
Inhibition of Vascular Smooth Muscle Cell Proliferation by ENPP1: The Role of CD73 and the Adenosine Signaling Axis

 , , , and
, , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals, Cells and Reagents
2.2. mRNA and Western Blot Analysis in Contractile and Synthetic Smooth Muscle Cells
2.3. Cell-Based Assays
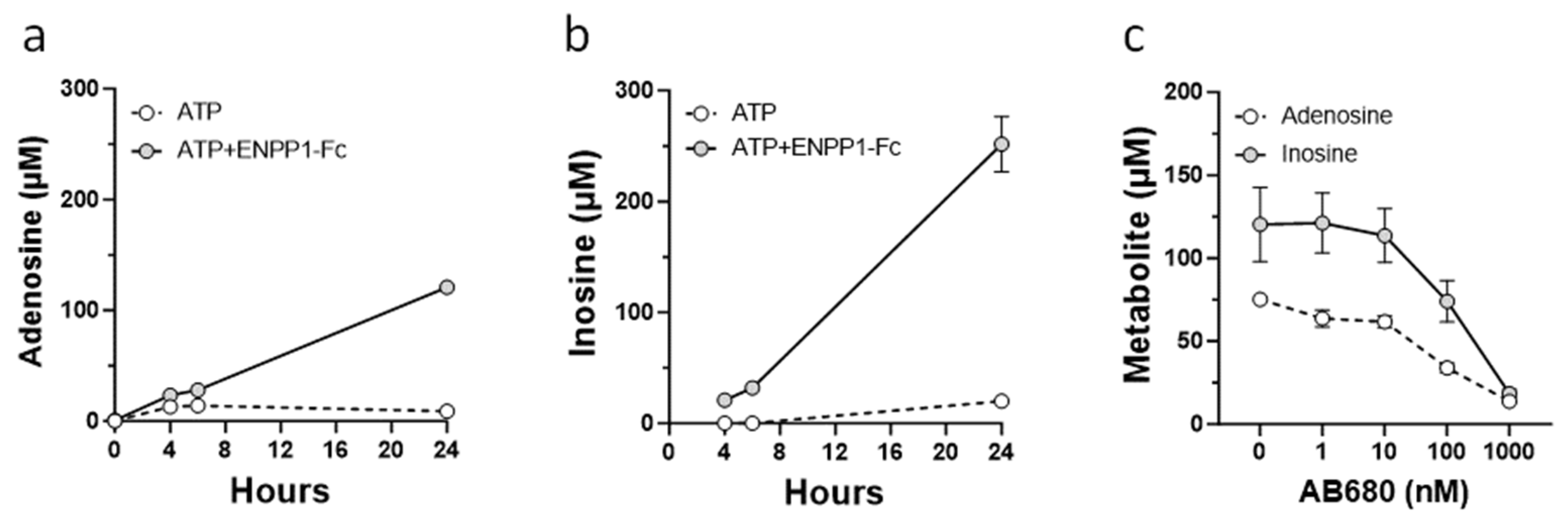
2.4. Analysis of Adenosine Metabolites
2.5. Carotid Artery Ligation Model
2.6. Histomorphometry and Immunohistochemistry
2.7. Statistical Analysis
3. Results
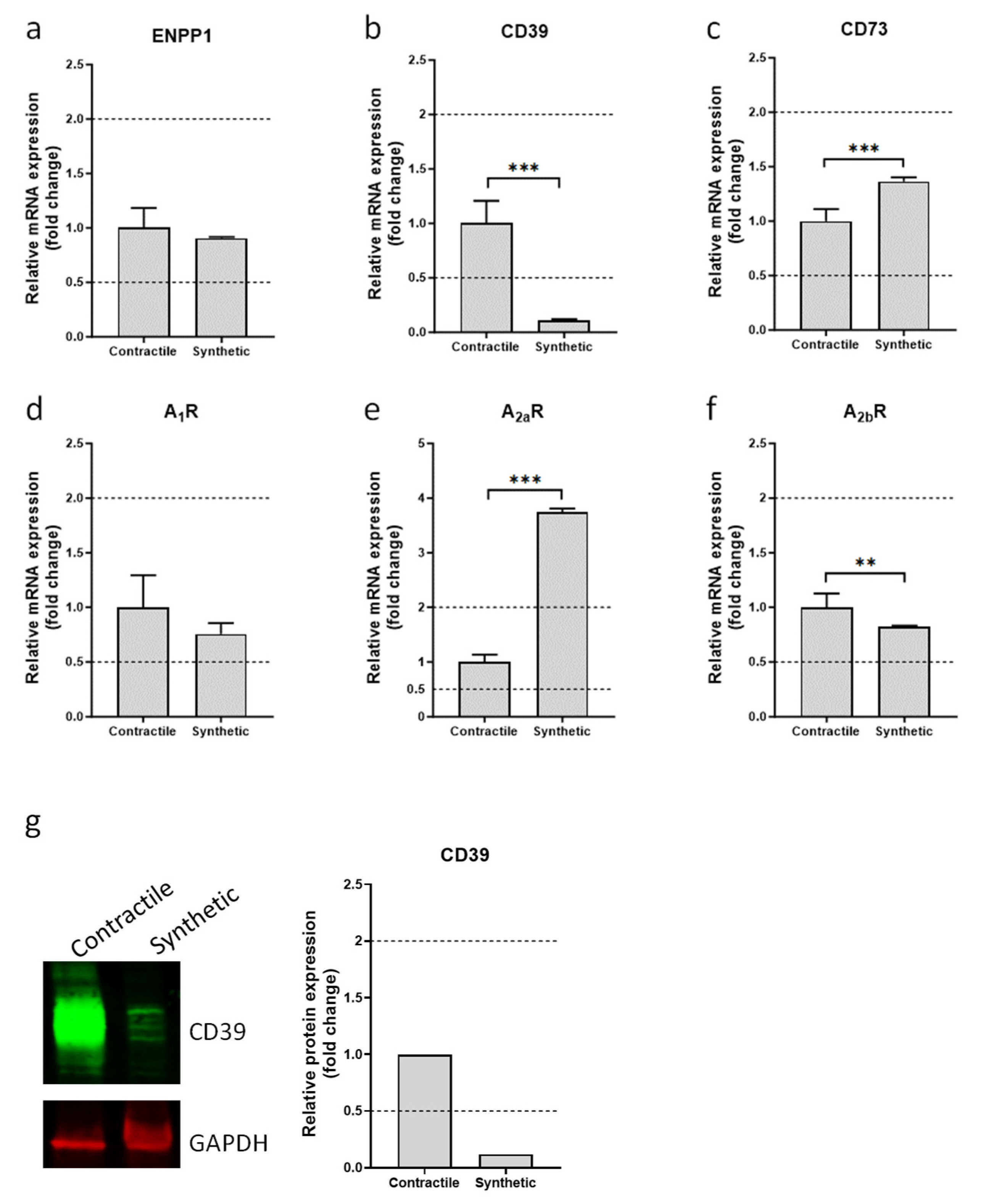
3.1. Expression of Ectonucleotidases and Adenosine Receptors in Contractile and Synthetic VSMCs
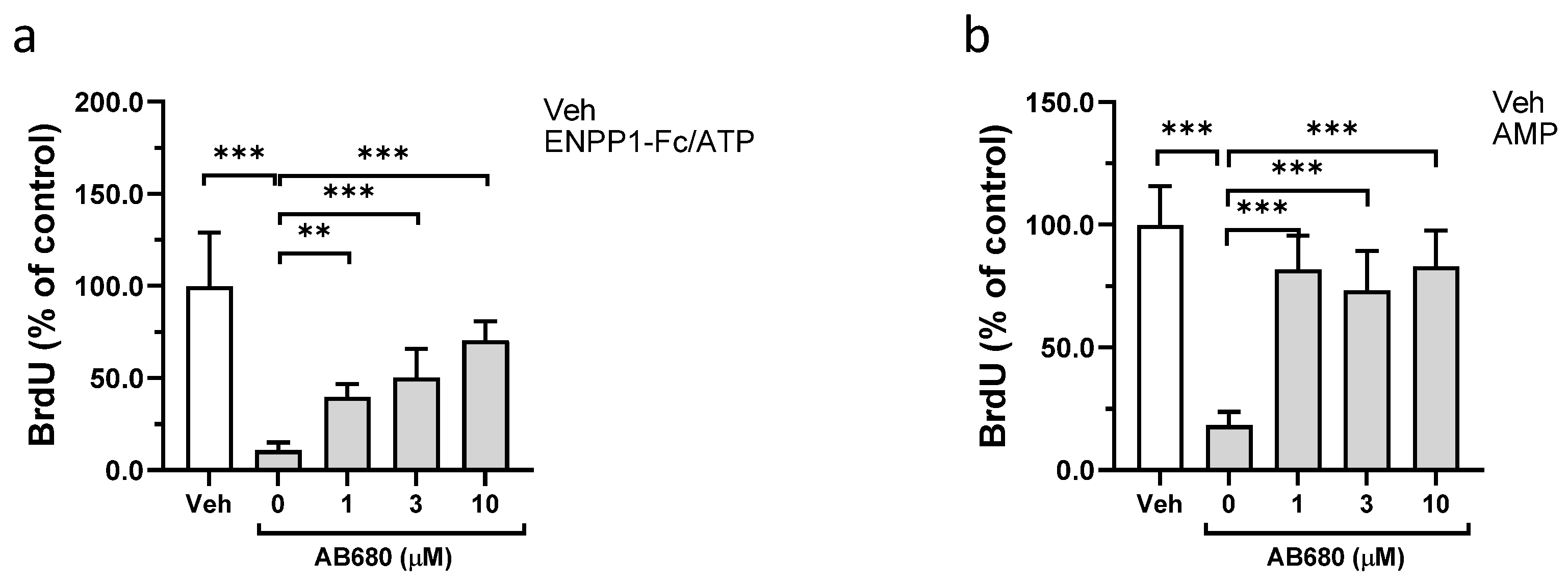
3.2. Effect of ENPP1/ATP, AMP, and Adenosine on Proliferation of VSMCs
3.3. Role of CD73 in Antiproliferative Effect of ENPP1/ATP
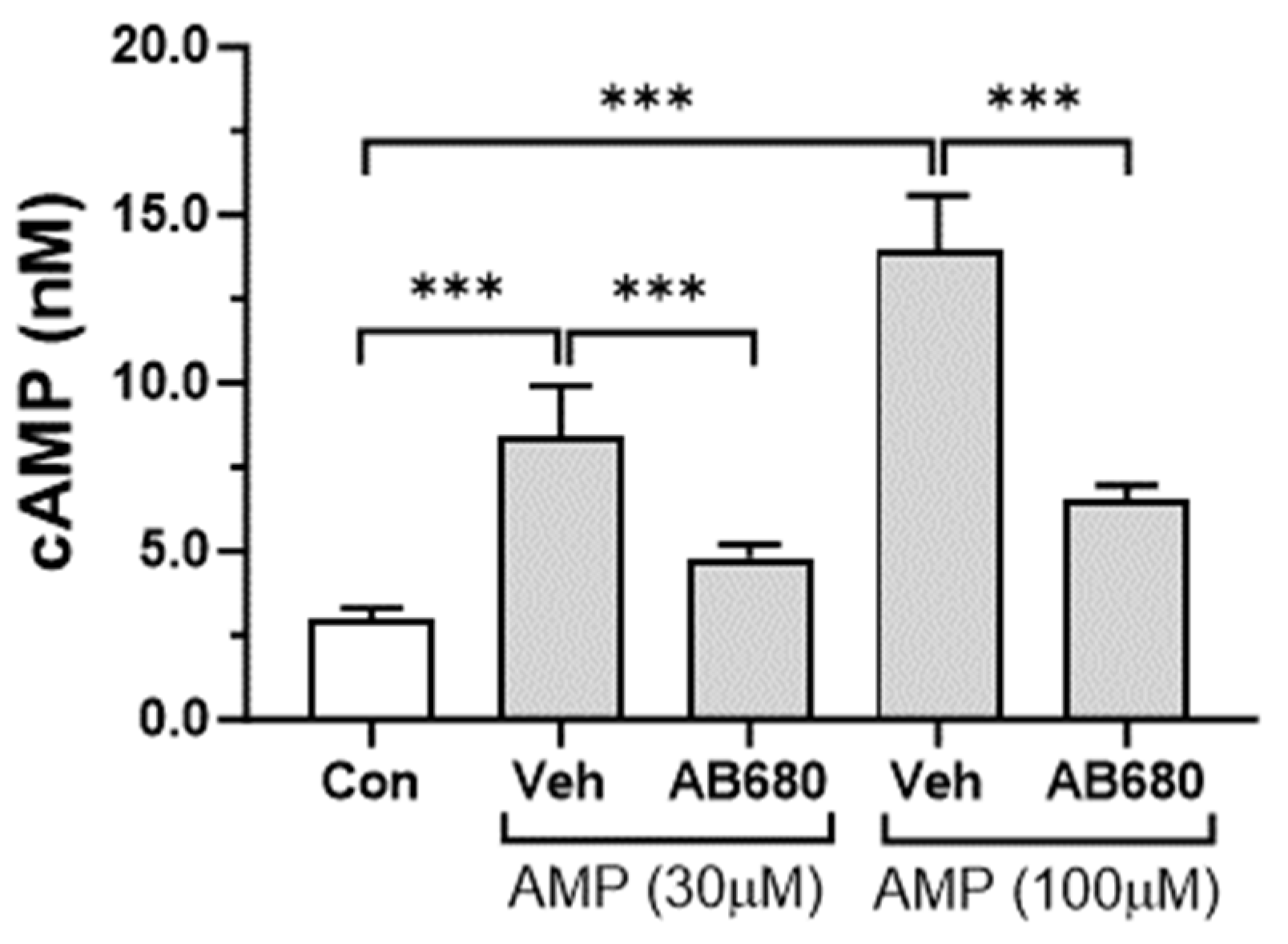
3.4. Activation of cAMP-PKA Signaling Pathway by Extracellular AMP
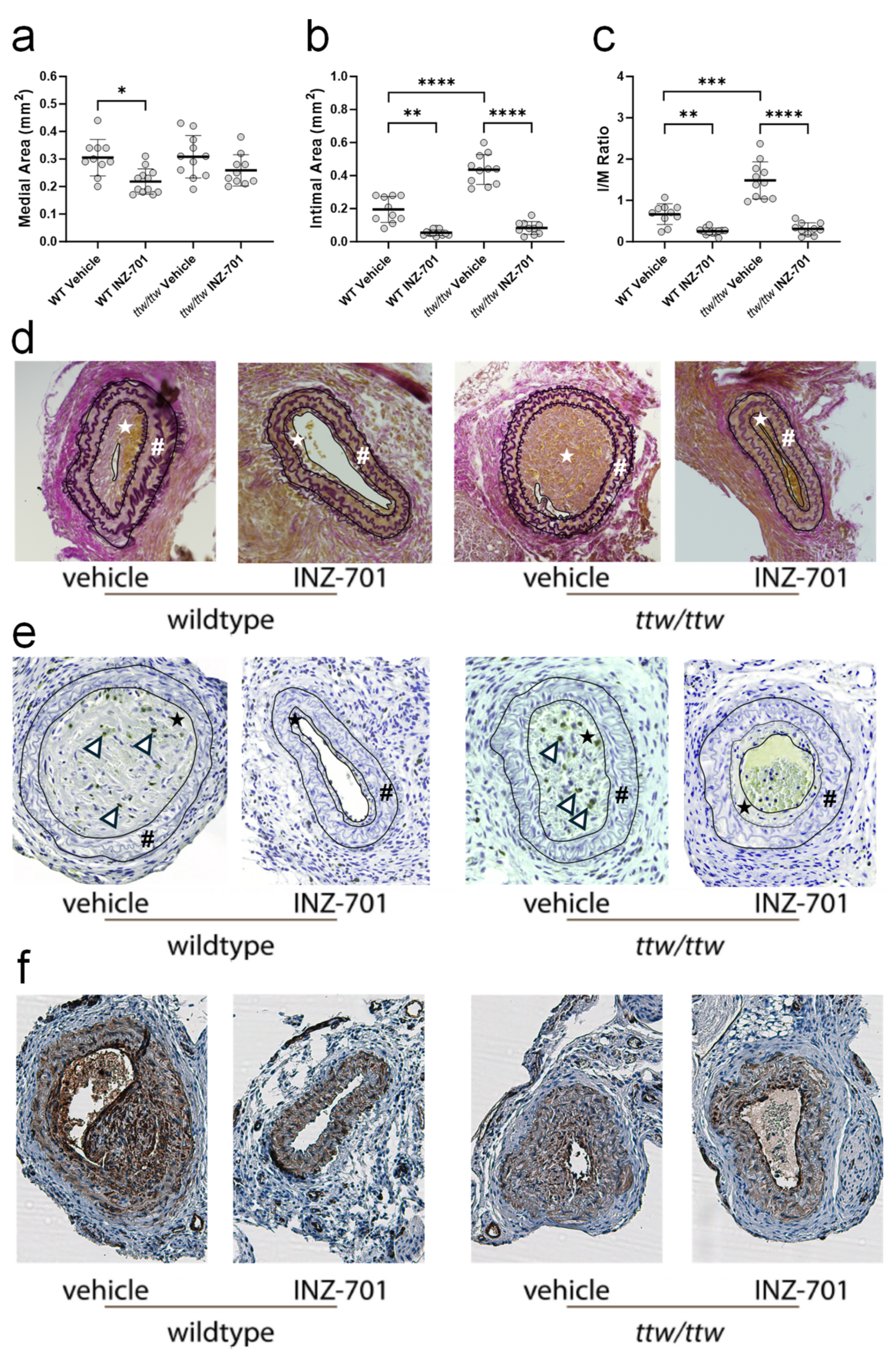
3.5. In Vivo Effect of ENPP-Fc Fusion Protein INZ-701 on Neointima Formation
4. Discussion
5. Conclusions
6. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Schermuly, R.T.; Ghofrani, H.A.; Wilkins, M.R.; Grimminger, F. Mechanisms of disease: Pulmonary arterial hypertension. Nat. Rev. Cardiol. 2011, 8, 443–455. [Google Scholar] [CrossRef]
- Sakao, S.; Voelkel, N.F.; Tatsumi, K. The vascular bed in COPD: Pulmonary hypertension and pulmonary vascular alterations. Eur. Respir. Rev. 2014, 23, 350–355. [Google Scholar] [CrossRef] [PubMed]
- Fox, B.M.; Dorschel, K.B.; Lawton, M.T.; Wanebo, J.E. Pathophysiology of Vascular Stenosis and Remodeling in Moyamoya Disease. Front. Neurol. 2021, 12, 661578. [Google Scholar] [CrossRef]
- Lee, T.; Roy-Chaudhury, P. Advances and new frontiers in the pathophysiology of venous neointimal hyperplasia and dialysis access stenosis. Adv. Chronic Kidney Dis. 2009, 16, 329–338. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.; Chauhan, V.; Krishnamoorthy, M.; Wang, Y.; Arend, L.; Mistry, M.J.; El-Khatib, M.; Banerjee, R.; Munda, R.; Roy-Chaudhury, P. Severe venous neointimal hyperplasia prior to dialysis access surgery. Nephrol. Dial. Transplant. 2011, 26, 2264–2270. [Google Scholar] [CrossRef]
