
Mild Hyperthermia-Induced Thermogenesis in the Endoplasmic Reticulum Defines Stress Response Mechanisms

 , , , , , ,
, , , , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines and Culture Conditions
2.2. Fluorescent Labeling for Measuring ER Temperature and Ca2+ Levels
2.2.1. Temperature Determination with ER Thermo Yellow (λex/λem 510/591 nm)
2.2.2. Nuclear Staining with Hoechst 33342 (λex/λem 350/461 nm)
2.2.3. Determination of ER Ca2+ Levels with Mag-Fluo-4 ER (λex/λem 493/517 nm)
2.3. Cell Fixation
2.4. Treatments
2.4.1. Heat-Shock Experiments
2.4.2. Treatment with ER Stressors
2.5. RNA Isolation and Real-Time Quantitative Polymerase Chain Reaction (RT-qPCR)
2.6. Flow Cytometry Analysis
2.6.1. Measuring XBP1 Protein Levels
2.6.2. Measuring ER Ca2+ Levels
2.7. Fluorescence Microscopy Assays
2.7.1. ER Temperature Measurements by ER Thermo Yellow Fluorescence
2.7.2. Super-Resolution Microscopy
3. Results
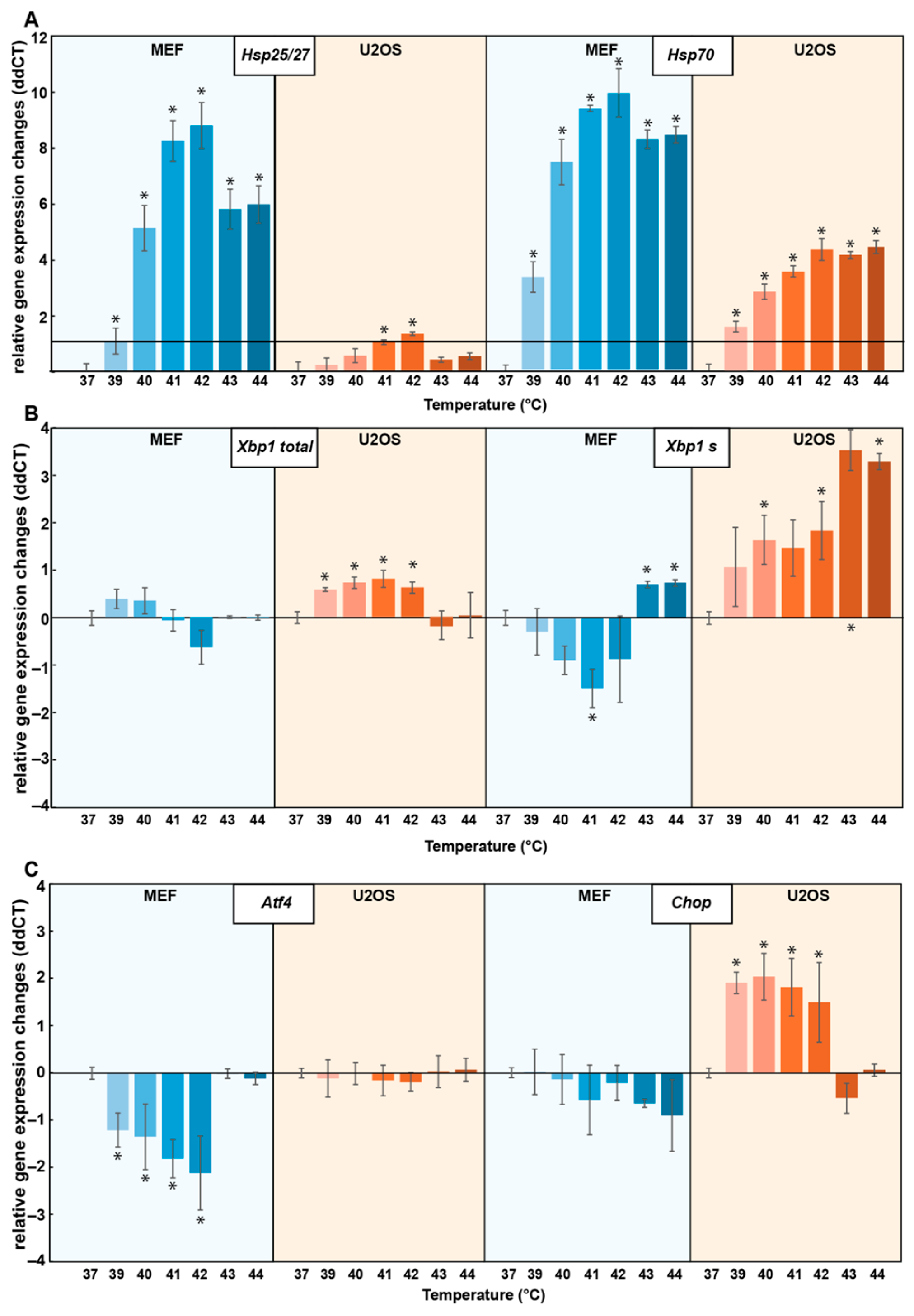
3.1. Distinct Cell Types Show Differential Stress Transcriptome Profiles upon Heat Treatment
3.2. IRE1 Clustering in Response to Mild Heat Treatment of 40 °C
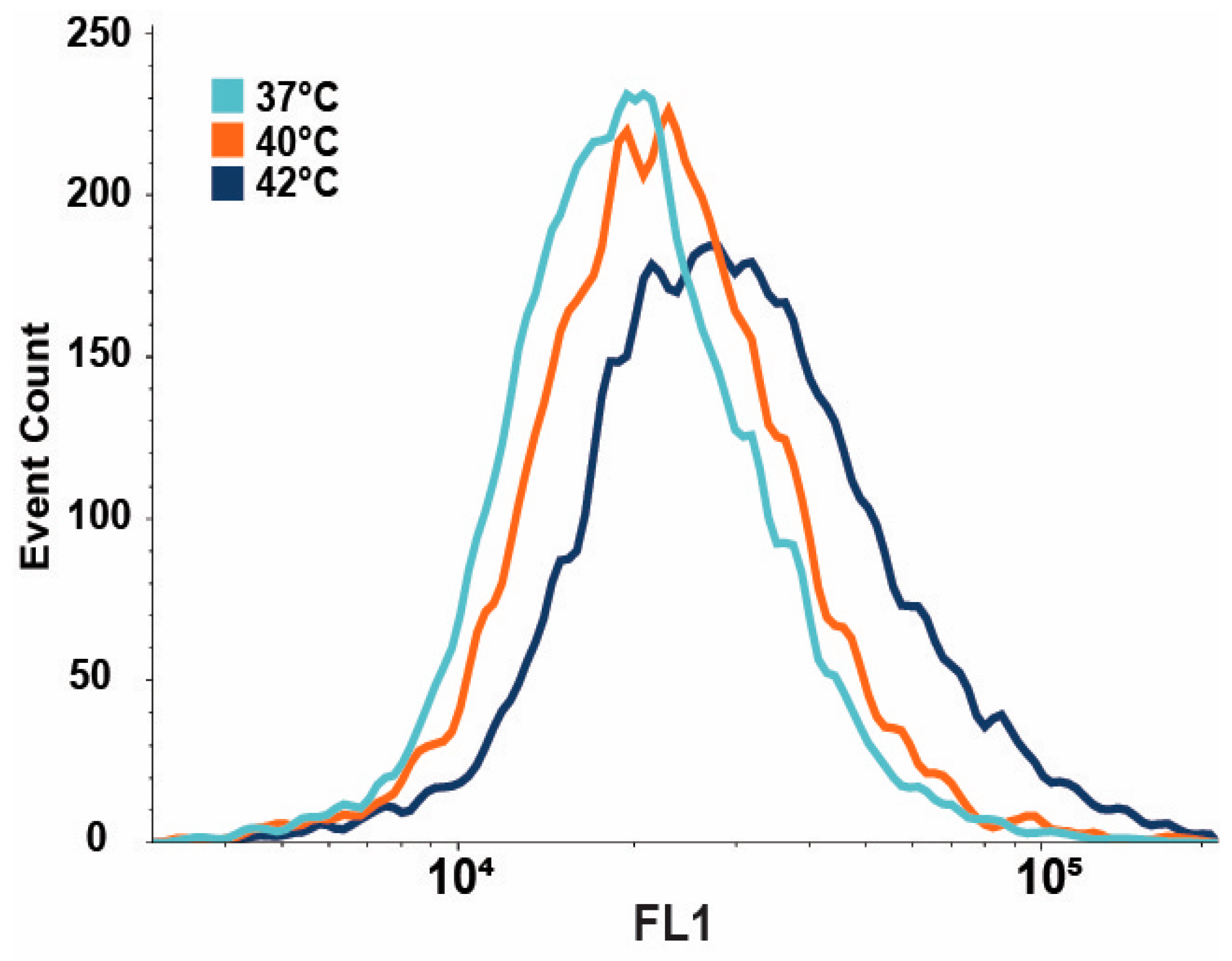
3.3. IRE1-Dependent XBP1 Expression Is Upregulated upon Mild Heat Treatment
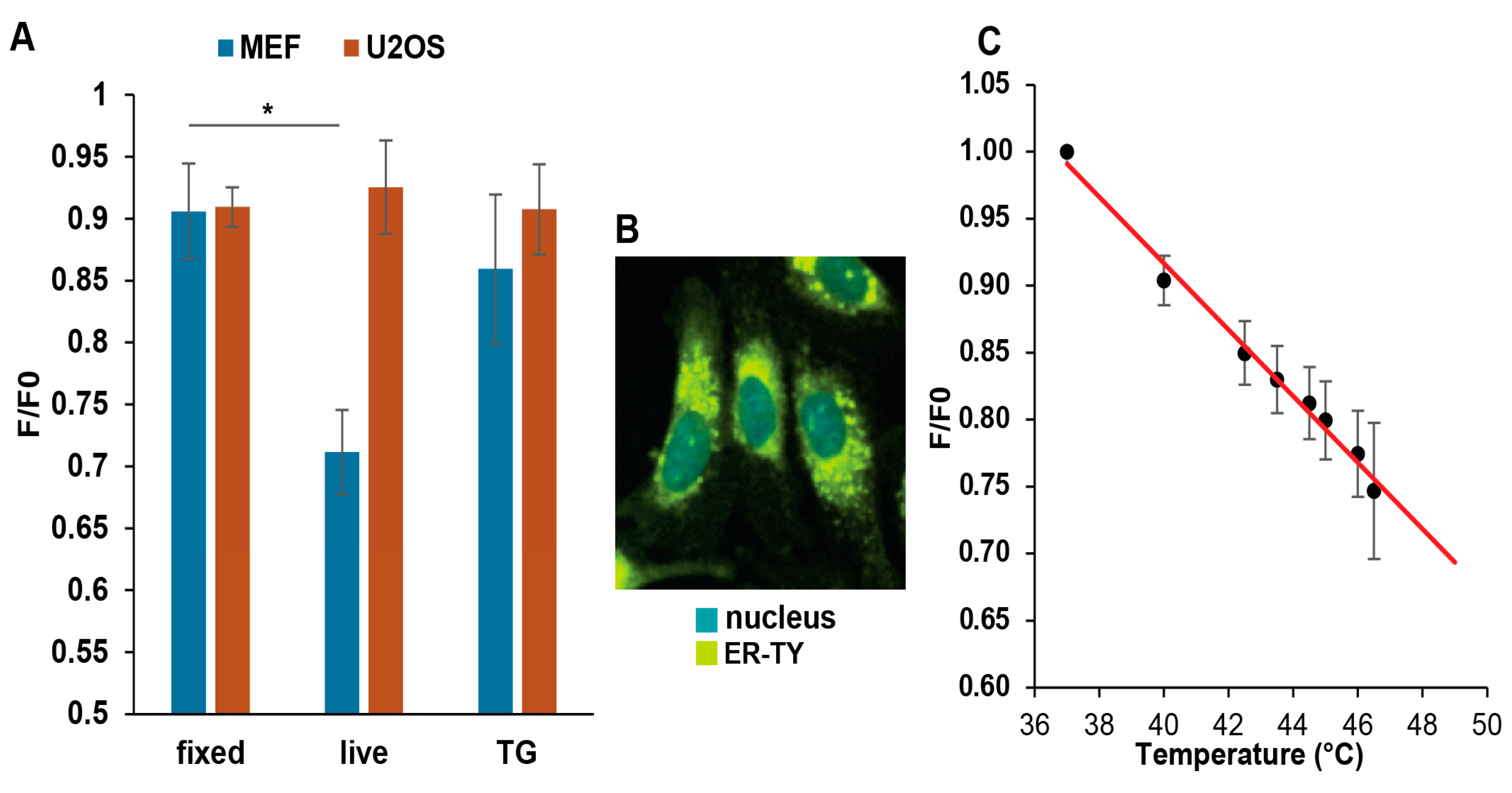
3.4. Mild Heat Could Induce Thermogenesis in the ER
3.5. ER Ca2+ Levels Decrease upon Mild Heat Treatment
4. Discussion
4.1. Interplay between the UPR and HSR upon Heat Stress
4.2. Mild Heat-Induced Intracellular Thermogenesis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Almanza, A.; Carlesso, A.; Chintha, C.; Creedican, S.; Doultsinos, D.; Leuzzi, B.; Luís, A.; McCarthy, N.; Montibeller, L.; More, S.; et al. Endoplasmic Reticulum Stress Signalling – from Basic Mechanisms to Clinical Applications. FEBS J. 2019, 286, 241–278. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Shi, C.; He, M.; Xiong, S.; Xia, X. Endoplasmic Reticulum Stress: Molecular Mechanism and Therapeutic Targets - Signal Transduction and Targeted Therapy. Signal Transduct. Target. Ther. 2023, 8, 352. [Google Scholar] [CrossRef] [PubMed]
