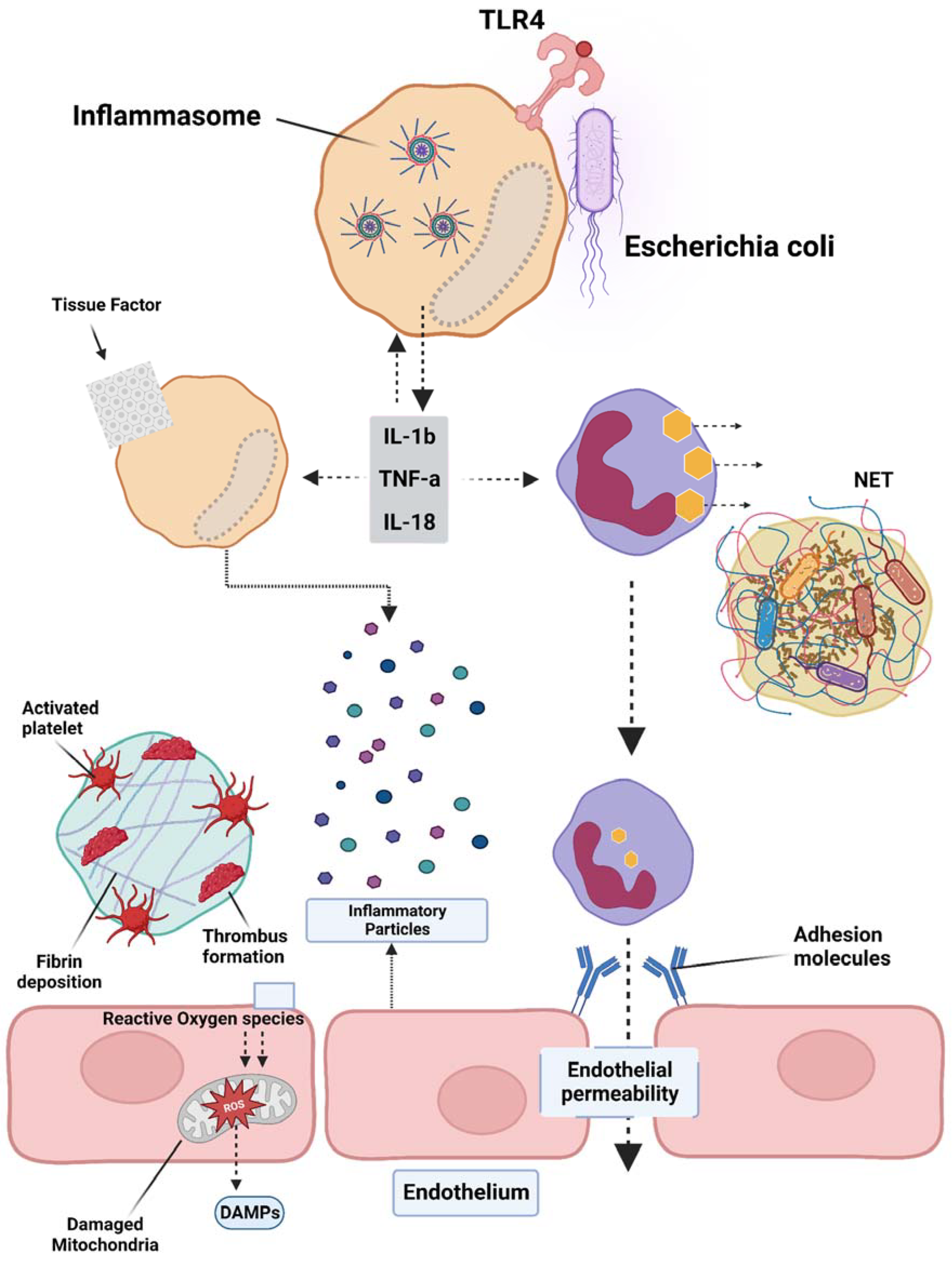
3. Inflammatory Mediators Associated with Pyroptosis and Their Role in Subsequent Coagulation Disorders
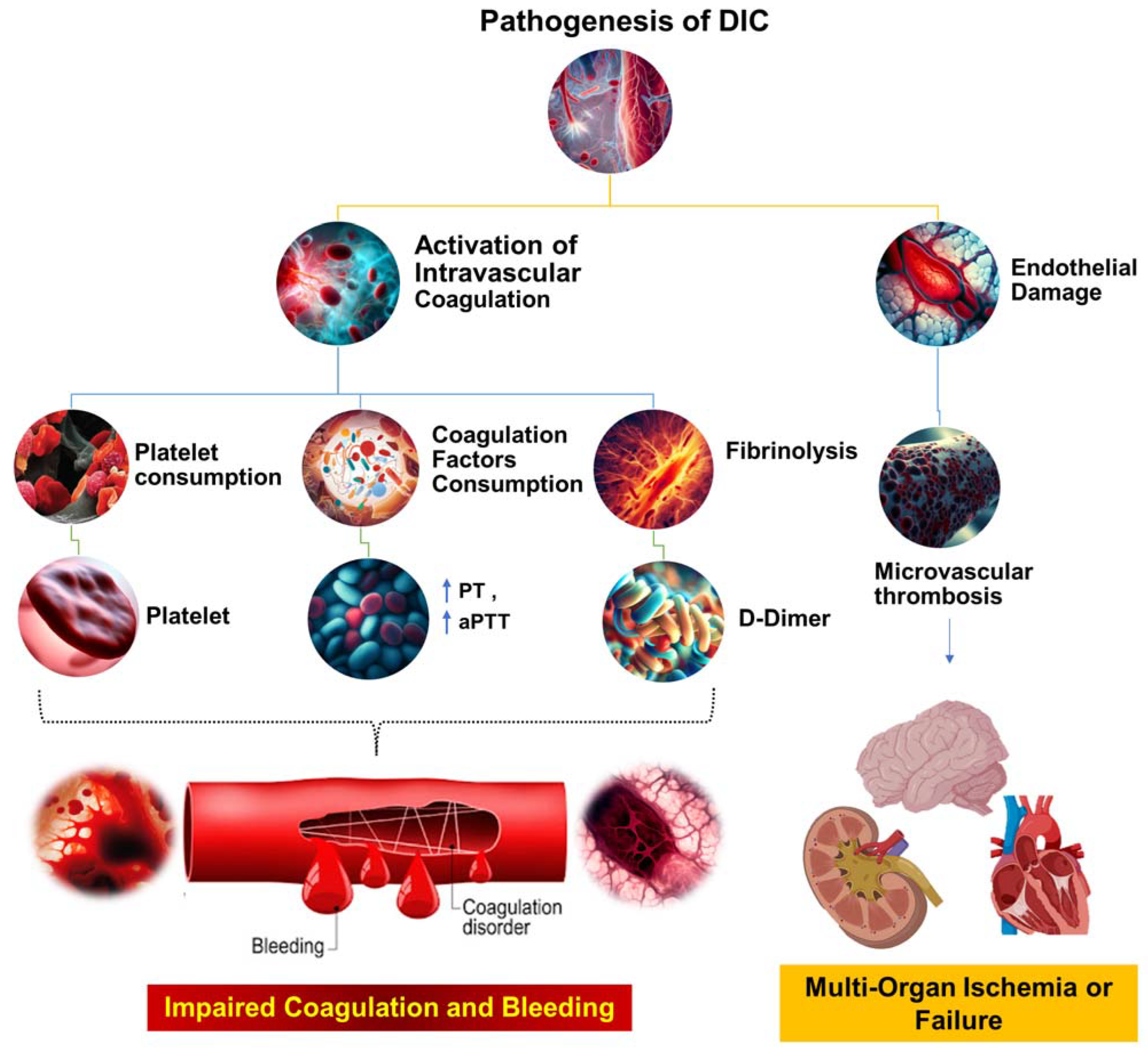
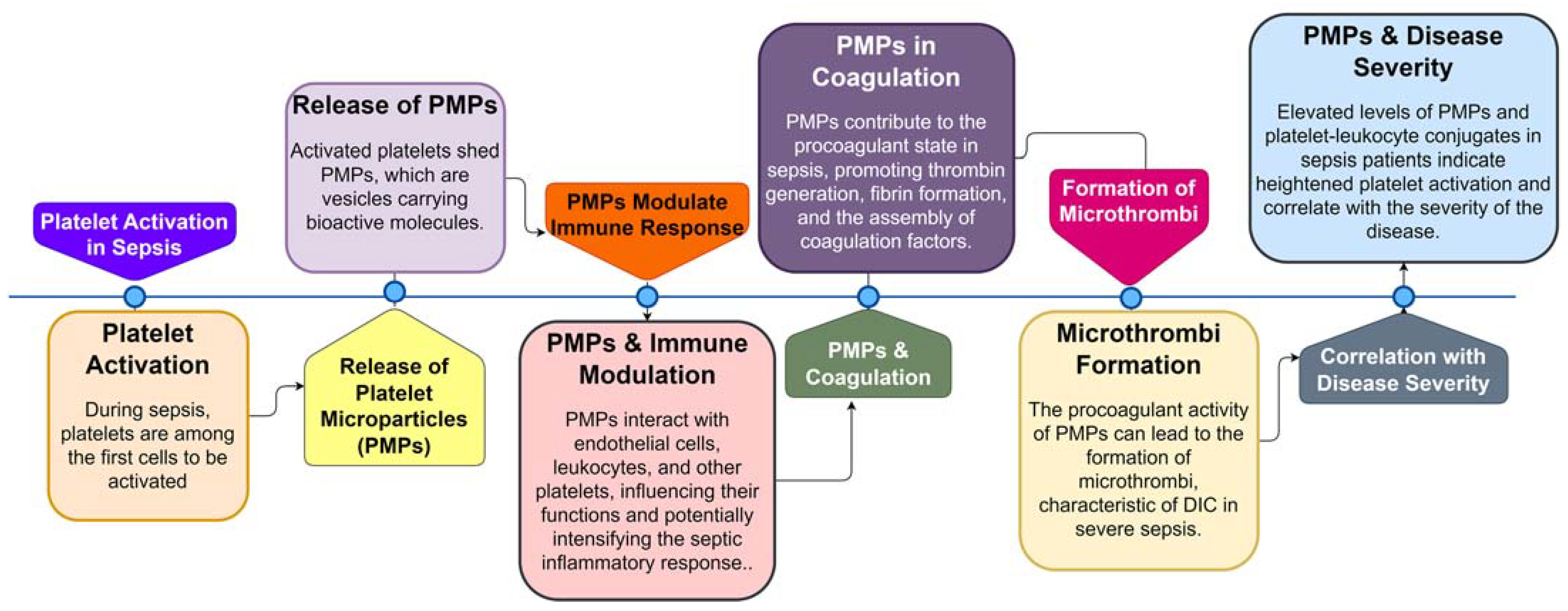
4. Platelet Activation and Its Effects on the Coagulation Cascade in Sepsis
5. Tissue Factor Pathway Inhibitor (TFPI) and Fibrin Deposition during Sepsis
6. Thrombin-Activated Fibrinolysis Inhibitor (TAFI) and Its Role in Sepsis
7. Epigenetic Alterations and Immunosuppressive Immune Cell Phenotypes
8. The Role of Extracellular Nuclear Products in Sepsis, Coagulation Disorders, and Thrombosis
9. Unraveling the Interplay of Sepsis, SARS-CoV-2, and Flaviviruses: A Comparative Analysis of Molecular Pathogenesis and Controversial Therapeutic Implications
10. Research Progress in the Treatment of Sepsis-related DIC
11. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Nesheim, M.E.; Tracy, R.P.; Mann, K.G. “Clotspeed,” a Mathematical Simulation of the Functional Properties of Prothrom-binase. J. Biol. Chem. 1984, 259, 1447–1453. [Google Scholar] [CrossRef] [PubMed]
- Martinod, K.; Wagner, D.D. Thrombosis: Tangled up in NETs. Blood 2014, 123, 2768–2776. [Google Scholar] [CrossRef]
- Ito, T.; Kawahara, K.; Nakamura, T.; Yamada, S.; Nakamura, T.; Abeyama, K.; Hashiguchi, T.; Maruyama, I. High-mobility group box 1 protein promotes development of microvascular thrombosis in rats. J. Thromb. Haemost. 2007, 5, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Lv, B.; Wang, H.; Tang, Y.; Fan, Z.; Xiao, X.; Chen, F. High-Mobility Group Box 1 Protein Induces Tissue Factor Expression in Vascular Endothelial Cells via Activation of NF-ΚB and Egr-1. Thromb. Haemost. 2009, 102, 352–359. [Google Scholar]
- Maugeri, N.; Campana, L.; Gavina, M.; Covino, C.; De Metrio, M.; Panciroli, C.; Maiuri, L.; Maseri, A.; D’Angelo, A.; Bianchi, M.E.; et al. Activated platelets present high mobility group box 1 to neutrophils, inducing autophagy and promoting the extrusion of neutrophil extracellular traps. J. Thromb. Haemost. 2014, 12, 2074–2088. [Google Scholar] [CrossRef] [PubMed]
- Deng, M.; Tang, Y.; Li, W.; Wang, X.; Zhang, R.; Zhang, X.; Zhao, X.; Liu, J.; Tang, C.; Liu, Z.; et al. The Endotoxin Delivery Protein HMGB1 Mediates Caspase-11-Dependent Lethality in Sepsis. Immunity 2018, 49, 740–753.e7. [Google Scholar] [CrossRef]
- Chang, J. COVID-19 Sepsis: Pathogenesis and Endothelial Molecular Mechanisms Based on “Two-Path Unifying Theory” of Hemostasis and Endotheliopathy-Associated Vascular Microthrombotic Disease, and Proposed Therapeutic Approach with Antimicrothrombotic Therapy. Vasc. Health Risk Manag. 2021, ume 17, 273–298. [Google Scholar] [CrossRef]
- Unar, A.; Imtiaz, M.; Trung, T.T.; Rafiq, M.; Fatmi, M.Q.; Jafar, T.H. Structural and Functional Analyses of SARS COV-2 RNA-Dependent RNA Polymerase Protein and Complementary vs. Synthetic Drugs against COVID-19 and the Exploration of Binding Sites for Docking, Molecular Dynamics Simulation, and Density Functional Theory Studies. Curr. Bioinform. 2022, 17, 632–656. [Google Scholar]
- Zelek, W.M.; Harrison, R.A. Complement and COVID-19: Three years on, what we know, what we don’t know, and what we ought to know. Immunobiology 2023, 228, 152393. [Google Scholar] [CrossRef] [PubMed]
- Aboudounya, M.M.; Heads, R.J. COVID-19 and Toll-Like Receptor 4 (TLR4): SARS-CoV-2 May Bind and Activate TLR4 to Increase ACE2 Expression, Facilitating Entry and Causing Hyperinflammation. Mediat. Inflamm. 2021, 2021, 1–18. [Google Scholar] [CrossRef]
- Florescu, D.F.; Kalil, A.C. Cytomegalovirus infections in non-immunocompromised and immunocompromised patients in the intensive care unit. Infect. Disord.-Drug Targets 2011, 11, 354–364. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, K.; Tamura, T.; Sawatsubashi, Y. Sepsis and disseminated intravascular coagulation. J. Intensiv. Care 2016, 4, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Wada, H.; Thachil, J.; Di Nisio, M.; Mathew, P.; Kurosawa, S.; Gando, S.; Kim, H.K.; Nielsen, J.D.; Dempfle, C.-E.; Levi, M.; et al. Guidance for diagnosis and treatment of DIC from harmonization of the recommendations from three guidelines. J. Thromb. Haemost. 2013, 11, 761–767. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.-L.; Wang, X.-Z.; Liu, X.-X.; Hao, D.; Jaladat, Y.; Lu, F.; Sun, T.; Lv, C.-J. Low-dose heparin as treatment for early disseminated intravascular coagulation during sepsis: A prospective clinical study. Exp. Ther. Med. 2013, 7, 604–608. [Google Scholar] [CrossRef] [PubMed]
- Aikawa, N.; Shimazaki, S.; Yamamoto, Y.; Saito, H.; Maruyama, I.; Ohno, R.; Hirayama, A.; Aoki, Y.; Aoki, N. Thrombomodulin Alfa in the Treatment of Infectious Patients Complicated by Disseminated Intravascular Coagulation: Subanalysis from the Phase 3 Trial. J. Shock 2011, 35, 349–354. [Google Scholar] [CrossRef] [PubMed]
- Iba, T.; Gando, S.; Thachil, J. Anticoagulant therapy for sepsis-associated disseminated intravascular coagulation: The view from Japan. J. Thromb. Haemost. 2014, 12, 1010–1019. [Google Scholar] [CrossRef] [PubMed]
