Mechanisms Underlying Hepatitis C Virus-Associated Hepatic Fibrosis



{kind=link}
{kind=link}
Abstract
:1. Introduction
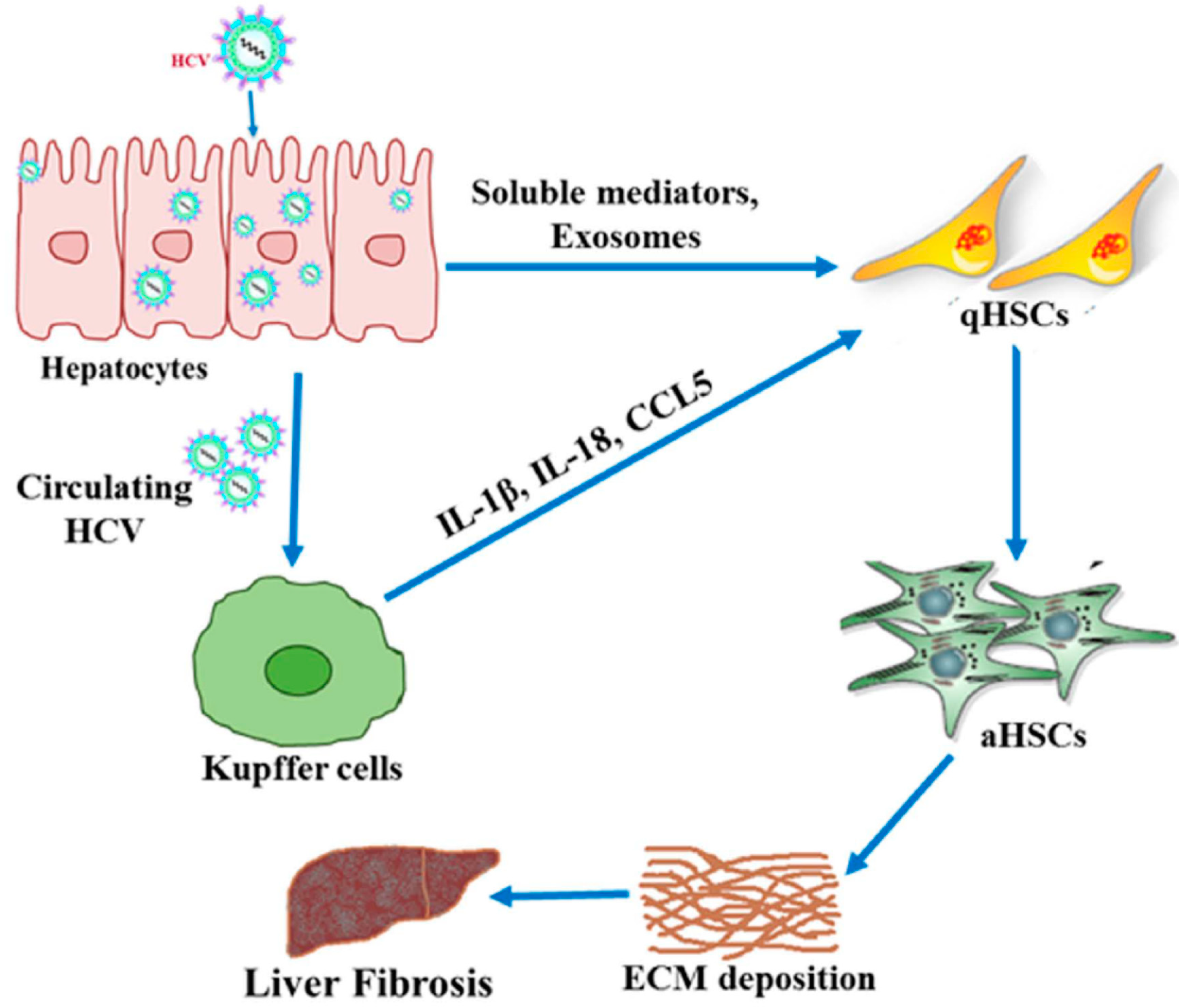
2. Mechanisms of HCV-Associated Liver Fibrosis
2.1. Inflammation
2.2. Role of microRNAs
2.3. Long Non-Coding RNAs
2.4. Exosomes
3. Fibrosis Markers
4. Therapies
5. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Ray, R.B.; Ray, R. Hepatitis C virus manipulates humans as its favorite host for long term relationship. Hepatology 2018, 69, 889–900. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization Fact Sheet. Available online: https://www.who.int/news-room/fact-sheets/detail/hepatitis-c (accessed on 12 October 2019).