- Vazquez-Padron, R.I.; Duque, J.C.; Tabbara, M.; Salman, L.H.; Martinez, L. Intimal Hyperplasia and Arteriovenous Fistula Failure: Looking Beyond Size Differences. Kidney360 2021, 2, 1360–1372. [Google Scholar] [CrossRef] [PubMed]
- Baby, D.; Upadhyay, M.; Joseph, M.D.; Asopa, S.J.; Choudhury, B.K.; Rajguru, J.P.; Gupta, S. Calciphylaxis and its diagnosis: A review. J. Family Med. Prim. Care 2019, 8, 2763–2767. [Google Scholar] [CrossRef]
- Peykar, S.; Angiolillo, D.J.; Bass, T.A.; Costa, M.A. Saphenous vein graft disease. Minerva Cardioangiol. 2004, 52, 379–390. [Google Scholar]
- Giustino, G.; Colombo, A.; Camaj, A.; Yasumura, K.; Mehran, R.; Stone, G.W.; Kini, A.; Sharma, S.K. Coronary In-Stent Restenosis: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2022, 80, 348–372. [Google Scholar] [CrossRef]
- Ramzy, D.; Rao, V.; Brahm, J.; Miriuka, S.; Delgado, D.; Ross, H.J. Cardiac allograft vasculopathy: A review. Can. J. Surg. 2005, 48, 319–327. [Google Scholar]
- Moran, J.J. Idiopathic arterial calcification of infancy: A clinicopathologic study. Pathol. Annu. 1975, 10, 393–417. [Google Scholar] [PubMed]
- Kawai, K.; Sato, Y.; Kawakami, R.; Sakamoto, A.; Cornelissen, A.; Mori, M.; Ghosh, S.; Kutys, R.; Virmani, R.; Finn, A.V. Generalized Arterial Calcification of Infancy (GACI): Optimizing Care with a Multidisciplinary Approach. J. Multidiscip. Healthc. 2022, 15, 1261–1276. [Google Scholar] [CrossRef] [PubMed]
- Goding, J.W.; Grobben, B.; Slegers, H. Physiological and pathophysiological functions of the ecto-nucleotide pyrophosphatase/phosphodiesterase family. Biochim. Biophys. Acta 2003, 1638, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Hamczyk, M.R.; Villa-Bellosta, R. Pyrophosphate metabolism and calcification. Aging 2018, 10, 3652–3653. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, C.R.; Carpenter, T.O.; Braddock, D.T. ENPP1 in Blood and Bone: Skeletal and Soft Tissue Diseases Induced by ENPP1 Deficiency. Annu. Rev. Pathol. 2023, 19, 507–540. [Google Scholar] [CrossRef] [PubMed]