- Dhainaut, J.F.; Yan, S.B.; Joyce, D.E.; Pettilä, V.; Basson, B.; Brandt, J.T.; Sundin, D.P.; Levi, M. Treatment effects of drotrecogin alfa (activated) in patients with severe sepsis with or without overt disseminated intravascular coagulation. J. Thromb. Haemost. 2004, 2, 1924–1933. [Google Scholar] [CrossRef]
- Di Nisio, M.; Baudo, F.; Cosmi, B.; D’Angelo, A.; De Gasperi, A.; Malato, A.; Schiavoni, M.; Squizzato, A. Diagnosis and treatment of disseminated intravascular coagulation: Guidelines of the Italian Society for Haemostasis and Thrombosis (SISET). Thromb. Res. 2012, 129, e177–e184. [Google Scholar] [CrossRef]
- Aoki, N.; Matsuda, T.; Saito, H.; Takatsuki, K.; Okajima, K.; Takahashi, H.; Takamatsu, J.; Asakura, H.; Ogawa, N. A Com-parative Double-Blind Randomized Trial of Activated Protein C and Unfractionated Heparin in the Treatment of Disseminated Intravascular Coagulation. Int. J. Hematol. 2002, 75, 540–547. [Google Scholar] [CrossRef]
- Warren, B.L.; Eid, A.; Singer, P.; Pillay, S.S.; Carl, P.; Novak, I.; Chalupa, P.; Atherstone, A.; Pénzes, I.; Kübler, A. High-Dose Antithrombin III in Severe Sepsis: A Randomized Controlled Trial. JAMA 2001, 286, 1869–1878. [Google Scholar] [CrossRef]
- Tagami, T.; Matsui, H.; Horiguchi, H.; Fushimi, K.; Yasunaga, H. Antithrombin and mortality in severe pneumonia patients with sepsis-associated disseminated intravascular coagulation: An observational nationwide study. J. Thromb. Haemost. 2014, 12, 1470–1479. [Google Scholar] [CrossRef] [PubMed]
- Wiedermann, C.J. Antithrombin Concentrate Use in Disseminated Intravascular Coagulation of Sepsis: Meta-analyses Revis-ited. J. Thromb. Haemost. 2018, 16, 455–457. [Google Scholar] [CrossRef]
- Nishida, O.; Ogura, H.; Egi, M.; Fujishima, S.; Hayashi, Y.; Iba, T.; Imaizumi, H.; Inoue, S.; Kakihana, Y.; Kotani, J.; et al. The Japanese Clinical Practice Guidelines for Management of Sepsis and Septic Shock 2016 (J-SSCG 2016). J. Intensiv. Care 2018, 6, 1–77. [Google Scholar] [CrossRef]
- Vincent, J.-L.; Francois, B.; Zabolotskikh, I.; Daga, M.K.; Lascarrou, J.-B.; Kirov, M.Y.; Pettilä, V.; Wittebole, X.; Meziani, F.; Mercier, E.; et al. Effect of a Recombinant Human Soluble Thrombomodulin on Mortality in Patients With Sepsis-Associated Coagulopathy. JAMA 2019, 321, 1993–2002. [Google Scholar] [CrossRef] [PubMed]
- Cruz, D.N.; Antonelli, M.; Fumagalli, R.; Foltran, F.; Brienza, N.; Donati, A.; Malcangi, V.; Petrini, F.; Volta, G.; Pallavicini, F.M.B. Early Use of Polymyxin B Hemoperfusion in Abdominal Septic Shock: The EUPHAS Randomized Controlled Trial. JAMA 2009, 301, 2445–2452. [Google Scholar] [CrossRef] [PubMed]
- Payen, D.M.; The ABDOMIX Group; Guilhot, J.; Launey, Y.; Lukaszewicz, A.C.; Kaaki, M.; Veber, B.; Pottecher, J.; Joannes-Boyau, O.; Martin-Lefevre, L.; et al. Early use of polymyxin B hemoperfusion in patients with septic shock due to peritonitis: A multicenter randomized control trial. Intensiv. Care Med. 2015, 41, 975–984. [Google Scholar] [CrossRef]
- Fu, S.; Yu, S.; Wang, L.; Ma, X.; Li, X. Unfractionated heparin improves the clinical efficacy in adult sepsis patients: A systematic review and meta-analysis. BMC Anesthesiol. 2022, 22, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Murata, A.; Okamoto, K.; Mayumi, T.; Muramatsu, K.; Matsuda, S. Recent Change in Treatment of Disseminated Intravascular Coagulation in Japan. Clin. Appl. Thromb. 2015, 22, 21–27. [Google Scholar] [CrossRef]
- Egi, M.; Ogura, H.; Yatabe, T.; Atagi, K.; Inoue, S.; Iba, T.; Kakihana, Y.; Kawasaki, T.; Kushimoto, S.; Kuroda, Y.; et al. The Japanese Clinical Practice Guidelines for Management of Sepsis and Septic Shock 2020 (J-SSCG 2020). Acute Med. Surg. 2021, 8. [Google Scholar] [CrossRef]
- Tagami, T. Antithrombin concentrate use in sepsis-associated disseminated intravascular coagulation: Re-evaluation of a ‘pendulum effect’ drug using a nationwide database. J. Thromb. Haemost. 2018, 16, 458–461. [Google Scholar] [CrossRef]
- Fitzgerald, J.R.; Foster, T.J.; Cox, D. The interaction of bacterial pathogens with platelets. Nat. Rev. Genet. 2006, 4, 445–457. [Google Scholar] [CrossRef] [PubMed]
- Taylor, F.B., Jr.; Toh, C.-H.; Hoots, K.W.; Wada, H.; Levi, M.; Scientific Subcommittee on Disseminated Intravascular Coagu-lation (DIC) of the International Society on Thrombosis and Haemostasis (ISTH). Towards Definition, Clinical and Laboratory Criteria, and a Scoring System for Disseminated Intravascular Coagulation. Thromb. Haemost. 2001, 86, 1327–1330. [Google Scholar] [CrossRef] [PubMed]
- Iba, T.; Connors, J.M.; Nagaoka, I.; Levy, J.H. Recent advances in the research and management of sepsis-associated DIC. Int. J. Hematol. 2021, 113, 24–33. [Google Scholar] [CrossRef] [PubMed]
- Singer, M.; Deutschman, C.S.; Seymour, C.W.; Shankar-Hari, M.; Annane, D.; Bauer, M.; Bellomo, R.; Bernard, G.R.; Chiche, J.-D.; Coopersmith, C.M.; et al. The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA 2016, 315, 801–810. [Google Scholar] [CrossRef] [PubMed]
- Assimakopoulos, S.F.; Eleftheriotis, G.; Lagadinou, M.; Karamouzos, V.; Dousdampanis, P.; Siakallis, G.; Marangos, M. SARS CoV-2-Induced Viral Sepsis: The Role of Gut Barrier Dysfunction. Microorganisms 2022, 10, 1050. [Google Scholar] [CrossRef]