- Ekpanyapong, S.; Reddy, K.R. Hepatitis C virus therapy in advanced liver disease: Outcomes and challenges. United Eur. Gastroenterol. J. 2019, 7, 642–650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsuchida, T.; Friedman, S.L. Mechanisms of hepatic stellate cell activation. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 397–411. [Google Scholar] [CrossRef] [PubMed]
- Sebastiani, G.; Gkouvatsos, K.; Pantopoulos, K. Chronic hepatitis C and liver fibrosis. World J. Gastroenterol. 2014, 20, 11033–11053. [Google Scholar] [CrossRef] [PubMed]
- Friedman, S.L. Mechanisms of Hepatic Fibrogenesis. Gastroenterology 2008, 134, 1655–1669. [Google Scholar] [CrossRef] [Green Version]
- Friedman, S.L. Mechanisms of disease: Mechanisms of hepatic fibrosis and therapeutic implications. Nat. Clin. Pr. Gastroenterol. Hepatol. 2004, 1, 98–105. [Google Scholar] [CrossRef]
- Salas-Villalobosa, T.B.; Lozano-Sepúlvedaa, S.A.; Rincón-Sánchezb, A.R.; Govea-Salasc, M.; Rivas-Estilla, A.M. Mechanisms involved in liver damage resolution after hepatitis C virus clearance. Med. Univ. 2017, 19, 100–107. [Google Scholar] [CrossRef]
- Jaroszewicz, J.; Flisiak-Jackiewicz, M.; Lebensztejn, D.; Flisiak, R. Current drugs in early development for treating hepatitis C virus-related hepatic fibrosis. Expert Opin. Investig. Drugs 2015, 24, 1229–1239. [Google Scholar] [CrossRef]
- Friedman, S.L. Evolving challenges in hepatic fibrosis. Nat. Rev. Gastroenterol. Hepatol. 2010, 7, 425–436. [Google Scholar] [CrossRef]
- Finkelmeier, F.; Dultz, G.; Peiffer, K.H.; Kronenberger, B.; Krauss, F.; Zeuzem, S.; Sarrazin, C.; Vermehren, J.; Waidmann, O. Risk of de novo hepatocellular carcinoma after HCV treatment with direct-acting antivirals. Liver Cancer 2018, 7, 190–204. [Google Scholar] [CrossRef]
- Waheed, Y. Ledipasvir and sofosbuvir: Interferon free therapy for hepatitis C virus genotype 1 infection. World J. Virol. 2015, 4, 33–35. [Google Scholar] [CrossRef] [PubMed]
- Kanwal, F.; Kramer, J.; Asch, S.M.; Chayanupatkul, M.; Cao, Y.; El-Serag, H.B. Risk of hepatocellular cancer in HCV patients treated with direct-acting antiviral agents. Gastroenterology 2017, 153, 996–1005. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Deng, H.; Cui, H.; Fang, J.; Zuo, Z.; Deng, J.; Li, Y.; Wang, X.; Zhao, L. Inflammatory responses and inflammation-associated diseases in organs. Oncotarget 2018, 9, 7204–7218. [Google Scholar] [CrossRef] [PubMed]
- Ghany, M.G.; Kleiner, D.E.; Alter, H.; Doo, E.; Khokar, F.; Promrat, K.; Herion, D.; Park, Y.; Liang, T.J.; Hoofnagle, J.H. Progression of fibrosis in chronic hepatitis C. Gastroenterology 2003, 124, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Dejan, B.; Vuk, V.; Suzana, P.; Danijela, J.; Slobodanka, M.; Predrag, D.; Dusko, A.; Aleksandra, A.; Dragic, B.; Jelena, C.; et al. Chronic hepatitis C: Conspectus of immunological events in the course of fibrosis evolution. PLoS ONE 2019, 14, e0219508. [Google Scholar]
- Shrivastava, S.; Mukherjee, A.; Ray, R.; Ray, R.B. Hepatitis C virus induces interleukin-1β (IL-1β)/IL-18 in circulatory and resident liver macrophages. J. Virol. 2013, 87, 12284–12290. [Google Scholar] [CrossRef]
- Capone, F.; Guerriero, E.; Colonna, G.; Maio, P.; Mangia, A.; Castello, G.; Costantini, S. Cytokinome profile evaluation in patients with hepatitis C virus infection. World J. Gastroenterol. 2014, 20, 9261–9269. [Google Scholar]