- Roberts, F.; Zhu, D.; Farquharson, C.; Macrae, V.E. ENPP1 in the Regulation of Mineralization and Beyond. Trends Biochem. Sci. 2019, 44, 616–628. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, C.R.; Hackbarth, M.E.; Ziegler, S.G.; Pan, K.S.; Roberts, M.S.; Rosing, D.R.; Whelpley, M.S.; Bryant, J.C.; Macnamara, E.F.; Wang, S.; et al. Prospective phenotyping of long-term survivors of generalized arterial calcification of infancy (GACI). Genet. Med. 2021, 23, 396–407. [Google Scholar] [CrossRef] [PubMed]
- Nitschke, Y.; Yan, Y.; Buers, I.; Kintziger, K.; Askew, K.; Rutsch, F. ENPP1-Fc prevents neointima formation in generalized arterial calcification of infancy through the generation of AMP. Exp. Mol. Med. 2018, 50, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Serrano, R.L.; Yu, W.; Terkeltaub, R. Mono-allelic and bi-allelic ENPP1 deficiency promote post-injury neointimal hyperplasia associated with increased C/EBP homologous protein expression. Atherosclerosis 2014, 233, 493–502. [Google Scholar] [CrossRef]
- Okawa, A.; Nakamura, I.; Goto, S.; Moriya, H.; Nakamura, Y.; Ikegawa, S. Mutation in Npps in a mouse model of ossification of the posterior longitudinal ligament of the spine. Nat. Genet. 1998, 19, 271–273. [Google Scholar] [CrossRef]
- Cheng, Z.; O’Brien, K.; Howe, J.; Sullivan, C.; Schrier, D.; Lynch, A.; Jungles, S.; Sabbagh, Y.; Thompson, D. INZ-701 Prevents Ectopic Tissue Calcification and Restores Bone Architecture and Growth in ENPP1-Deficient Mice. J. Bone Miner. Res. 2021, 36, 1594–1604. [Google Scholar] [CrossRef] [PubMed]
- Dall’Acqua, W.F.; Woods, R.M.; Ward, E.S.; Palaszynski, S.R.; Patel, N.K.; Brewah, Y.A.; Wu, H.; Kiener, P.A.; Langermann, S. Increasing the affinity of a human IgG1 for the neonatal Fc receptor: Biological consequences. J. Immunol. 2002, 169, 5171–5180. [Google Scholar] [CrossRef] [PubMed]
- Rensen, S.S.; Doevendans, P.A.; van Eys, G.J. Regulation and characteristics of vascular smooth muscle cell phenotypic diversity. Neth. Heart J. 2007, 15, 100–108. [Google Scholar] [CrossRef]