- Talan, L.; Kalkan, I.A.; Altıntaş, N.D.; Yörük, F. Cytomegalovirus Reactivation in Critically-ill COVID-19 Patients. Balk. Med. J. 2022, 39, 301–302. [Google Scholar] [CrossRef]
- Walton, A.H.; Muenzer, J.T.; Rasche, D.; Boomer, J.S.; Sato, B.; Brownstein, B.H.; Pachot, A.; Brooks, T.L.; Deych, E.; Shannon, W.D.; et al. Reactivation of Multiple Viruses in Patients with Sepsis. PLoS ONE 2014, 9, e98819. [Google Scholar] [CrossRef]
- Almansa, R.; Eiros, J.M.; de Gonzalo-Calvo, D.; Lopez-Izquierdo, R.; Moncusí-Moix, A.; Gort-Paniello, C.; de la Fuente, A.; Gonzalez-Gonzalez, L.; Rodríguez-Jara, F.; Jorge, N.; et al. Antigenemia Is Associated to Viral Sepsis and Mortality in COVID-19. SSRN Electron. J. 2021. [Google Scholar] [CrossRef]
- Barrows, N.J.; Campos, R.K.; Liao, K.-C.; Prasanth, K.R.; Soto-Acosta, R.; Yeh, S.-C.; Schott-Lerner, G.; Pompon, J.; Sessions, O.M.; Bradrick, S.S.; et al. Biochemistry and Molecular Biology of Flaviviruses. Chem. Rev. 2018, 118, 4448–4482. [Google Scholar] [CrossRef]
- Anh, N.T.; Chi Minh City, H.; Nam, V. Viral Etiology of Central Nervous System Infections and Community-Acquired Sepsis in Southeast Asia: Unravelling the Unknown. Ph.D. Thesis, The Open University, Milton Keynes, UK, 2021. [Google Scholar]
- van der Poll, T.; van de Veerdonk, F.L.; Scicluna, B.P.; Netea, M.G. The Immunopathology of Sepsis and Potential Therapeutic Targets. Nat. Rev. Immunol. 2017, 17, 407–420. [Google Scholar] [CrossRef]
- Cunningham, R.; Cockayne, A.; Humphreys, H. Clinical and molecular aspects of the pathogenesis of Staphylococcus aureus bone and joint infections. J. Med Microbiol. 1996, 44, 157–164. [Google Scholar] [CrossRef]
- Werdan, K.; Dietz, S.; Loeffler, B.; Niemann, S.; Bushnaq, H.; Silber, R.-E.; Peters, G.; Mueller-Werdan, U. Mechanisms of Infective Endocarditis: Pathogen–Host Interaction and Risk States. Nat. Rev. Cardiol. 2014, 11, 35–50. [Google Scholar] [CrossRef] [PubMed]
- Langerak, T.; Van Gorp, E.C.; Levi, M.; Cate, H.T. Crosstalk of Inflammation and Coagulation in Infectious Disease and Their Roles in Disseminated Intravascular Coagulation. In Consultative Hemostasis and Thrombosis; Elsevier: Amsterdam, The Netherlands, 2018; pp. 226–240. [Google Scholar] [CrossRef]
- Prazanowski, M.; Kur, B.; Barańska, M.; Lutz, W.; Piłacik, B.; Kolaciński, Z. The Leiden Mutation and Activated Protein C Resistance as Risk Factors for Disseminated Intravascular Coagulation in Acutely Poisoned Patients. Clin. Toxicol. 2006, 44, 53–57. [Google Scholar] [CrossRef] [PubMed]
- Rhodes, A.; Evans, L.E.; Alhazzani, W.; Levy, M.M.; Antonelli, M.; Ferrer, R.; Kumar, A.; Sevransky, J.E.; Sprung, C.L.; Nunnally, M.E.; et al. Surviving Sepsis Campaign: International Guidelines for Management of Sepsis and Septic Shock: 2016. Intensive Care Med. 2017, 43, 304–377. [Google Scholar] [CrossRef] [PubMed]
- Yildirim, F.; Yildirim, M. Is It Sufficient to Evaluate COVID-19 Infected Critically Ill Patients Only in Terms of VTE Risk Factors? What about Disease Severity? Clin. Respir. J. 2022, 16, 533. [Google Scholar] [CrossRef] [PubMed]
- Hofstra, J.-J.; Schouten, M.; Levi, M. Thrombophilia as a Risk Factor for Outcome in Sepsis. In Intensive Care Medicine: Annual Update; Springer: New York, NY, USA, 2008; pp. 713–720. [Google Scholar]
- Tikkanen, M. Placental abruption: Epidemiology, risk factors and consequences. Acta Obstet. Gynecol. Scand. 2011, 90, 140–149. [Google Scholar] [CrossRef]
- Cohen, J.; Vincent, J.-L.; Adhikari, N.K.J.; Machado, F.R.; Angus, D.C.; Calandra, T.; Jaton, K.; Giulieri, S.; Delaloye, J.; Opal, S.; et al. Sepsis: A roadmap for future research. Lancet Infect. Dis. 2015, 15, 581–614. [Google Scholar] [CrossRef]
- Liesenborghs, L.; Verhamme, P.; Vanassche, T. Staphylococcus aureus, master manipulator of the human hemostatic system. J. Thromb. Haemost. 2018, 16, 441–454. [Google Scholar] [CrossRef]
- Lowy, F.D. Staphylococcus aureus Infections. N. Engl. J. Med. 1998, 339, 520–532. [Google Scholar] [CrossRef]
- Chang, J.C. Disseminated intravascular coagulation: New identity as endotheliopathy-associated vascular microthrombotic disease based on in vivo hemostasis and endothelial molecular pathogenesis. Thromb. J. 2020, 18, 1–21. [Google Scholar] [CrossRef]
- Gille-Johnson, P. Diagnostic and Prognostic Markers in Sepsis; Karolinska Institutet: Stockholm, Sweden, 2013; ISBN 9798744401733. [Google Scholar]
- Ottolina, D.; Ferrari, M.; Zazzeron, L.; Scotti, E.; Stanziano, M.; Rovati, C.; Marenghi, C.; Gattinoni, L.; Caironi, P. Strong ion difference and arterial bicarbonate concentration as cornerstones of the impact of fluid therapy on acid-base balance. Crit. Care 2013, 17, P378. [Google Scholar] [CrossRef][Green Version]
- Chun, T.T.; Potz, B.A.; Young, W.A.; Ayala, A. Overview of the Molecular Pathways and Mediators of Sepsis. In Sepsis: Definitions, Pathophysiology and the Challenge of Bedside Management; Springer: Berlin/Heidelberg, Germany, 2017; pp. 47–69. [Google Scholar] [CrossRef]