- Ron, D.; Walter, P. Signal Integration in the Endoplasmic Reticulum Unfolded Protein Response. Nat. Rev. Mol. Cell Biol. 2007, 8, 519–529. [Google Scholar] [CrossRef]
- Peksel, B.; Gombos, I.; Péter, M.; Vigh, L.; Tiszlavicz, Á.; Brameshuber, M.; Balogh, G.; Schütz, G.J.; Horváth, I.; Vigh, L.; et al. Mild Heat Induces a Distinct “Eustress” Response in Chinese Hamster Ovary Cells but Does Not Induce Heat Shock Protein Synthesis. Sci. Rep. 2017, 7, 15643. [Google Scholar] [CrossRef] [PubMed]
- Tiszlavicz, Á.; Gombos, I.; Péter, M.; Hegedűs, Z.; Hunya, Á.; Dukic, B.; Nagy, I.; Peksel, B.; Balogh, G.; Horváth, I.; et al. Distinct Cellular Tools of Mild Hyperthermia-Induced Acquired Stress Tolerance in Chinese Hamster Ovary Cells. Biomedicines 2022, 10, 1172. [Google Scholar] [CrossRef] [PubMed]
- Bettaieb, A.; Averill-Bates, D.A. Thermotolerance Induced at a Mild Temperature of 40°C Alleviates Heat Shock-Induced ER Stress and Apoptosis in HeLa Cells. Biochim. Biophys. Acta BBA-Mol. Cell Res. 2015, 1853, 52–62. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Gupta, S.; Hu, W.; McGrath, B.C.; Cavener, D.R. Hyperthermia Induces the ER Stress Pathway. PLoS ONE 2011, 6, e23740. [Google Scholar] [CrossRef] [PubMed]
- Adams, C.J.; Kopp, M.C.; Larburu, N.; Nowak, P.R.; Ali, M.M.U. Structure and Molecular Mechanism of ER Stress Signaling by the Unfolded Protein Response Signal Activator IRE1. Front. Mol. Biosci. 2019, 6. [Google Scholar] [CrossRef]
- Lindholm, D.; Korhonen, L.; Eriksson, O.; Kõks, S. Recent Insights into the Role of Unfolded Protein Response in ER Stress in Health and Disease. Front. Cell Dev. Biol. 2017, 5, 48. [Google Scholar] [CrossRef]
- Radanović, T.; Ernst, R. The Unfolded Protein Response as a Guardian of the Secretory Pathway. Cells 2021, 10, 2965. [Google Scholar] [CrossRef]
- Ernst, R.; Ballweg, S.; Levental, I. Cellular Mechanisms of Physicochemical Membrane Homeostasis. Curr. Opin. Cell Biol. 2018, 53, 44–51. [Google Scholar] [CrossRef] [PubMed]