- Liu, M.; Gomez, D. Smooth Muscle Cell Phenotypic Diversity. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 1715–1723. [Google Scholar] [CrossRef]
- Sorokin, V.; Vickneson, K.; Kofidis, T.; Woo, C.C.; Lin, X.Y.; Foo, R.; Shanahan, C.M. Role of Vascular Smooth Muscle Cell Plasticity and Interactions in Vessel Wall Inflammation. Front. Immunol. 2020, 11, 599415. [Google Scholar] [CrossRef]
- Enjyoji, K.; Sevigny, J.; Lin, Y.; Frenette, P.S.; Christie, P.D.; Esch, J.S., 2nd; Imai, M.; Edelberg, J.M.; Rayburn, H.; Lech, M.; et al. Targeted disruption of cd39/ATP diphosphohydrolase results in disordered hemostasis and thromboregulation. Nat. Med. 1999, 5, 1010–1017. [Google Scholar] [CrossRef]
- Koziak, K.; Sevigny, J.; Robson, S.C.; Siegel, J.B.; Kaczmarek, E. Analysis of CD39/ATP diphosphohydrolase (ATPDase) expression in endothelial cells, platelets and leukocytes. Thromb. Haemost. 1999, 82, 1538–1544. [Google Scholar] [CrossRef]
- Kansas, G.S.; Wood, G.S.; Tedder, T.F. Expression, distribution, and biochemistry of human CD39. Role in activation-associated homotypic adhesion of lymphocytes. J. Immunol. 1991, 146, 2235–2244. [Google Scholar] [CrossRef]
- Kaczmarek, E.; Koziak, K.; Sevigny, J.; Siegel, J.B.; Anrather, J.; Beaudoin, A.R.; Bach, F.H.; Robson, S.C. Identification and characterization of CD39/vascular ATP diphosphohydrolase. J. Biol. Chem. 1996, 271, 33116–33122. [Google Scholar] [CrossRef] [PubMed]
- Marcus, A.J.; Broekman, M.J.; Drosopoulos, J.H.; Islam, N.; Alyonycheva, T.N.; Safier, L.B.; Hajjar, K.A.; Posnett, D.N.; Schoenborn, M.A.; Schooley, K.A.; et al. The endothelial cell ecto-ADPase responsible for inhibition of platelet function is CD39. J. Clin. Invest. 1997, 99, 1351–1360. [Google Scholar] [CrossRef]
- Antonioli, L.; Pacher, P.; Vizi, E.S.; Hasko, G. CD39 and CD73 in immunity and inflammation. Trends Mol. Med. 2013, 19, 355–367. [Google Scholar] [CrossRef]
- Deaglio, S.; Robson, S.C. Ectonucleotidases as regulators of purinergic signaling in thrombosis, inflammation, and immunity. Adv. Pharmacol. 2011, 61, 301–332. [Google Scholar] [CrossRef]
