1. Introduction
Liver fibrosis is characterized by the intrahepatic accumulation of connective tissue as a consequence of chronic liver injury. This process occurs in most types of chronic liver diseases, including immune-, viral-, alcoholic- and metabolic-mediated forms [
1]. Liver fibrosis may progress to cirrhosis, where the liver architecture is disrupted due to the accumulation of extracellular matrix (ECM) and may ultimately lead to liver failure [
1]. Moreover, patients with cirrhosis are at increased risk to develop hepatocellular carcinoma. Liver transplantation is then the only available therapy left [
2]. Fortunately, accumulating evidence reveals that liver fibrosis in early stages is largely reversible [
2] and major efforts are being made to develop new drug-based therapies to reverse liver fibrosis and/or prevent progression to cirrhosis.
The main cell type involved in liver fibrosis is the hepatic stellate cell (HSC). HSCs are responsible for producing most of the ECM proteins, like collagens and fibronectins that accumulate in the chronically injured liver. HSCs in the healthy liver are considered “quiescent” (qHSCs), reside in the space of Disse, store vitamin A in large cytoplasmic lipid droplets and express high levels of the lipogenic transcription factor peroxisome proliferator-activated receptor gamma (PPAR-γ) [
3]. The qHSCs contain the largest reserves of vitamin A in the human body and are considered key regulators of systemic distribution to vitamin A-requiring peripheral tissues [
4]. Upon liver injury, however, qHSCs lose their vitamin A and lipid stores, turn down PPAR-γ expression and transdifferentiate into myofibroblast-like cells (e.g., activated HSCs (aHSCs)) that are highly proliferative, contractile, migratory and produce excessive amounts of extracellular matrix proteins, including collagen I and III [
5]. The aHSCs express alpha-smooth muscle actin (α-SMA) and produce pro-inflammatory and pro-fibrogenic cytokines, like TGF-β [
3,
6]. Hepatic stellate cell activation is central to the development of fibrosis and thus an important process to target in the quest to treat liver fibrosis.
Furthermore, inhibiting these metabolic pathways has been shown to prevent the undergoing activation in vitro [
7,
8]. It has been recently demonstrated that plastic-cultured aHSCs have enhanced oxidative phosphorylation and glycolysis compared to less activated matrigel-cultured aHSCs [
9]. However, functional analyses to quantify metabolic processes that differentiate freshly isolated qHSCs versus culture-activated HSCs have not been reported yet. Here, we hypothesize that HSC transdifferentiation is not only accompanied by a shift to glycolysis and glutaminolysis, but also an induction of mitochondrial oxidative phosphorylation (OXPHOS) to meet the high energy demand of aHSCs. Thus, the objectives of this study were to investigate the contribution of mitochondrial tic metabolism in the activation process of HSCs and identify new mitochondrial targets for the treatment of liver fibrosis.
2. Materials and Methods
2.1. Reagents
Fully activated rat and human HSCs (at least 7 days in culture) were treated for 3 days with 2.5 mmol/L 2-Deoxy-D-glucose (2DG, D8375, Sigma, Saint Luis, MO, USA) or 5 µmol/L GLS1 inhibitor (CB-839, 5.33717, EMD Millipore, Burlington, NJ, USA) to inhibit glycolysis or glutaminolysis, respectively. OXPHOS was inhibited by either 5 µmol/L rotenone (R8875, Sigma) or 2 mmol/L metformin (317240, EMD Millipore).
2.2. Rat and Human HSC Isolation, Cell Culture and Treatments
Primary rat hepatic stellate cells were isolated by a two-step perfusion of the liver with pronase (Merck, Amsterdam, The Netherlands) and collagenase-P (Roche, Almere, The Netherlands) and further purified by Nycodenz (Axis-ShieldPOC, Oslo, Norway) gradient centrifugation, as described before [
10]. HSCs were cultured in Iscove’s modified Dulbecco’s medium (IMDM) with Glutamax (Invitrogen, Breda, The Netherlands) supplemented with 20% heat-inactivated fetal calf serum (FCS, Invitrogen), 1 mmol/L sodium pyruvate (Invitrogen), 1x MEM non-essential amino acids (Invitrogen), 50 µg/mL gentamicin (Invitrogen) and penicillin–streptomycin–fungizone (PSF) in a humidified incubator at 37 °C with 5% CO
2. Freshly isolated quiescent rat HSCs (r-qHSCs) were allowed to attach to culture plates for 4 or 24 h and harvested (r-qHSCs), or cultured for 7 days to become fully activated rHSCs (r-aHSCs). After 7 days, cells were trypsinized and seeded for experiments.
Primary human HSCs were isolated from macroscopically normal liver specimens obtained from fresh tumor resections by an “all-in-one” liver cell purification procedure, as described previously [
11]. Freshly isolated quiescent human HSCs were cultured for two weeks as part of the purification protocol and to become fully activated HSCs (h-aHSCs). h-aHSCs were passaged by trypsinization and passages two to five were used for experiments.
LX-2 cells were obtained from Merk Millipore (SCC064) and used in passages 22–30. Dulbecco’s modified Eagle’s medium (DMEM) with high glucose supplemented with 10% fecal calf serum and 1% antibiotics was used for culturing them. Cells were passed by trypsinization and medium was refreshed every 3 days or when necessary. Cells were grown in 5% CO2 and 37 °C in ambient air.
2.3. Real-Time Monitoring of Cell Proliferation
Proliferation was assessed using the xCELLigence Real Time Cell Analyzing (RCTA) system (RTCA DP; ACEA Biosciences, Inc., San Diego, CA, USA). Primary human or rat aHSCs were plated in E-plates to record cell proliferation, according to the manufacturer’s instructions [
12]. Treatment was started 24 h after attachment and finished after an additional 72 h of treatment. Results were recorded and analyzed by RTCA software.