- Fun, X.H.; Thibault, G. Lipid Bilayer Stress and Proteotoxic Stress-Induced Unfolded Protein Response Deploy Divergent Transcriptional and Non-Transcriptional Programmes. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2020, 1865, 158449. [Google Scholar] [CrossRef] [PubMed]
- Gianfrancesco, M.A.; Paquot, N.; Piette, J.; Legrand-Poels, S. Lipid Bilayer Stress in Obesity-Linked Inflammatory and Metabolic Disorders. Biochem. Pharmacol. 2018, 153, 168–183. [Google Scholar] [CrossRef] [PubMed]
- Halbleib, K.; Pesek, K.; Covino, R.; Hofbauer, H.F.; Wunnicke, D.; Hänelt, I.; Hummer, G.; Ernst, R. Activation of the Unfolded Protein Response by Lipid Bilayer Stress. Mol. Cell 2017, 67, 673–684.e8. [Google Scholar] [CrossRef] [PubMed]
- Tam, A.B.; Roberts, L.S.; Chandra, V.; Rivera, I.G.; Nomura, D.K.; Forbes, D.J.; Niwa, M. The UPR Activator ATF6 Responds to Proteotoxic and Lipotoxic Stress by Distinct Mechanisms. Dev. Cell 2018, 46, 327–343.e7. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Taubert, S. Beyond Proteostasis: Lipid Metabolism as a New Player in Er Homeostasis. Metabolites 2021, 11, 52. [Google Scholar] [CrossRef]
- Beignon, F.; Gueguen, N.; Tricoire-Leignel, H.; Mattei, C.; Lenaers, G. The Multiple Facets of Mitochondrial Regulations Controlling Cellular Thermogenesis. Cell. Mol. Life Sci. 2022, 79, 525. [Google Scholar] [CrossRef] [PubMed]
- Chrétien, D.; Bénit, P.; Ha, H.-H.; Keipert, S.; El-Khoury, R.; Chang, Y.-T.; Jastroch, M.; Jacobs, H.T.; Rustin, P.; Rak, M. Mitochondria Are Physiologically Maintained at Close to 50 °C. PLoS Biol. 2018, 16, e2003992. [Google Scholar] [CrossRef] [PubMed]
- Terzioglu, M.; Veeroja, K.; Montonen, T.; Ihalainen, T.O.; Salminen, T.S.; Bénit, P.; Rustin, P.; Chang, Y.-T.; Nagai, T.; Jacobs, H.T. Mitochondrial Temperature Homeostasis Resists External Metabolic Stresses; elife, 2023; 12, RP89232.