- Negash, A.A.; Ramos, H.J.; Crochet, N.; Lau, D.T.; Doehle, B.; Papic, N.; Delker, D.A.; Jo, J.; Bertoletti, A.; Hagedorn, C.H.; et al. IL-1β production through the NLRP3 inflammasome by hepatic macrophages links hepatitis C virus infection with liver inflammation and disease. PLoS Pathog. 2013, 9, e1003330. [Google Scholar] [CrossRef]
- Schroder, K.; Tschopp, J. The inflammasomes. Cell 2010, 140, 821–832. [Google Scholar] [CrossRef]
- van de Veerdonk, F.L.; Netea, M.G.; Dinarello, C.A.; Joosten, L.A. Inflammasome activation and IL-1β and IL-18 processing during infection. Trends Immunol. 2011, 32, 110–116. [Google Scholar] [CrossRef]
- Schulze-Krebs, A.; Preimel, D.; Popov, Y.; Bartenschlager, R.; Lohmann, V.; Pinzani, M.; Schuppan, D. Hepatitis C virus-replicating hepatocytes induce fibrogenic activation of hepatic stellate cells. Gastroenterology 2005, 129, 246–258. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, R.; Devhare, P.B.; Steele, R.; Ray, R.; Ray, R.B. Hepatitis C virus induced CCL5 secretion from macrophages activates hepatic stellate cells. Hepatology 2017, 66, 746–757. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, R.; Devhare, P.B.; Ray, R.B.; Ray, R. Hepatitis c virus induced tumor initiating cancer stem-like cells activate stromal fibroblasts in xenograft tumor model. Hepatology 2017, 66, 1766–1778. [Google Scholar] [CrossRef] [PubMed]
- Luan, J.; Ju, D. Inflammasome: A double-edged sword in liver diseases. Front. Immunol. 2018, 9, 2201. [Google Scholar] [CrossRef]
- Lin, W.; Weinberg, E.M.; Chung, R.T. Pathogenesis of accelerated fibrosis in HIV/HCV co-infection. J. Infect. Dis. 2013, 207, S13–S18. [Google Scholar] [CrossRef]
- Lin, W.; Tsai, W.; Shao, R.; Wu, G.; Peng, L.F.; Barlow, L.L.; Chung, W.J.; Zhang, L.; Zhao, H.; Jang, J. HCV regulates TGF-β1 production through the generation of reactive oxygen species in an NFκB-dependent manner. Gastroenterology 2010, 138, 2509–2518. [Google Scholar] [CrossRef]
- Lin, W.; Wu, G.; Li, S.; Weinberg, E.M.; Kumthip, K.; Peng, L.F.; Me’ndez-Navarro, J.; Chen, W.; Jilg, W.; Zhao, H. HIV and HCV cooperatively promote hepatic fibrogenesis via induction of reactive oxygen species and NFĸB. J. Biol. Chem. 2011, 286, 2665–2674. [Google Scholar] [CrossRef]
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef]
- Shrivastava, S.; Petrone, J.; Steele, R.; Lauer, G.M.; Di Bisceglie, A.M.; Ray, R.B. Upregulation of circulating miR-20a is correlated with hepatitis C virus-mediated liver disease progression. Hepatology 2013, 58, 863–871. [Google Scholar] [CrossRef]
- Matsuura, K.; Giorgi, V.D.; Schechterly, C.; Wang, R.Y.; Farci, P.; Tanaka, Y.; Alter, H.J. Circulating let-7 levels in plasma and extracellular vesicles correlate with hepatic fibrosis progression in chronic hepatitis C. Hepatology 2016, 64, 732–745. [Google Scholar] [CrossRef]
- Li, H.; Jiang, J.D.; Peng, Z.G. MicroRNA-mediated interactions between host and hepatitis C virus. World J. Gastroenterol. 2016, 22, 1487–1496. [Google Scholar] [CrossRef]
- Coll, M.; El Taghdouini, A.; Perea, L.; Mannaerts, I.; Vila-Casadesus, M.; Blaya, D.; Rodrigo-Torres, D.; Affò, S.; Morales-Ibanez, O.; Graupera, I.; et al. Integrative miRNA and gene expression profiling analysis of human quiescent hepatic stellate cells. Sci. Rep. 2015, 5, 11549. [Google Scholar] [CrossRef]
- Yu, F.; Lu, Z.; Huang, K.; Wang, X.; Xu, Z.; Chen, B.; Dong, P.; Zheng, J. MicroRNA-17-5p-activated Wnt/beta-catenin pathway contributes to the progression of liver fibrosis. Oncotarget 2016, 7, 81–93. [Google Scholar]