- Kanthi, Y.M.; Sutton, N.R.; Pinsky, D.J. CD39: Interface between vascular thrombosis and inflammation. Curr. Atheroscler. Rep. 2014, 16, 425. [Google Scholar] [CrossRef]
- Eltzschig, H.K.; Ibla, J.C.; Furuta, G.T.; Leonard, M.O.; Jacobson, K.A.; Enjyoji, K.; Robson, S.C.; Colgan, S.P. Coordinated adenine nucleotide phosphohydrolysis and nucleoside signaling in posthypoxic endothelium: Role of ectonucleotidases and adenosine A2B receptors. J. Exp. Med. 2003, 198, 783–796. [Google Scholar] [CrossRef] [PubMed]
- Marcus, A.J.; Broekman, M.J.; Drosopoulos, J.H.; Olson, K.E.; Islam, N.; Pinsky, D.J.; Levi, R. Role of CD39 (NTPDase-1) in thromboregulation, cerebroprotection, and cardioprotection. Semin. Thromb. Hemost. 2005, 31, 234–246. [Google Scholar] [CrossRef]
- Eltzschig, H.K.; Kohler, D.; Eckle, T.; Kong, T.; Robson, S.C.; Colgan, S.P. Central role of Sp1-regulated CD39 in hypoxia/ischemia protection. Blood 2009, 113, 224–232. [Google Scholar] [CrossRef]
- Helenius, M.H.; Vattulainen, S.; Orcholski, M.; Aho, J.; Komulainen, A.; Taimen, P.; Wang, L.; de Jesus Perez, V.A.; Koskenvuo, J.W.; Alastalo, T.P. Suppression of endothelial CD39/ENTPD1 is associated with pulmonary vascular remodeling in pulmonary arterial hypertension. Am. J. Physiol. Lung Cell Mol. Physiol. 2015, 308, L1046–L1057. [Google Scholar] [CrossRef] [PubMed]
- Roy, C.; Tabiasco, J.; Caillon, A.; Delneste, Y.; Merot, J.; Favre, J.; Guihot, A.L.; Martin, L.; Nascimento, D.C.; Ryffel, B.; et al. Loss of vascular expression of nucleoside triphosphate diphosphohydrolase-1/CD39 in hypertension. Purinergic Signal 2018, 14, 73–82. [Google Scholar] [CrossRef]
- Kanthi, Y.; Hyman, M.C.; Liao, H.; Baek, A.E.; Visovatti, S.H.; Sutton, N.R.; Goonewardena, S.N.; Neral, M.K.; Jo, H.; Pinsky, D.J. Flow-dependent expression of ectonucleotide tri(di)phosphohydrolase-1 and suppression of atherosclerosis. J. Clin. Investig. 2015, 125, 3027–3036. [Google Scholar] [CrossRef]
- Hao, H.; Ma, X.; Chen, H.; Zhu, L.; Xu, Z.; Li, Q.; Xu, C.; Zhang, Y.; Peng, Z.; Wang, M. The cyclic adenosine monophosphate elevating medicine, forskolin, reduces neointimal formation and atherogenesis in mice. J. Cell Mol. Med. 2020, 24, 9638–9645. [Google Scholar] [CrossRef]