- Zhang, P.; Zou, B.; Liou, Y.-C.; Huang, C. The pathogenesis and diagnosis of sepsis post burn injury. Burn. Trauma 2021, 9, tkaa047. [Google Scholar] [CrossRef] [PubMed]
- Wada, H.; Matsumoto, T.; Yamashita, Y. Diagnosis and treatment of disseminated intravascular coagulation (DIC) according to four DIC guidelines. J. Intensiv. Care 2014, 2, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Iba, T.; Arakawa, M.; Levy, J.H.; Yamakawa, K.; Koami, H.; Hifumi, T.; Sato, K. Sepsis-Induced Coagulopathy and Japanese Association for Acute Medicine DIC in Coagulopathic Patients with Decreased Antithrombin and Treated by Antithrombin. Clin. Appl. Thromb. Hemost. 2018, 24, 1020–1026. [Google Scholar] [CrossRef] [PubMed]
- Wakabayashi, I.; Mambo, N.; Ueda, T.; Nonaka, D.; Lee, L.-J.; Tanaka, K.; Kotani, J. New Biomarkers for Prediction of Dis-seminated Intravascular Coagulation in Patients with Sepsis. Clin. Appl. Thromb. Hemost. 2018, 24, 223S–229S. [Google Scholar] [CrossRef] [PubMed]
- Giarratano, A. Sepsis-induced coagulopathy and disseminated intravascular coagulation. Aboutopen 2022, 9, 58–60. [Google Scholar] [CrossRef]
- Iba, T.; Di Nisio, M.; Levy, J.H.; Kitamura, N.; Thachil, J. New criteria for sepsis-induced coagulopathy (SIC) following the revised sepsis definition: A retrospective analysis of a nationwide survey. BMJ Open 2017, 7, e017046. [Google Scholar] [CrossRef] [PubMed]
- Le Balc’h, P.; Pinceaux, K.; Pronier, C.; Seguin, P.; Tadié, J.-M.; Reizine, F. Herpes simplex virus and cytomegalovirus reactivations among severe COVID-19 patients. Crit. Care 2020, 24, 1–3. [Google Scholar] [CrossRef]
- Mahmoudi, S.; Rezaei, M.; Mansouri, N.; Marjani, M.; Mansouri, D. Immunologic Features in Coronavirus Disease 2019: Functional Exhaustion of T Cells and Cytokine Storm. J. Clin. Immunol. 2020, 40, 974–976. [Google Scholar] [CrossRef]
- Chanihoon, G.Q.; Afridi, H.I.; Unar, A.; Talpur, F.N.; Kalochi, H.B.; Nassani, R.; Laghari, N.; Uddin, N.; Ghulam, A.; Chandio, A.U.R. Selenium and mercury concentrations in biological samples from patients with COVID-19. J. Trace Elements Med. Biol. 2022, 73, 127038. [Google Scholar] [CrossRef]
- Iuliano, A.D.; Roguski, K.M.; Chang, H.H.; Muscatello, D.J.; Palekar, R.; Tempia, S.; Cohen, C.; Gran, J.M.; Schanzer, D.; Cowling, B.J.; et al. Global Seasonal Influenza-associated Mor-tality Collaborator Network. Estimates of global seasonal influenza-associated respiratory mortality: A modelling study. Lancet 2018, 391, 1285–1300. [Google Scholar] [CrossRef] [PubMed]
- Prapty, C.N.B.S.; Rahmat, R.; Araf, Y.; Shounak, S.K.; Afrin, N.A.; Rahaman, T.I.; Hosen, M.J.; Zheng, C.; Hossain, G. SARS-CoV-2 and dengue virus co-infection: Epidemiology, pathogenesis, diagnosis, treatment, and management. Rev. Med Virol. 2022, 33, e2340. [Google Scholar] [CrossRef]
- Moriyama, K.; Nishida, O. Targeting Cytokines, Pathogen-Associated Molecular Patterns, and Damage-Associated Molecular Patterns in Sepsis via Blood Purification. Int. J. Mol. Sci. 2021, 22, 8882. [Google Scholar] [CrossRef] [PubMed]
- Gando, S.; Levi, M.; Toh, C.-H. Disseminated Intravascular Coagulation. Nat. Rev. Dis. Primers 2016, 2, 16037. [Google Scholar] [CrossRef] [PubMed]
- Iba, T.; Umemura, Y.; Wada, H.; Levy, J.H. Roles of Coagulation Abnormalities and Microthrombosis in Sepsis: Pathophysiology, Diagnosis, and Treatment. Arch. Med. Res. 2021, 52, 788–797. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wu, Y.F.; Jiang, H.W.; Khan, R.; Han, Q.Q.; Iqbal, F.; Jiang, X.H.; Shi, Q.H. The Molecular Control of Meiotic Dou-ble-Strand Break (DSB) Formation and Its Significance in Human Infertility. Asian J. Androl. 2021, 23, 555–561. [Google Scholar]
- Mackman, N.; Tilley, R.E.; Key, N.S. Role of the extrinsic pathway of blood coagulation in hemostasis and thrombosis. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 1687–1693. [Google Scholar] [CrossRef]
- Parikh, V.; Tucci, V.; Galwankar, S. Infections of the nervous system. Int. J. Crit. Illn. Inj. Sci. 2012, 2, 82–97. [Google Scholar] [CrossRef]
- Wright, E.J.; Brew, B.J.; Wesselingh, S.L. Pathogenesis and Diagnosis of Viral Infections of the Nervous System. Neurol. Clin. 2008, 26, 617–633. [Google Scholar] [CrossRef]
- Friedrich, F.; Geusau, A.; Greisenegger, S.; Ossege, M.; Aigner, M. Manifest psychosis in neurosyphilis. Gen. Hosp. Psychiatry 2009, 31, 379–381. [Google Scholar] [CrossRef]
- Simmons, J.; Pittet, J.-F. The coagulopathy of acute sepsis. Curr. Opin. Anaesthesiol. 2015, 28, 227–236. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J. The immunopathogenesis of sepsis. Nature 2002, 420, 885–891. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zhang, Z.; Ruan, J.; Pan, Y.; Magupalli, V.G.; Wu, H.; Lieberman, J. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature 2016, 535, 153–158. [Google Scholar] [CrossRef]
- Rathinam, V.A.K.; Zhao, Y.; Shao, F. Innate immunity to intracellular LPS. Nat. Immunol. 2019, 20, 527–533. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Allen, H.; Banerjee, S.; Franklin, S.; Herzog, L.; Johnston, C.; McDowell, J.; Paskind, M.; Rodman, L.; Salfeld, J.; et al. Mice deficient in IL-1β-converting enzyme are defective in production of mature IL-1β and resistant to endotoxic shock. Cell 1995, 80, 401–411. [Google Scholar] [CrossRef]