- Tzur, G.; Israel, A.; Levy, A.; Benjamin, H.; Meiri, E.; Shufaro, Y.; Meir, K.; Khvalevsky, E.; Spector, Y.; Rojansky, N.; et al. Comprehensive gene and microRNA expression profiling reveals a role for microRNAs in human liver development. PLoS ONE 2009, 4, e7511. [Google Scholar] [CrossRef]
- Venugopal, S.K.; Jiang, J.; Kim, T.H.; Li, Y.; Wang, S.S.; Torok, N.J.; Wu, J.; Zern, M.A. Liver fibrosis causes downregulation of miRNA-150 and miRNA-194 in hepatic stellate cells, and their overexpression causes decreased stellate cell activation. Am. J. Physiol. Gastrointest. Liver Physiol. 2010, 298, G101–G106. [Google Scholar] [CrossRef] [Green Version]
- Ji, J.; Zhang, J.; Huang, G.; Qian, J.; Wang, X.; Mei, S. Over-expressed microRNA-27a and 27b influence fat accumulation and cell proliferation during rat hepatic stellate cell activation. FEBS Lett. 2009, 583, 759–766. [Google Scholar] [CrossRef] [Green Version]
- Bandyopadhyay, S.; Friedman, R.C.; Marquez, R.T. Hepatitis C virus infection and hepatic stellate cell activation downregulate miR-29: miR-29 overexpression reduces hepatitis C viral abundance in culture. J. Infect. Dis. 2011, 203, 1753–1762. [Google Scholar] [CrossRef]
- Sarma, N.J.; Tiriveedhi, V.; Crippin, J.S.; Chapman, W.C.; Mohanakumar, T. Hepatitis C virus-induced changes in microRNA 107 (miRNA-107) and miRNA-449a modulate CCL2 by targeting the interleukin-6 receptor complex in hepatitis. J. Virol. 2014, 88, 3733–3743. [Google Scholar] [CrossRef]
- Sarma, N.J.; Tiriveedhi, V.; Subramanian, V. Hepatitis C virus mediated changes in miRNA-449a modulates inflammatory biomarker YKL40 through components of the NOTCH signaling pathway. PLoS ONE 2012, 7, e50826. [Google Scholar] [CrossRef]
- Shaker, O.G.; Senousy, M.A. Serum microRNAs as predictors for liver fibrosis staging in hepatitis C virus-associated chronic liver disease patients. J. Viral Hepat. 2017, 24, 636–644. [Google Scholar] [CrossRef]
- Abdel-Al, A.; El-Ahwany, E.; Zoheiry, M.; Hassan, M.; Ouf, A.; Abu-Taleb, H.; Abdel Rahim, A.; El-Talkawy, M.D.; Zada, S. miRNA-221 and miRNA-222 are promising biomarkers for progression of liver fibrosis in HCV Egyptian patients. Virus Res. 2018, 253, 135–139. [Google Scholar] [CrossRef]
- Halász, T.; Horváth, G.; Pár, G.; Werling, K.; Kiss, A.; Schaff, Z.; Lendvai, G. miR-122 negatively correlates with liver fibrosis as detected by histology and FibroScan. World J. Gastroenterol. 2015, 21, 7814–7823. [Google Scholar] [CrossRef]
- Khanizadeh, S.; Ravanshad, M.; Hosseini, S.Y.; Davoodian, P.; Zadeh, A.N.; Sabahi, F.; Sarvari, J.; Khanlari, Z.; Hasani-Azad, M. The possible role of NS3 protease activity of hepatitis C virus on fibrogenesis and miR-122 expression in hepatic stellate cells. Acta Virol. 2016, 60, 242–248. [Google Scholar] [CrossRef] [Green Version]
- Lee, T.H.; Matta, B.; King, B.D.; Hodges, M.R.; Tillmann, H.L.; Patel, K. MicroRNA-122 associates with serum apolipoprotein B but not liver fibrosis markers in CHC genotype 1 infection. J. Med. Virol. 2015, 87, 1722–1726. [Google Scholar] [CrossRef]
- Marquez, R.T.; Bandyopadhyay, S.; Wendlandt, E.B. Correlation between microRNA expression levels and clinical parameters associated with chronic hepatitis C viral infection in humans. Lab Invest. 2010, 90, 1727–1736. [Google Scholar] [CrossRef] [Green Version]
- Zhu, B.; Wei, X.X.; Wang, T.B.; Zhou, Y.C.; Liu, A.M.; Zhang, G.W. Increased miR-16 expression induced by hepatitis C virus infection promotes liver fibrosis through downregulation of hepatocyte growth factor and Smad7. Arch. Virol. 2015, 160, 2043–2050. [Google Scholar] [CrossRef]