- Souness, J.E.; Hassall, G.A.; Parrott, D.P. Inhibition of pig aortic smooth muscle cell DNA synthesis by selective type III and type IV cyclic AMP phosphodiesterase inhibitors. Biochem. Pharmacol. 1992, 44, 857–866. [Google Scholar] [CrossRef] [PubMed]
- Sadhu, D.N.; Ramos, K.S. Cyclic AMP inhibits c-Ha-ras protooncogene expression and DNA synthesis in rat aortic smooth muscle cells. Experientia 1993, 49, 567–570. [Google Scholar] [CrossRef] [PubMed]
- Indolfi, C.; Avvedimento, E.V.; Di Lorenzo, E.; Esposito, G.; Rapacciuolo, A.; Giuliano, P.; Grieco, D.; Cavuto, L.; Stingone, A.M.; Ciullo, I.; et al. Activation of cAMP-PKA signaling in vivo inhibits smooth muscle cell proliferation induced by vascular injury. Nat. Med. 1997, 3, 775–779. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.C.; Hudson, C.A.; Kimura, T.E.; White, S.J.; Sala-Newby, G.B.; Newby, A.C.; Bond, M. Divergent Regulation of Actin Dynamics and Megakaryoblastic Leukemia-1 and -2 (Mkl1/2) by cAMP in Endothelial and Smooth Muscle Cells. Sci. Rep. 2017, 7, 3681. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Koupenova, M.; McCrann, D.J.; Kopeikina, K.J.; Kagan, H.M.; Schreiber, B.M.; Ravid, K. The A2b adenosine receptor protects against vascular injury. Proc. Natl. Acad. Sci. USA 2008, 105, 792–796. [Google Scholar] [CrossRef] [PubMed]
- Dubey, R.K.; Gillespie, D.G.; Mi, Z.; Jackson, E.K. Adenosine inhibits growth of human aortic smooth muscle cells via A2B receptors. Hypertension 1998, 31 Pt 2, 516–521. [Google Scholar] [CrossRef] [PubMed]
- Dubey, R.K.; Fingerle, J.; Gillespie, D.G.; Mi, Z.; Rosselli, M.; Imthurn, B.; Jackson, E.K. Adenosine Attenuates Human Coronary Artery Smooth Muscle Cell Proliferation by Inhibiting Multiple Signaling Pathways That Converge on Cyclin D. Hypertension 2015, 66, 1207–1219. [Google Scholar] [CrossRef] [PubMed]