- Arai, S.; Lee, S.-C.; Zhai, D.; Suzuki, M.; Chang, Y.T. A Molecular Fluorescent Probe for Targeted Visualization of Temperature at the Endoplasmic Reticulum. Sci. Rep. 2014, 4, 6701. [Google Scholar] [CrossRef] [PubMed]
- Macherel, D.; Haraux, F.; Guillou, H.; Bourgeois, O. The Conundrum of Hot Mitochondria. Biochim. Biophys. Acta - Bioenerg. 2021, 1862, 148348. [Google Scholar] [CrossRef]
- Belyy, V.; Zuazo-Gaztelu, I.; Alamban, A.; Ashkenazi, A.; Walter, P. Endoplasmic Reticulum Stress Activates Human IRE1α through Reversible Assembly of Inactive Dimers into Small Oligomers. eLife 2022, 11, e74342. [Google Scholar] [CrossRef]
- Nougarède, A.; Tesnière, C.; Ylanko, J.; Rimokh, R.; Gillet, G.; Andrews, D.W. Improved IRE1 and PERK Pathway Sensors for Multiplex Endoplasmic Reticulum Stress Assay Reveal Stress Response to Nuclear Dyes Used for Image Segmentation. Assay Drug Dev. Technol. 2018, 16, 350–360. [Google Scholar] [CrossRef]
- Lebeau, P.F.; Platko, K.; Byun, J.H.; Austin, R.C. Calcium as a Reliable Marker for the Quantitative Assessment of Endoplasmic Reticulum Stress in Live Cells. J. Biol. Chem. 2021, 296, 100779. [Google Scholar] [CrossRef]
- Rees, E.J.; Erdelyi, M.; Schierle, G.S.K.; Knight, A.; Kaminski, C.F. Elements of Image Processing in Localization Microscopy. J. Opt. 2013, 15, 094012. [Google Scholar] [CrossRef]
- Belyy, V.; Tran, N.-H.; Walter, P. Quantitative Microscopy Reveals Dynamics and Fate of Clustered IRE1α. Proc. Natl. Acad. Sci. USA 2020, 117, 1533–1542. [Google Scholar] [CrossRef]
- Meis, L. Ca2+-ATPases (SERCA): Energy Transduction and Heat Production in Transport ATPases. J. Membr. Biol. 2002, 188, 1–9. [Google Scholar] [CrossRef]
- Rutkowski, D.T.; Hegde, R.S. Regulation of Basal Cellular Physiology by the Homeostatic Unfolded Protein Response. J. Cell Biol. 2010, 189, 783–794. [Google Scholar] [CrossRef]
- Marcu, M.G.; Doyle, M.; Bertolotti, A.; Ron, D.; Hendershot, L.; Neckers, L. Heat Shock Protein 90 Modulates the Unfolded Protein Response by Stabilizing IRE1alpha. Mol. Cell. Biol. 2002, 22, 8506–8513. [Google Scholar] [CrossRef]