- Ramachandran, S.; Ilias-Basha, H.; Sarma, N.J. Hepatitis C virus induced miR200c down modulates FAP-1, a negative regulator of Src signaling and promotes hepatic fibrosis. PLoS ONE 2013, 8, e70744. [Google Scholar] [CrossRef]
- Birney, E.; Stamatoyannopoulos, J.A.; Dutta, A.; Guigó, R.; Gingeras, T.R.; Margulies, E.H.; Weng, Z.; Snyder, M.; Dermitzakis, E.T.; Thurman, R.E.; et al. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature 2007, 447, 799–816. [Google Scholar] [Green Version]
- Ponting, C.P.; Oliver, P.L.; Reik, W. Evolution and functions of long noncoding RNAs. Cell 2009, 136, 629–641. [Google Scholar] [CrossRef]
- Barriocanal, M.; Fortes, P. Long non-coding RNAs in hepatitis C virus-infected cells. Front. Microbiol. 2017, 8, 1833. [Google Scholar] [CrossRef]
- Hudson, W.H.; Prokhnevska, N.; Gensheimer, J.; Akondy, R.; McGuire, D.J.; Ahmed, R.; Kissick, H.T. Expression of novel long noncoding RNAs defines virus-specific effector and memory CD8+ T cells. Nat. Commun. 2019, 10, 196. [Google Scholar] [CrossRef]
- Yuan, J.H.; Yang, F.; Wang, F.; Ma, J.Z.; Guo, Y.J.; Tao, Q.F.; Liu, F.; Pan, W.; Wang, T.T.; Zhou, C.C.; et al. A long noncoding RNA activated by TGF-β promotes the invasion-metastasis cascade in hepatocellular carcinoma. Cancer Cell 2014, 25, 666–681. [Google Scholar] [CrossRef]
- Qian, X.; Xu, C.; Zhao, P.; Qi, Z. Long non-coding RNA GAS5 inhibited hepatitis C virus replication by binding viral NS3 protein. Virology 2016, 492, 155–165. [Google Scholar] [CrossRef]
- Plissonnier, M.L.; Herzog, K.; Levrero, M.; Zeisel, M.B. Non-coding RNAs and hepatitis C virus-induced hepatocellular carcinoma. Viruses 2018, 10, E591. [Google Scholar] [CrossRef]
- Sur, S.; Sasaki, R.; Devhare, P.; Steele, R.; Ray, R.; Ray, R.B. Association between microRNA-373 and long noncoding RNA NORAD in hepatitis C virus-infected hepatocytes impairs Wee1 expression for growth promotion. J. Virol. 2018, 92, e01215–e01218. [Google Scholar] [CrossRef]
- Fu, N.; Zhao, S.X.; Kong, L.B.; Du, J.H.; Ren, W.G.; Han, F.; Zhang, Q.S.; Li, W.C.; Cui, P.; Wang, R.Q.; et al. LncRNA-ATB/microRNA-200a/β-catenin regulatory axis involved in the progression of HCV-related hepatic fibrosis. Gene 2017, 618, 1–7. [Google Scholar] [CrossRef]
- Fu, N.; Niu, X.; Wang, Y.; Du, H.; Wang, B.; Du, J.; Li, Y.; Wang, R.; Zhang, Y.; Zhao, S.; et al. Role of lncRNA-activated by transforming growth factor beta in the progression of hepatitis C virus-related liver fibrosis. Discov. Med. 2016, 22, 29–42. [Google Scholar]
- Théry, C.; Zitvogel, L.; Amigorena, S. Exosomes: Composition, biogenesis and function. Nat. Rev. Immunol. 2002, 2, 569–579. [Google Scholar] [CrossRef]
- Schorey, J.S.; Cheng, Y.; Singh, P.P.; Smith, V.L. Exosomes and other extracellular vesicles in host-pathogen interactions. EMBO Rep. 2015, 16, 24–43. [Google Scholar] [CrossRef]
- Devhare, P.B.; Ray, R.B. Extracellular vesicles: Novel mediator for cell to cell communications in liver pathogenesis. Mol. Asp. Med. 2018, 60, 115–122. [Google Scholar] [CrossRef]