- Dubey, R.K.; Baruscotti, I.; Stiller, R.; Fingerle, J.; Gillespie, D.G.; Mi, Z.; Leeners, B.; Imthurn, B.; Rosselli, M.; Jackson, E.K. Adenosine, Via A(2B) Receptors, Inhibits Human (P-SMC) Progenitor Smooth Muscle Cell Growth. Hypertension 2020, 75, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Ying, L.; Jin, K.K.; Nan, Y.; Hu, S.; Wu, X.; Qi, R.; Luo, X.; Wang, L. Adenosine A(2A) receptor activation reverses hypoxia-induced rat pulmonary artery smooth muscle cell proliferation via cyclic AMP-mediated inhibition of the SDF1-CXC4 signaling pathway. Int. J. Mol. Med. 2018, 42, 607–614. [Google Scholar] [CrossRef]
- Zernecke, A.; Bidzhekov, K.; Ozuyaman, B.; Fraemohs, L.; Liehn, E.A.; Luscher-Firzlaff, J.M.; Luscher, B.; Schrader, J.; Weber, C. CD73/ecto-5′-nucleotidase protects against vascular inflammation and neointima formation. Circulation 2006, 113, 2120–2127. [Google Scholar] [CrossRef]
- McPherson, J.A.; Barringhaus, K.G.; Bishop, G.G.; Sanders, J.M.; Rieger, J.M.; Hesselbacher, S.E.; Gimple, L.W.; Powers, E.R.; Macdonald, T.; Sullivan, G.; et al. Adenosine A(2A) receptor stimulation reduces inflammation and neointimal growth in a murine carotid ligation model. Arterioscler. Thromb. Vasc. Biol. 2001, 21, 791–796. [Google Scholar] [CrossRef] [PubMed]
- Bot, I.; de Vries, H.; Korporaal, S.J.; Foks, A.C.; Bot, M.; van Veldhoven, J.; Ter Borg, M.N.; van Santbrink, P.J.; van Berkel, T.J.; Kuiper, J.; et al. Adenosine A(2)B receptor agonism inhibits neointimal lesion development after arterial injury in apolipoprotein E-deficient mice. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 2197–2205. [Google Scholar] [CrossRef] [PubMed]
- Ji, Y.; Adeola, O.; Strawn, T.L.; Jeong, S.S.; Chen, R.; Fay, W.P. Recombinant soluble apyrase APT102 inhibits thrombosis and intimal hyperplasia in vein grafts without adversely affecting hemostasis or re-endothelialization. J. Thromb. Haemost. 2017, 15, 814–825. [Google Scholar] [CrossRef] [PubMed]
- Pinsky, D.J. Cd39 as a Critical Ectonucleotidase Defense against Pathological Vascular Remodeling. Trans. Am. Clin. Climatol. Assoc. 2018, 129, 132–139. [Google Scholar]
- Koziak, K.; Bojakowska, M.; Robson, S.C.; Bojakowski, K.; Soin, J.; Csizmadia, E.; Religa, P.; Gaciong, Z.; Kaczmarek, E. Overexpression of CD39/nucleoside triphosphate diphosphohydrolase-1 decreases smooth muscle cell proliferation and prevents neointima formation after angioplasty. J. Thromb. Haemost. 2008, 6, 1191–1197. [Google Scholar] [CrossRef]