- Wang, S.; Miura, M.; Jung, Y.-K.; Zhu, H.; Li, E.; Yuan, J. Murine Caspase-11, an ICE-Interacting Protease, Is Essential for the Activation of ICE. Cell 1998, 92, 501–509. [Google Scholar] [CrossRef]
- Kang, S.-J.; Wang, S.; Hara, H.; Peterson, E.P.; Namura, S.; Amin-Hanjani, S.; Huang, Z.; Srinivasan, A.; Tomaselli, K.J.; Thornberry, N.A.; et al. Dual Role of Caspase-11 in Mediating Activation of Caspase-1 and Caspase-3 under Pathological Conditions. J. Cell Biol. 2000, 149, 613–622. [Google Scholar] [CrossRef]
- Kayagaki, N.; Warming, S.; Lamkanfi, M.; Walle, L.V.; Louie, S.; Dong, J.; Newton, K.; Qu, Y.; Liu, J.; Heldens, S.; et al. Non-canonical inflammasome activation targets caspase-11. Nature 2011, 479, 117–121. [Google Scholar] [CrossRef]
- Kayagaki, N.; Wong, M.T.; Stowe, I.B.; Ramani, S.R.; Gonzalez, L.C.; Akashi-Takamura, S.; Miyake, K.; Zhang, J.; Lee, W.P.; Muszyński, A.; et al. Noncanonical Inflammasome Activation by Intracellular LPS Independent of TLR4. Science 2013, 341, 1246–1249. [Google Scholar] [CrossRef]
- Hagar, J.A.; Powell, D.A.; Aachoui, Y.; Ernst, R.K.; Miao, E.A. Cytoplasmic LPS Activates Caspase-11: Implications in TLR4-Independent Endotoxic Shock. Science 2013, 341, 1250–1253. [Google Scholar] [CrossRef]
- Wu, C.; Lu, W.; Zhang, Y.; Zhang, G.; Shi, X.; Hisada, Y.; Grover, S.P.; Zhang, X.; Li, L.; Xiang, B.; et al. Inflammasome Activation Triggers Blood Clotting and Host Death through Pyroptosis. Immunity 2019, 50, 1401–1411.e4. [Google Scholar] [CrossRef] [PubMed]
- Pawlinski, R.; Pedersen, B.; Schabbauer, G.; Tencati, M.; Holscher, T.; Boisvert, W.; Andrade-Gordon, P.; Frank, R.D.; Mackman, N. Role of tissue factor and protease-activated receptors in a mouse model of endotoxemia. Blood 2004, 103, 1342–1347. [Google Scholar] [CrossRef]
- Pawlinski, R.; Wang, J.-G.; Owens, A.P.; Williams, J.; Antoniak, S.; Tencati, M.; Luther, T.; Rowley, J.W.; Low, E.N.; Weyrich, A.S.; et al. Hematopoietic and nonhematopoietic cell tissue factor activates the coagulation cascade in endotoxemic mice. Blood 2010, 116, 806–814. [Google Scholar] [CrossRef] [PubMed]
- Ammollo, C.T.; Semeraro, F.; Colucci, M.; Semeraro, N. Coagulopathy of Acute Sepsis. Semin. Thromb. Hemost. 2015, 41, 650–658. [Google Scholar] [CrossRef] [PubMed]
- Østerud, B.; Bjørklid, E. The Tissue Factor Pathway in Disseminated Intravascular Coagulation. Semin. Thromb. Hemost. 2001, 27, 605–618. [Google Scholar] [CrossRef] [PubMed]
- van Hinsbergh, V.W.M. Endothelium—Role in Regulation of Coagulation and Inflammation. Semin. Immunopathol. 2012, 34, 93–106. [Google Scholar] [CrossRef]
- Levi, M.; Schultz, M.; van der Poll, T. Sepsis and thrombosis. Semin. Thromb. Hemost. 2013, 39, 559–566. [Google Scholar] [PubMed]
- Opal, S.M.; Kessler, C.M.; Roemisch, J.; Knaub, S. Antithrombin, heparin, and heparan sulfate. Crit. Care Med. 2002, 30, S325–S331. [Google Scholar] [CrossRef] [PubMed]
- Fogagnolo, A.; Campo, G.C.; Mari, M.; Pompei, G.; Pavasini, R.; Volta, C.A.; Spadaro, S. The Underestimated Role of Platelets in Severe Infection a Narrative Review. Cells 2022, 11, 424. [Google Scholar] [CrossRef]
- Dewitte, A.; Lepreux, S.; Villeneuve, J.; Rigothier, C.; Combe, C.; Ouattara, A.; Ripoche, J. Blood platelets and sepsis pathophysiology: A new therapeutic prospect in critical ill patients? Ann. Intensiv. Care 2017, 7, 1–18. [Google Scholar] [CrossRef]
- Hamzeh-Cognasse, H.; Damien, P.; Chabert, A.; Pozzetto, B.; Cognasse, F.; Garraud, O. Platelets and Infections – Complex Interactions with Bacteria. Front. Immunol. 2015, 6, 82. [Google Scholar] [CrossRef]
- Assinger, A.; Schrottmaier, W.C.; Salzmann, M.; Rayes, J. Platelets in Sepsis: An Update on Experimental Models and Clinical Data. Front. Immunol. 2019, 10, 1687. [Google Scholar] [CrossRef] [PubMed]
- Soriano, A.O.; Jy, W.; Chirinos, J.A.; Valdivia, M.A.; Velasquez, H.S.; Jimenez, J.J.; Horstman, L.L.; Kett, D.H.; Schein, R.M.H.; Ahn, Y.S. Levels of endothelial and platelet microparticles and their interactions with leukocytes negatively correlate with organ dysfunction and predict mortality in severe sepsis. Crit. Care Med. 2005, 33, 2540–2546. [Google Scholar] [CrossRef]
- Giustozzi, M.; Ehrlinder, H.; Bongiovanni, D.; Borovac, J.A.; Guerreiro, R.A.; Gąsecka, A.; Papakonstantinou, P.E.; Parker, W.A. Coagulopathy and sepsis: Pathophysiology, clinical manifestations and treatment. Blood Rev. 2021, 50, 100864. [Google Scholar] [CrossRef] [PubMed]
- Naime, A.C.A.; Ganaes, J.O.F.; Lopes-Pires, M.E. Sepsis: The Involvement of Platelets and the Current Treatments. Curr. Mol. Pharmacol. 2018, 11, 261–269. [Google Scholar] [CrossRef] [PubMed]
- Oikonomou, E.; Stampouloglou, P.K.; Siasos, G.; Bletsa, E.; Vogiatzi, G.; Kalogeras, K.; Katsianos, E.; Vavuranakis, M.-A.; Souvaliotis, N.; Vavuranakis, M. The Role of Cell-derived Microparticles in Cardiovascular Diseases: Current Concepts. Curr. Pharm. Des. 2022, 28, 1745–1757. [Google Scholar] [CrossRef]
- Reid, V.; Webster, N. Role of microparticles in sepsis. Br. J. Anaesth. 2012, 109, 503–513. [Google Scholar] [CrossRef]