- Ramakrishnaiah, V.; Thumann, C.; Fofana, I.; Habersetzer, F.; Pan, Q.; de Ruiter, P.E.; Willemsen, R.; Demmers, J.A.; Stalin Raj, V.; Jenster, G.; et al. Exosome-mediated transmission of hepatitis C virus between human hepatoma Huh7.5 cells. Proc. Natl. Acad. Sci. USA 2013, 110, 13109–13113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giugliano, S.; Kriss, M.; Golden-Mason, L.; Dobrinskikh, E.; Stone, A.E.; Soto-Gutierrez, A.; Mitchell, A.; Khetani, S.R.; Yamane, D.; Stoddard, M.; et al. Hepatitis C virus infection induces autocrine interferon signaling by human liver endothelial cells and release of exosomes, which inhibits viral replication. Gastroenterology 2015, 148, 392–402. [Google Scholar] [CrossRef] [PubMed]
- Shrivastava, S.; Devhare, P.; Sujijantarat, N.; Steele, R.; Kwon, Y.C.; Ray, R.; Ray, R.B. Knockdown of autophagy inhibits infectious hepatitis C virus release by the exosomal pathway. J. Virol. 2015, 90, 1387–1396. [Google Scholar] [CrossRef] [PubMed]
- Devhare, P.B.; Sasaki, R.; Shrivastava, S.; Di Bisceglie, A.M.; Ray, R.; Ray, R.B. Exosome-mediated intercellular communication between hepatitis C virus-infected hepatocytes and hepatic stellate cells. J. Virol. 2017, 91, e02216–e02225. [Google Scholar] [CrossRef]
- Kim, J.H.; Lee, C.H.; Lee, S.W. Hepatitis C virus infection stimulates transforming growth factor-β1 expression through upregulating miR-192. J. Microbiol. 2016, 54, 520–526. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Lee, C.H.; Lee, S.W. Exosomal transmission of microRNA from HCV replicating cells stimulates transdifferentiation in hepatic stellate cells. Mol. Ther. Nucleic Acids 2019, 14, 483–497. [Google Scholar] [CrossRef] [PubMed]
- Karsdal, M.A.; Krarup, H.; Sand, J.M.; Christensen, P.B.; Gerstoft, J.; Leeming, D.J.; Weis, N.; Schaffalitzky de Muckadell, O.B.; Krag, A. Review article: The efficacy of biomarkers in chronic fibroproliferative diseases-early diagnosis and prognosis, with liver fibrosis as an exemplar. Aliment. Pharmacol. Ther. 2014, 40, 233–249. [Google Scholar] [CrossRef]
- Ripoll, C.; Groszmann, R.; Garcia-Tsao, G.; Grace, N.; Burroughs, A.; Planas, R.; Escorsell, A.; Garcia-Pagan, J.C.; Makuch, R.; Patch, D.; et al. Hepatic venous pressure gradient predicts clinical decompensation in patients with compensated cirrhosis. Portal hypertension collaborative group. Gastroenterology 2007, 133, 481–488. [Google Scholar] [CrossRef]
- Asselah, T.; Marcellin, P.; Bedossa, P. Improving performance of liver biopsy in fibrosis assessment. J. Hepatol. 2014, 61, 193–195. [Google Scholar] [CrossRef] [Green Version]
- Bravo, A.A.; Sheth, S.G.; Chopra, S. Liver biopsy. N. Engl. J. Med. 2001, 344, 495–500. [Google Scholar] [CrossRef]
- Baranova, A.; Lal, P.; Birerdinc, A.; Younossi, Z.M. Non-invasive markers for hepatic fibrosis. BMC Gastroenterol. 2011, 11, 91. [Google Scholar] [CrossRef] [PubMed]
- Forns, X.; Ampurdanès, S.; Llovet, J.M.; Aponte, J.; Quintó, L.; Martínez-Bauer, E.; Bruguera, M.; Sánchez-Tapias, J.M.; Rodés, J. Identification of chronic hepatitis C patients without hepatic fibrosis by a simple predictive model. Hepatology 2002, 36, 986–992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giannini, E.; Risso, D.; Botta, F.; Chiarbonello, B.; Fasoli, A.; Malfatti, F.; Romagnoli, P.; Testa, E.; Ceppa, P.; Testa, R. Validity and clinical utility of the aspartate aminotransferase-alanine aminotransferase ratio in assessing disease severity and prognosis in patients with hepatitis C virus-related chronic liver disease. Arch. Int. Med. 2003, 163, 218–224. [Google Scholar] [CrossRef] [PubMed]