- Drosopoulos, J.H.; Kraemer, R.; Shen, H.; Upmacis, R.K.; Marcus, A.J.; Musi, E. Human solCD39 inhibits injury-induced development of neointimal hyperplasia. Thromb. Haemost. 2010, 103, 426–434. [Google Scholar] [CrossRef]
- Tinton, S.; Buc-Calderon, P. Inhibition of protein synthesis induced by adenine nucleotides requires their metabolism into adenosine. Biochem. Pharmacol. 1995, 50, 481–488. [Google Scholar] [CrossRef]
- Dubey, R.K.; Gillespie, D.G.; Jackson, E.K. Adenosine inhibits collagen and total protein synthesis in vascular smooth muscle cells. Hypertension 1999, 33, 190–194. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tchernychev, B.; Nitschke, Y.; Chu, D.; Sullivan, C.; Flaman, L.; O’Brien, K.; Howe, J.; Cheng, Z.; Thompson, D.; Ortiz, D.; et al. Inhibition of Vascular Smooth Muscle Cell Proliferation by ENPP1: The Role of CD73 and the Adenosine Signaling Axis. Cells 2024, 13, 1128. https://doi.org/10.3390/cells13131128
Tchernychev B, Nitschke Y, Chu D, Sullivan C, Flaman L, O’Brien K, Howe J, Cheng Z, Thompson D, Ortiz D, et al. Inhibition of Vascular Smooth Muscle Cell Proliferation by ENPP1: The Role of CD73 and the Adenosine Signaling Axis. Cells. 2024; 13(13):1128. https://doi.org/10.3390/cells13131128
Chicago/Turabian StyleTchernychev, Boris, Yvonne Nitschke, Di Chu, Caitlin Sullivan, Lisa Flaman, Kevin O’Brien, Jennifer Howe, Zhiliang Cheng, David Thompson, Daniel Ortiz, and et al. 2024. "Inhibition of Vascular Smooth Muscle Cell Proliferation by ENPP1: The Role of CD73 and the Adenosine Signaling Axis" Cells 13, no. 13: 1128. https://doi.org/10.3390/cells13131128
APA StyleTchernychev, B., Nitschke, Y., Chu, D., Sullivan, C., Flaman, L., O’Brien, K., Howe, J., Cheng, Z., Thompson, D., Ortiz, D., Rutsch, F., & Sabbagh, Y. (2024). Inhibition of Vascular Smooth Muscle Cell Proliferation by ENPP1: The Role of CD73 and the Adenosine Signaling Axis. Cells, 13(13), 1128. https://doi.org/10.3390/cells13131128