- Somogyvári, M.; Khatatneh, S.; Sőti, C. Hsp90: From Cellular to Organismal Proteostasis. Cells 2022, 11, 2479. [Google Scholar] [CrossRef]
- Walczak, A.; Gradzik, K.; Kabzinski, J.; Przybylowska-Sygut, K.; Majsterek, I. The Role of the ER-Induced UPR Pathway and the Efficacy of Its Inhibitors and Inducers in the Inhibition of Tumor Progression. Oxid. Med. Cell. Longev. 2019, 2019, 5729710. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.; Tang, H.; Liu, Z.; Österlund, T.; Nielsen, J.; Petranovic, D. Management of the Endoplasmic Reticulum Stress by Activation of the Heat Shock Response in Yeast. FEMS Yeast Res. 2014, 14, 481–494. [Google Scholar] [CrossRef] [PubMed]
- Arvan, P.; Zhao, X.; Ramos-Castaneda, J.; Chang, A. Secretory Pathway Quality Control Operating in Golgi, Plasmalemmal, and Endosomal Systems. Traffic Cph. Den. 2002, 3, 771–780. [Google Scholar] [CrossRef] [PubMed]
- Alagar Boopathy, L.R.; Jacob-Tomas, S.; Alecki, C.; Vera, M. Mechanisms Tailoring the Expression of Heat Shock Proteins to Proteostasis Challenges. J. Biol. Chem. 2022, 298, 101796. [Google Scholar] [CrossRef] [PubMed]
- Shyu, P.; Ng, B.S.H.; Ho, N.; Chaw, R.; Seah, Y.L.; Marvalim, C.; Thibault, G. Membrane Phospholipid Alteration Causes Chronic ER Stress through Early Degradation of Homeostatic ER-Resident Proteins. Sci. Rep. 2019, 9, 8637. [Google Scholar] [CrossRef] [PubMed]
- Celik, C.; Lee, S.Y.T.; Yap, W.S.; Thibault, G. Endoplasmic Reticulum Stress and Lipids in Health and Diseases. Prog. Lipid Res. 2023, 89, 101198. [Google Scholar] [CrossRef] [PubMed]
- Navarro-Tapia, E.; Querol, A.; Pérez-Torrado, R. Membrane Fluidification by Ethanol Stress Activates Unfolded Protein Response in Yeasts. Microb. Biotechnol. 2018, 11, 465–475. [Google Scholar] [CrossRef] [PubMed]
- Renne, M.F.; Ernst, R. Membrane Homeostasis beyond Fluidity: Control of Membrane Compressibility. Trends Biochem. Sci. 2023, 48, 963–977. [Google Scholar] [CrossRef]