- Iba, T.; Levy, J.H. Inflammation and thrombosis: Roles of neutrophils, platelets and endothelial cells and their interactions in thrombus formation during sepsis. J. Thromb. Haemost. 2018, 16, 231–241. [Google Scholar] [CrossRef]
- Morrissey, J.H.; Smith, S.A. Polyphosphate as modulator of hemostasis, thrombosis, and inflammation. J. Thromb. Haemost. 2015, 13, S92–S97. [Google Scholar] [CrossRef]
- Taylor, F.B., Jr.; Peer, G.T.; Lockhart, M.S.; Ferrell, G.; Esmon, C.T. Endothelial Cell Protein C Receptor Plays an Important Role in Protein C Activation in Vivo. Blood 2001, 97, 1685–1688. [Google Scholar] [CrossRef]
- Levi, M.; Dorffler-Melly, J.; Reitsma, P.; Buller, H.; Florquin, S.; van der Poll, T.; Carmeliet, P. Aggravation of endotoxin-induced disseminated intravascular coagulation and cytokine activation in heterozygous protein-C–deficient mice. Blood 2003, 101, 4823–4827. [Google Scholar] [CrossRef]
- Liaw, P.C.Y.; Esmon, C.T.; Kahnamoui, K.; Schmidt, S.; Kahnamoui, S.; Ferrell, G.; Beaudin, S.; Julian, J.A.; Weitz, J.I.; Crowther, M.; et al. Patients with severe sepsis vary markedly in their ability to generate activated protein C. Blood 2004, 104, 3958–3964. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Ivanciu, L.; Popescu, N.; Peer, G.; Hack, E.; Lupu, C.; Taylor Jr, F.B.; Lupu, F. Sepsis-Induced Coagulation in the Baboon Lung Is Associated with Decreased Tissue Factor Pathway Inhibitor. Am. J. Pathol. 2007, 171, 1066–1077. [Google Scholar] [CrossRef] [PubMed]
- Gando, S. Role of Fibrinolysis in Sepsis. Semin. Thromb. Hemost. 2013, 39, 392–399. [Google Scholar] [CrossRef] [PubMed]
- Huebner, B.R.; Moore, E.E.; Moore, H.B.; Gonzalez, E.; Kelher, M.R.; Sauaia, A.; Banerjee, A.; Silliman, C.C. Thrombin stimulates increased plasminogen activator inhibitor-1 release from liver compared to lung endothelium. J. Surg. Res. 2018, 225, 1–5. [Google Scholar] [CrossRef]
- Foley, J.H.; Kim, P.Y.; Mutch, N.J.; Gils, A. Insights into thrombin activatable fibrinolysis inhibitor function and regulation. J. Thromb. Haemost. 2013, 11, 306–315. [Google Scholar] [CrossRef] [PubMed]
- Boomer, J.S.; To, K.; Chang, K.C.; Takasu, O.; Osborne, D.F.; Walton, A.H.; Bricker, T.L.; Jarman, S.D.; Kreisel, D., 2nd; Krupnick, A.S.; et al. Immunosuppression in Patients Who Die of Sepsis and Multiple Organ Failure. JAMA 2011, 306, 2594–2605. [Google Scholar] [CrossRef]
- Hotchkiss, R.S.; Monneret, G.; Payen, D. Sepsis-induced immunosuppression: From cellular dysfunctions to immunotherapy. Nat. Rev. Immunol. 2013, 13, 862–874. [Google Scholar] [CrossRef]
- Huang, X.; Venet, F.; Wang, Y.L.; Lepape, A.; Yuan, Z.; Chen, Y.; Swan, R.; Kherouf, H.; Monneret, G.; Chung, C.-S.; et al. PD-1 expression by macrophages plays a pathologic role in altering microbial clearance and the innate inflammatory response to sepsis. Proc. Natl. Acad. Sci. USA 2009, 106, 6303–6308. [Google Scholar] [CrossRef]
- Scumpia, P.O.; Delano, M.J.; Kelly-Scumpia, K.M.; Weinstein, J.S.; Wynn, J.L.; Winfield, R.D.; Xia, C.; Chung, C.S.; Ayala, A.; Atkinson, M.A.; et al. Treatment with GITR agonistic antibody corrects adaptive immune dysfunction in sepsis. Blood 2007, 110, 3673–3681. [Google Scholar] [CrossRef]
- Chan, C.; Li, L.; McCall, C.E.; Yoza, B.K. Endotoxin Tolerance Disrupts Chromatin Remodeling and NF-ΚB Transactivation at the IL-1β Promoter. J. Immunol. 2005, 175, 461–468. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann, V.; Reichard, U.; Goosmann, C.; Fauler, B.; Uhlemann, Y.; Weiss, D.S.; Weinrauch, Y.; Zychlinsky, A. Neutrophil extracellular traps kill bacteria. Science 2004, 303, 1532–1535. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann, V.; Zychlinsky, A. Neutrophil Extracellular Traps: Is Immunity the Second Function of Chromatin? J. Cell Biol. 2012, 198, 773–783. [Google Scholar] [CrossRef] [PubMed]
- Alhamdi, Y.; Toh, C.-H. Recent Advances in Pathophysiology of Disseminated Intravascular Coagulation: The Role of Circu-lating Histones and Neutrophil Extracellular Traps. F1000Research 2017, 6, 2143. [Google Scholar] [CrossRef]
- Sunden-Cullberg, J.; Norrby-Teglund, A.; Treutiger, C.J. The role of high mobility group box-1 protein in severe sepsis. Curr. Opin. Infect. Dis. 2006, 19, 231–236. [Google Scholar] [CrossRef] [PubMed]
- McDonald, B.; Davis, R.P.; Kim, S.-J.; Tse, M.; Esmon, C.T.; Kolaczkowska, E.; Jenne, C.N. Platelets and neutrophil extracellular traps collaborate to promote intravascular coagulation during sepsis in mice. Blood 2017, 129, 1357–1367. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Zhang, H.; Yin, Y.-L.; Guo, W.-Z.; Ma, Y.-Q.; Wang, Y.-B.; Shu, C.; Dong, L.-Q. Role of interleukin-6 to differentiate sepsis from non-infectious systemic inflammatory response syndrome. Cytokine 2016, 88, 126–135. [Google Scholar] [CrossRef]
- Xu, J.; Zhang, X.; Pelayo, R.; Monestier, M.; Ammollo, C.T.; Semeraro, F.; Taylor, F.B.; Esmon, N.L.; Lupu, F.; Esmon, C.T. Extracellular histones are major mediators of death in sepsis. Nat. Med. 2009, 15, 1318–1321. [Google Scholar] [CrossRef]