- Wai, C.T.; Greenson, J.K.; Fontana, R.J.; Kalbfleisch, J.D.; Marrero, J.A.; Conjeevaram, H.S.; Lok, A.S. A simple noninvasive index can predict both significant fibrosis and cirrhosis in patients with chronic hepatitis C. Hepatology 2003, 38, 518–526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vallet-Pichard, A.; Mallet, V.; Nalpas, B.; Verkarre, V.; Nalpas, A.; Dhalluin-Venier, V.; Fontaine, H.; Pol, S. FIB-4: An inexpensive and accurate marker of fibrosis in HCV infection. Comparison with liver biopsy and fibrotest. Hepatology 2007, 46, 32–36. [Google Scholar] [CrossRef] [PubMed]
- Chou, R.; Wasson, N. Blood tests to diagnose fibrosis or cirrhosis in patients with chronic hepatitis C virus infection: A systematic review. Ann. Intern. Med. 2013, 158, 807–820. [Google Scholar] [CrossRef]
- Alboraie, M.A.; Afifi, M.E.; Elghamry, F.G.; Shalaby, H.A.; Elshennawy, G.E.; Abdelaziz, A.A.; Shaheen, M.U.; Abo El-Seoud, A.R. Egy-score predicts severe hepatic fibrosis and cirrhosis in Egyptians with chronic liver diseases: A pilot study. Hepatol. Mon. 2013, 13, e10810. [Google Scholar] [CrossRef]
- Alboraie, M.; Khairy, M.; Elsharkawy, M.; Asem, N.; Elsharkawy, A.; Esmat, G. Value of Egy-Score in diagnosis of significant, advanced hepatic fibrosis and cirrhosis compared to aspartate aminotransferase-to-platelet ratio index, FIB-4 and Forns’ index in chronic hepatitis C virus. Hepatol. Res. 2015, 45, 560–570. [Google Scholar] [CrossRef]
- Friedrich-Rust, M.; Ong, M.F.; Martens, S.; Sarrazin, C.; Bojunga, J.; Zeuzem, S.; Herrmann, E. Performance of transient elastography for the staging of liver fibrosis: A meta-analysis. Gastroenterology 2008, 134, 960–974. [Google Scholar] [CrossRef]
- Cardoso, A.C.; Carvalho-Filho, R.J.; Stern, C.; Dipumpo, A.; Giuily, N.; Ripault, M.P.; Asselah, T.; Boyer, N.; Lada, O.; Castelnau, C.; et al. Direct comparison of diagnostic performance of transient elastography in patients with chronic hepatitis B and chronic hepatitis C. Liver Int. 2012, 32, 612–621. [Google Scholar] [CrossRef]
- Gorka-Dynysiewicz, J.; Pazgan-Simon, M.; Zuwala-Jagiello, J. Pentraxin 3 detects clinically significant fibrosis in patients with chronic viral hepatitis C. Biomed. Res. Int. 2019, 2019, 2639248. [Google Scholar] [CrossRef] [PubMed]
- Calvaruso, V.; Craxı, A. Regression of fibrosis after HBV antiviral therapy. Is cirrhosis reversible? Liver Int. 2014, 34, 85–90. [Google Scholar] [CrossRef] [PubMed]
- Van der Meer, A.J.; Berenguer, M. Reversion of disease manifestations after HCV eradication. J. Hepatol. 2016, 65, S95–S108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manns, M.P.; McHutchison, J.G.; Gordon, S.C.; Rustgi, V.K.; Shiffman, M.; Reindollar, R.; Goodman, Z.D.; Koury, K.; Ling, M.; Albrecht, J.K. Peginterferon alfa-2b plus ribavirin compared with interferon alfa-2b plus ribavirin for initial treatment of chronic hepatitis C: A randomised trial. Lancet 2001, 358, 958–965. [Google Scholar] [CrossRef]
- Poynard, T.; McHutchison, J.; Manns, M.; Trepo, C.; Lindsay, K.; Goodman, Z.; Ling, M.H.; Albrecht, J. Impact of pegylated interferon alfa- 2b and ribavirin on liver fibrosis in patients with chronic hepatitis C. Gastroenterology 2002, 122, 1303–1313. [Google Scholar] [CrossRef]
- Debes, J.D.; Smith, C.I. NS5A: A new target for antiviral drugs in the treatment of hepatitis C virus infection. Hepatology 2012, 56, 797–799. [Google Scholar] [CrossRef]
- Pawlotsky, J.M.; Feld, J.J.; Zeuzem, S.; Hoofnagle, J.H. From non-A, non-B hepatitis to hepatitis C virus cure. J. Hepatol. 2015, 62, S87–S99. [Google Scholar] [CrossRef] [Green Version]