- Kučerka, N.; Nieh, M.-P.; Katsaras, J. Fluid Phase Lipid Areas and Bilayer Thicknesses of Commonly Used Phosphatidylcholines as a Function of Temperature. Biochim. Biophys. Acta BBA - Biomembr. 2011, 1808, 2761–2771. [Google Scholar] [CrossRef]
- Pan, J.; Tristram-Nagle, S.; Kučerka, N.; Nagle, J.F. Temperature Dependence of Structure, Bending Rigidity, and Bilayer Interactions of Dioleoylphosphatidylcholine Bilayers. Biophys. J. 2008, 94, 117–124. [Google Scholar] [CrossRef]
- Kono, N.; Amin-Wetzel, N.; Ron, D. Generic Membrane-Spanning Features Endow IRE1α with Responsiveness to Membrane Aberrancy. Mol. Biol. Cell 2017, 28, 2318–2332. [Google Scholar] [CrossRef]
- Fedorenko, A.; Lishko, P.V.; Kirichok, Y. Mechanism of Fatty-Acid-Dependent UCP1 Uncoupling in Brown Fat Mitochondria. Cell 2012, 151, 400–413. [Google Scholar] [CrossRef] [PubMed]
- Bal, N.C.; Maurya, S.K.; Sopariwala, D.H.; Sahoo, S.K.; Gupta, S.C.; Shaikh, S.A.; Pant, M.; Rowland, L.A.; Bombardier, E.; Goonasekera, S.A.; et al. Sarcolipin Is a Newly Identified Regulator of Muscle-Based Thermogenesis in Mammals. Nat. Med. 2012, 18, 1575–1579. [Google Scholar] [CrossRef] [PubMed]
- Bal, N.C.; Periasamy, M. Uncoupling of Sarcoendoplasmic Reticulum Calcium ATPase Pump Activity by Sarcolipin as the Basis for Muscle Non-Shivering Thermogenesis. Philos. Trans. R. Soc. B Biol. Sci. 2020, 375, 20190135. [Google Scholar] [CrossRef] [PubMed]
- Block, B.A. Thermogenesis in Muscle. Annu. Rev. Physiol. 1994, 56, 535–577. [Google Scholar] [CrossRef] [PubMed]
- Andersen, O.S.; Koeppe, R.E. Bilayer Thickness and Membrane Protein Function: An Energetic Perspective. Annu. Rev. Biophys. Biomol. Struct. 2007, 36, 107–130. [Google Scholar] [CrossRef] [PubMed]
- Caffrey, M.; Feigenson, G.W. Fluorescence Quenching in Model Membranes. 3. Relationship between Calcium Adenosinetriphosphatase Enzyme Activity and the Affinity of the Protein for Phosphatidylcholines with Different Acyl Chain Characteristicst. Biochemistry 1981, 20, 1949–1961. [Google Scholar] [CrossRef] [PubMed]