- Hell, L.; Thaler, J.; Martinod, K.; Ay, C.; Posch, F.; Wagner, D.D.; Pabinger, I. OC-16-Neutrophil Extracellular Traps and Tissue Factor-Bearing Microvesicles: A Liaison Dangereuse Causing Overt DIC in Cancer Patients? Thromb. Res. 2016, 140, S174–S175. [Google Scholar] [CrossRef]
- Swystun, L.L.; Mukherjee, S.; Liaw, P.C. Breast cancer chemotherapy induces the release of cell-free DNA, a novel procoagulant stimulus. J. Thromb. Haemost. 2011, 9, 2313–2321. [Google Scholar] [CrossRef]
- Kim, J.E.; Yoo, H.J.; Gu, J.Y.; Kim, H.K. Histones Induce the Procoagulant Phenotype of Endothelial Cells through Tissue Factor Up-Regulation and Thrombomodulin Down-Regulation. PLoS ONE 2016, 11, e0156763. [Google Scholar] [CrossRef] [PubMed]
- Barranco-Medina, S.; Pozzi, N.; Vogt, A.D.; Di Cera, E. Histone H4 Promotes Prothrombin Autoactivation. J. Biol. Chem. 2013, 288, 35749–35757. [Google Scholar] [CrossRef]
- Varjú, I.; Longstaff, C.; Szabó, L.; Farkas, Z.; Varga-Szabó, V.J.; Tanka-Salamon, A.; Machovich, R.; Kolev, K. DNA, histones and neutrophil extracellular traps exert anti-fibrinolytic effects in a plasma environment. Thromb. Haemost. 2015, 113, 1289–1298. [Google Scholar] [CrossRef] [PubMed]
- Abrams, S.T.; Zhang, N.; Manson, J.; Liu, T.; Dart, C.; Baluwa, F.; Wang, S.S.; Brohi, K.; Kipar, A.; Yu, W.; et al. Circulating Histones Are Mediators of Trauma-associated Lung Injury. Am. J. Respir. Crit. Care Med. 2013, 187, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.K.; Joshi, M.B.; Philippova, M.; Erne, P.; Hasler, P.; Hahn, S.; Resink, T.J. Activated Endothelial Cells Induce Neu-trophil Extracellular Traps and Are Susceptible to NETosis-Mediated Cell Death. FEBS Lett. 2010, 584, 3193–3197. [Google Scholar] [CrossRef] [PubMed]
- Stakos, D.A.; Kambas, K.; Konstantinidis, T.; Mitroulis, I.; Apostolidou, E.; Arelaki, S.; Tsironidou, V.; Giatromanolaki, A.; Skendros, P.; Konstantinides, S.; et al. Expression of functional tissue factor by neutrophil extracellular traps in culprit artery of acute myocardial infarction. Eur. Heart J. 2015, 36, 1405–1414. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Condition | Definition | Pathogenesis | Causes | Clinical Features | Diagnosis | Treatment | Reference |
|---|---|---|---|---|---|---|---|
| Sepsis | Life-threatening response to infection, causing organ dysfunction. | Dysregulated immune response, leading to inflammation. | Bacterial, viral, fungal infections. | Fever, chills, organ dysfunction. | Clinical symptoms, blood cultures. | Antibiotics, supportive care. | [4] |
| DIC | Widespread clotting in blood vessels. | Triggered by conditions like infections, trauma. | Sepsis, trauma, malignancies. | Bleeding, thrombosis, organ dysfunction. | Coagulation tests, low platelets. | Address cause, blood transfusions. | [2,3] |
| SIC | DIC subset linked to sepsis. | Interaction between inflammation and coagulation. | Prolonged PT, aPTT, decreased platelets. | Organ dysfunction, clot formation. | Focus on organ dysfunction. | Anticoagulants. | [2,3] |
| Septic Shock | Severe sepsis subset with high mortality risk. | Acute circulatory failure. | Elevated lactate, decreased platelets. | Hypotension, altered mental state. | Sepsis-3 definition. | Corticosteroids, immunomodulatory drugs. | [4] |
| SARS-CoV-2 | Respiratory infection by SARS-CoV-2. | Virus targets ACE2 receptors; can cause ARDS. | SARS-CoV-2 transmission via droplets. | Respiratory symptoms, ARDS. | PCR, chest imaging, serological tests. | Symptomatic relief, antivirals. | [5,6,7,8] |
| Flaviviruses | Diseases from viruses like Zika, dengue. | Infection of immune cells, causing imbalanced response. | Mosquito-borne or direct contact. | Fever, rash, potential organ failure. | PCR, serological assays, culturing. | Supportive care, antivirals/antibiotics. | [7,9,10] |
| Pathology | Molecular Mechanism and Pathways | Clinical Manifestations | Diagnostic Markers | Therapeutic Approaches | Relation to Sepsis-3 Controversy | Recent Research Findings | Reference |
|---|---|---|---|---|---|---|---|
| Sepsis | Systemic inflammatory response to infection, characterized by PAMPs release and activation of PRRs such as TLRs, triggering NF-κB and MAPK pathways. This results in cytokine storm and potential secondary infections. | Fever, tachycardia, dyspnea, hypotension, altered cognition, organ dysfunction. | Elevated proinflammatory cytokines, leukocytosis, thrombocytopenia, hyperlactatemia, organ dysfunction (SOFA score). | Broad-spectrum antibiotics, fluid resuscitation, vasopressors, corticosteroids, supportive care. | Sepsis-3 emphasizes organ dysfunction, potentially overlooking early sepsis without organ dysfunction. | Focus on early biomarkers and immune response role in sepsis progression. | [4,25,26] |
| DIC | Coagulopathy triggered by conditions, including sepsis. Characterized by widespread coagulation activation, microthrombi formation, organ dysfunction, ischemia, and bleeding manifestations. | Bleeding, purpura, petechiae, organ dysfunction. | Prolonged PT and aPTT, thrombocytopenia, increased FDPs, decreased fibrinogen. | Treatment of underlying cause, blood product transfusion, anticoagulants. | Sepsis-3 may not capture DIC complexity in sepsis due to organ dysfunction focus. | Exploration of DIC mechanisms in sepsis and potential coagulation cascade modulation. | [3,22,27,28,29] |