- Foster, G.R.; Afdhal, N.; Roberts, S.K.; Brau, N.; Gane, E.J.; Pianko, S.; Lawitz, E.; Thompson, A.; Shiffman, M.L.; Cooper, C.; et al. Sofosbuvir and velpatasvir for HCV genotype 2 and 3 infection. N. Engl. J. Med. 2015, 373, 2608–2617. [Google Scholar] [CrossRef]
- Veldt, B.J.; Heathcote, E.J.; Wedemeyer, H.; Reichen, J.; Hofmann, W.P.; Zeuzem, S.; Manns, M.P.; Hansen, B.E.; Schalm, S.W.; Janssen, H.L. Sustained virologic response and clinical outcomes in patients with chronic hepatitis C and advanced fibrosis. Ann. Int. Med. 2007, 147, 677–684. [Google Scholar] [CrossRef]
- Simmons, B.; Saleem, J.; Heath, K.; Cooke, G.S.; Hill, A. Long-term treatment outcomes of patients infected with hepatitis C virus: A systematic review and meta-analysis of the survival benefit of achieving a sustained virological response. Clin. Infect. Dis. 2015, 61, 730–740. [Google Scholar] [CrossRef]
- Wedemeyer, H.; Berg, T.; Flamm, S.L.; Foster, G.R.; Craxi, A.; Larrey, D.; Morgan, T.R.; Fried, M.W.; Poordad, F.; Trinh, R.; et al. Improvement in liver function and non-invasive estimates of liver fibrosis 48 weeks after treatment with ombitasvir/paritaprevir/r, dasabuvir and ribavirin in HCV genotype 1 patients with cirrhosis. J. Hepatol. 2015, 62, S637. [Google Scholar] [CrossRef]
- Fehily, S.R.; Papaluca, T.; Thompson, A.J. Long-term impact of direct-acting antiviral agent therapy in HCV cirrhosis: Critical review. Semin. Liver Dis. 2019, 39, 341–353. [Google Scholar] [CrossRef] [PubMed]
- Van der Meer, A.J.; Veldt, B.J.; Feld, J.J.; Wedemeyer, H.; Dufour, J.F.; Lammert, F.; Duarte-Rojo, A.; Heathcote, E.J.; Manns, M.P.; Kuske, L.; et al. Association between sustained virological response and all-cause mortality among patients with chronic hepatitis C and advanced hepatic fibrosis. JAMA 2012, 308, 2584–2593. [Google Scholar] [CrossRef] [PubMed]
- Bruno, S.; Crosignani, A.; Roffi, L.; De Lisi, S.; Rossi, S.; Boccaccio, V.; Zermiani, P.; Mondelli, M.; Maisonneuve, P. SVR is associated with no risk reduction of HCC development in patients with HCV-related cirrhosis. A prospective, up to 23 years, cohort follow-up study. In Proceedings of the EASL Meeting, London, UK, 9–13 April 2014; p. 465. [Google Scholar]
- Cheung, M.C.; Walker, A.J.; Hudson, B.E.; Verma, S.; McLauchlan, J.; Mutimer, D.J.; Brown, A.; Gelson, W.T.H.; MacDonald, D.C.; Agarwal, K.; et al. Outcomes after successful direct-acting antiviral therapy for patients with chronic hepatitis C and decompensated cirrhosis. J. Hepatol. 2016, 65, 741–747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Rao, H.; Chi, X.; Li, B.; Liu, H.; Wu, L.; Zhang, H.; Liu, S.; Zhou, G.; Li, N.; et al. Detection of residual HCV-RNA in patients who have achieved sustained virological response is associated with persistent histological abnormality. EBioMedicine 2019, 46, 227–235. [Google Scholar] [CrossRef] [Green Version]

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khatun, M.; Ray, R.B. Mechanisms Underlying Hepatitis C Virus-Associated Hepatic Fibrosis. Cells 2019, 8, 1249. https://doi.org/10.3390/cells8101249
Khatun M, Ray RB. Mechanisms Underlying Hepatitis C Virus-Associated Hepatic Fibrosis. Cells. 2019; 8(10):1249. https://doi.org/10.3390/cells8101249
Chicago/Turabian StyleKhatun, Mousumi, and Ratna B. Ray. 2019. "Mechanisms Underlying Hepatitis C Virus-Associated Hepatic Fibrosis" Cells 8, no. 10: 1249. https://doi.org/10.3390/cells8101249
APA StyleKhatun, M., & Ray, R. B. (2019). Mechanisms Underlying Hepatitis C Virus-Associated Hepatic Fibrosis. Cells, 8(10), 1249. https://doi.org/10.3390/cells8101249