- Karlovská, J.; Uhríková, D.; Kučerka, N.; Teixeira, J.; Devínsky, F.; Lacko, I.; Balgavý, P. Influence of N-Dodecyl-N,N-Dimethylamine N-Oxide on the Activity of Sarcoplasmic Reticulum Ca2+-Transporting ATPase Reconstituted into Diacylphosphatidylcholine Vesicles: Effects of Bilayer Physical Parameters. Biophys. Chem. 2006, 119, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Lepock, J.R. How Do Cells Respond to Their Thermal Environment? Int. J. Hyperthermia 2005, 21, 681–687. [Google Scholar] [CrossRef] [PubMed]
- Kriszt, R.; Arai, S.; Itoh, H.; Lee, M.H.; Goralczyk, A.G.; Ang, X.M.; Cypess, A.M.; White, A.P.; Shamsi, F.; Xue, R.; et al. Optical Visualisation of Thermogenesis in Stimulated Single-Cell Brown Adipocytes. Sci. Rep. 2017, 7, 1383. [Google Scholar] [CrossRef]
- Ikeda, K.; Kang, Q.; Yoneshiro, T.; Camporez, J.P.; Maki, H.; Homma, M.; Shinoda, K.; Chen, Y.; Lu, X.; Maretich, P.; et al. UCP1-Independent Signaling Involving SERCA2b-Mediated Calcium Cycling Regulates Beige Fat Thermogenesis and Systemic Glucose Homeostasis. Nat. Med. 2017, 23, 1454–1465. [Google Scholar] [CrossRef]
- Török, Z.; Crul, T.; Maresca, B.; Schütz, G.J.; Viana, F.; Dindia, L.; Piotto, S.; Brameshuber, M.; Balogh, G.; Péter, M.; et al. Plasma Membranes as Heat Stress Sensors: From Lipid-Controlled Molecular Switches to Therapeutic Applications. Biochim. Biophys. Acta 2014, 1838, 1594–1618. [Google Scholar] [CrossRef] [PubMed]
- Oka, T. Stress-Induced Hyperthermia and Hypothermia. Handb. Clin. Neurol. 2018, 157, 599–621. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Chang, A. Heat Shock Response Relieves ER Stress. EMBO J. 2008, 27, 1049–1059. [Google Scholar] [CrossRef] [PubMed]
- Balogh, G.; Péter, M.; Glatz, A.; Gombos, I.; Török, Z.; Horváth, I.; Harwood, J.L.; Vígh, L. Key Role of Lipids in Heat Stress Management. FEBS Lett. 2013, 587, 1970–1980. [Google Scholar] [CrossRef] [PubMed]
- Vigh, L.; Maresca, B.; Harwood, J.L. Does the Membrane’s Physical State Control the Expression of Heat Shock and Other Genes? Trends Biochem. Sci. 1998, 23, 369–374. [Google Scholar] [CrossRef]