| SIC | DIC subset associated with sepsis. Characterized by coagulation activation, fibrinolysis inhibition, clot formation, and potential organ dysfunction. | Similar to DIC, including bleeding, purpura, petechiae, organ dysfunction. | Prolonged PT and aPTT, thrombocytopenia, increased D-dimer, decreased antithrombin III. | Treatment of underlying sepsis, potential anticoagulants. | Sepsis-3 may capture SIC patients but may not reflect underlying coagulation abnormalities. | Exploration of SIC mechanisms and potential therapeutic strategies, including anticoagulants. | [30,31] |
| Septic Shock | Sepsis subset with profound circulatory, cellular, and metabolic abnormalities. Characterized by persistent hypotension unresponsive to fluid resuscitation, requiring vasopressors. | Persistent hypotension, altered cognition, oliguria, tachycardia, dyspnea, cool and clammy skin. | Hyperlactatemia, thrombocytopenia, increased D-dimer, increased procalcitonin, organ dysfunction (SOFA score). | Vasopressors, antibiotics, fluid resuscitation, corticosteroids, supportive care. | Sepsis-3 includes septic shock as a subset with increased mortality. Criticized for complexity and need for laboratory results. | Exploration of septic shock pathophysiology and potential therapeutic strategies, including corticosteroids. and immunomodulatory drugs. | [4,25,26,32] |
| SARS-CoV-2 | Virus enters host cells via ACE2 receptors, leading to viral replication and immune response activation. In severe cases, cytokine storm leads to severe inflammation and lung tissue damage. | Fever, cough, dyspnea, anosmia, fatigue, organ dysfunction in severe cases. | Positive RT-PCR for SARS-CoV-2, elevated proinflammatory cytokines, abnormal chest imaging. | Antivirals, corticosteroids, monoclonal antibodies, supportive care. | COVID-19 can lead to sepsis-like syndrome. Sepsis-3 may not capture unique aspects of COVID-19-related sepsis. | Focus on understanding severe COVID-19 pathophysiology, immune response role, and potential therapeutic targets. | [5,6,7,8,33,34,35] |
| Flaviviruses and Other Microorganisms | Different molecular mechanisms for host cell infection. Flaviviruses infect immune cells, leading to imbalanced immune response. Other microorganisms may produce toxins or virulent factors. | Symptoms vary, may include fever, rash, arthralgia, nausea, vomiting, diarrhea, cough, dyspnea. | Varies, may include positive culture or PCR, elevated proinflammatory cytokines, abnormal imaging. | Varies, may include antibiotics, antivirals, antifungals, supportive care. | Infections can lead to sepsis, but Sepsis-3 may not capture unique aspects of sepsis caused by these pathogens. | Exploration of pathogenesis of sepsis caused by these pathogens and potential therapeutic strategies. | [7,9,10,36,37,38] |
| Bacteria | GP | CA | SD | HE | SL | CP | SL | BF | RE |
|---|---|---|---|---|---|---|---|---|---|
| Staphylococcus aureus | + | + | + | + | + | + | + | + | FAN |
| Coagulase-negative staph | + | + | + | + | + | + | + | + | FAN |
| Streptococcus pneumonia | + | + | + | + | + | + | + | + | FAN |
| Haemophilus influenza b | + | + | + | + | + | + | + | + | MA |
| Neisseria meningitidis | + | + | + | + | + | + | + | + | FAN |
| Klebsiella pneumonia | + | + | + | + | + | + | + | + | FAN |
| Enterococcus faecalis | + | + | + | + | + | + | + | + | FAN |
| Acinetobacter baumanii | + | + | + | + | + | + | + | + | A |
| Escherichia coli | + | + | + | + | + | + | + | + | FAN |
| Salmonella enterica | + | + | + | + | + | + | + | + | FAN |
| Shigella dysenteriae | + | + | + | + | + | + | + | + | FAN |
| Citrobacter freundii | + | + | + | + | + | + | + | + | FAN |
| Serratia marcescens | + | + | + | + | + | + | + | + | FAN |
| Proteus mirabilis | + | + | + | + | + | + | + | + | FAN |
| Pseudomonas aeruginosa | + | + | + | + | + | + | + | + | FAN |
| Bacteroides fragilis | + | + | + | + | + | + | + | + | OAN |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Unar, A.; Bertolino, L.; Patauner, F.; Gallo, R.; Durante-Mangoni, E. Pathophysiology of Disseminated Intravascular Coagulation in Sepsis: A Clinically Focused Overview. Cells 2023, 12, 2120. https://doi.org/10.3390/cells12172120
Unar A, Bertolino L, Patauner F, Gallo R, Durante-Mangoni E. Pathophysiology of Disseminated Intravascular Coagulation in Sepsis: A Clinically Focused Overview. Cells. 2023; 12(17):2120. https://doi.org/10.3390/cells12172120
Chicago/Turabian StyleUnar, Ahsanullah, Lorenzo Bertolino, Fabian Patauner, Raffaella Gallo, and Emanuele Durante-Mangoni. 2023. "Pathophysiology of Disseminated Intravascular Coagulation in Sepsis: A Clinically Focused Overview" Cells 12, no. 17: 2120. https://doi.org/10.3390/cells12172120
APA StyleUnar, A., Bertolino, L., Patauner, F., Gallo, R., & Durante-Mangoni, E. (2023). Pathophysiology of Disseminated Intravascular Coagulation in Sepsis: A Clinically Focused Overview. Cells, 12(17), 2120. https://doi.org/10.3390/cells12172120