- Horváth, I.; Multhoff, G.; Sonnleitner, A.; Vígh, L. Membrane-Associated Stress Proteins: More than Simply Chaperones. Biochim. Biophys. Acta BBA-Biomembr. 2008, 1778, 1653–1664. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C.; Saxena, S. ER Stress and the Unfolded Protein Response in Neurodegeneration. Nat. Rev. Neurol. 2017, 13, 477–491. [Google Scholar] [CrossRef] [PubMed]
- Cnop, M.; Foufelle, F.; Velloso, L.A. Endoplasmic Reticulum Stress, Obesity and Diabetes. Trends Mol. Med. 2012, 18, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Fajardo, V.A.; Trojanowski, N.; Castelli, L.M.; Miotto, P.M.; Amoye, F.; Ward, W.E.; Tupling, A.R.; LeBlanc, P.J. Saturation of SERCA’s Lipid Annulus May Protect against Its Thermal Inactivation. Biochem. Biophys. Res. Commun. 2017, 484, 456–460. [Google Scholar] [CrossRef]
- Kunze, C.; Luijckx, P.; Jackson, A.L.; Donohue, I. Alternate Patterns of Temperature Variation Bring about Very Different Disease Outcomes at Different Mean Temperatures. eLife 2022, 11, e72861. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dukic, B.; Ruppert, Z.; Tóth, M.E.; Hunya, Á.; Czibula, Á.; Bíró, P.; Tiszlavicz, Á.; Péter, M.; Balogh, G.; Erdélyi, M.; et al. Mild Hyperthermia-Induced Thermogenesis in the Endoplasmic Reticulum Defines Stress Response Mechanisms. Cells 2024, 13, 1141. https://doi.org/10.3390/cells13131141
Dukic B, Ruppert Z, Tóth ME, Hunya Á, Czibula Á, Bíró P, Tiszlavicz Á, Péter M, Balogh G, Erdélyi M, et al. Mild Hyperthermia-Induced Thermogenesis in the Endoplasmic Reticulum Defines Stress Response Mechanisms. Cells. 2024; 13(13):1141. https://doi.org/10.3390/cells13131141
Chicago/Turabian StyleDukic, Barbara, Zsófia Ruppert, Melinda E. Tóth, Ákos Hunya, Ágnes Czibula, Péter Bíró, Ádám Tiszlavicz, Mária Péter, Gábor Balogh, Miklós Erdélyi, and et al. 2024. "Mild Hyperthermia-Induced Thermogenesis in the Endoplasmic Reticulum Defines Stress Response Mechanisms" Cells 13, no. 13: 1141. https://doi.org/10.3390/cells13131141