2.4. Real-Time Imaging Monitoring of Cell Dynamics
Cell migration, morphology and proliferation were assessed by the Live-Cell Analysis System Incucyte (Sartorius, Biopharma, Göttingen, Germany). Primary human or rat activated HSCs were seeded and after 24 h of attachment cells were treated and taken to the IncuCyte ZOOM® platform, which was housed inside a cell incubator at 37 °C/7.5% CO2, for 72 h. Nine images per well from three technical replicates were taken every 2 h using a 10× objective lens and then analyzed using IncuCyte™ Basic Software.
2.5. RNA Isolation, cDNA Synthesis and Real-Time Quantitative PCR
A quantitative real-time reverse transcription polymerase chain reaction (qRT-PCR) was performed as previously described [
13]. Briefly, RNA was isolated from cell cultures using TRIzol
® reagent according to the supplier’s instructions (Thermo Fisher Scientific, Breda, The Netherlands). RNA quality and quantity were determined using a NanoDrop 2000c UV–vis spectrophotometer (Thermo Fisher Scientific). The cDNA was synthesized from 2.5 µg RNA using random nanomers and M-MLV reverse transcriptase (Invitrogen, Carlsbad CA, USA). Taqman primers and probes were designed using Primer Express 3.0.1 and are shown in
Supplementary Table S1. All target genes were amplified using the Q-PCR core kit master mix (Eurogentec, Liège, Belgium) on a 7900HT Fast Q-PCR system (Thermo Fisher Scientific). SDSV2.4.1 (Thermo Fisher Scientific) software was used to analyze the data. Expression of genes is presented in 2-delta CT and normalized to 18S.
2.6. Protein Isolation, Quantification and Western Blot Analysis
Protein samples were prepared for Western blot analysis as described previously [
14]. Protein concentrations were quantified using the Bio-Rad protein assay (Bio-Rad, Hercules, CA, USA) with bovine serum albumin (BSA) as a standard. Thirty micrograms of protein were separated on Mini-PROTEAN
® TGX™ precast 4–15% gradient gels (Bio-Rad) and transferred to nitrocellulose membranes using the Trans-Blot Turbo transfer system, (Bio-Rad). Primary antibody details and dilutions are listed in
Supplementary Table S2 and appropriate horseradish peroxidase (HRP)-conjugated secondary antibodies (1:2000; P0448, DAKO) were used for detection. Proteins were detected using the Pierce ECL Western blotting kit (Thermo Fisher Scientific). Images were captured using the ChemiDoc XRS system and Image Lab version 3.0 (Bio-Rad).
2.7. Quantification of Glycolysis and OXPHOS
Extracellular acidification rates (ECARs) and oxygen consumption rates (OCRs) were measured using glycolysis stress kits and mito stress kits, respectively, in the extracellular flux analyzer Seahorse (Agilent Technologies, Santa Clara, CA, USA) following the manufacturer’s instructions and as described previously (14). Briefly, respiratory chain inhibitors (Oligo: oligomycin, a complex V inhibitor, FCCP: carbonyl cyanide-p-trifluoromethox- yphenyl-hydrazon, a protonophore and lastly A+R: antimycin A and rotenone, inhibitors of complex III and I) and glycolysis stimulators/inhibitors (glucose, to stimulate glycolysis, oligomycin, to inhibit mitochondrial metabolism and help reach glycolysis to its maximum, and 2-Deoxyglucose, glycolysis inhibitor) were added sequentially as described previously [
15] and following manufacturer’s instructions. OCR and ECAR were measured at 37 °C.
2.8. Enzyme Activity and Capacity (Vmax)
Enzymatic activity assays were performed in cellular extracts and all parameters were adapted from yeast to mammalian cells, as previously described [
16]. Briefly, all enzyme activities were measured in freshly prepared extracts at 37 °C in a Synergy H4 plate reader (BioTek, Winooski, VT, USA) as described previously [
17]. For all assays, the reaction mixtures without start reagent were pre-warmed at 37 °C. The conditions used for each enzymatic reaction are described here: Hexokinase (HK; EC2.7.1.1)—1.2 mmol/L NADP+, 10 mmol/L glucose, 1.8 U/mL glucose-6-phosphate dehydrogenase (EC1.1.1.49), and 10 mmol/L ATP as start reagent. Phosphoglucose isomerase (GPI; EC5.3.1.9)—0.4 mmol/L NADP+, 1.8 U/mL glucose-6-phosphate dehydrogenase (EC1.1.1.49), and 2 mmol/L fructose 6-phosphate as start reagent. Phosphofructokinase (PFK; EC2.7.1.11)—0.15 mmol/L NADH, 1 mmol/L ATP, 0.5 U/mL aldolase (EC4.1.2.13), 0.6 U/mL glycerol-3P-dehydrogenase (EC1.1.1.8), 1.8 U/mL triosephosphate isomerase (EC5.3.1.1), and 10 mmol/L fructose 6-phosphate as start reagent. Aldolase (ALD; EC4.1.2.13)—0.15 mmol/L NADH, 0.6 U/mL glycerol-3P-dehydrogenase (EC1.1.1.8), 1.8 U/mL triosephosphate isomerase (EC5.3.1.1), and 2 mmol/L fructose 1,6-bisphosphate as start reagent. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH; EC1.2.1.12)—0.15 mmol/L NADH, 1 mmol/L ATP, 24 U/mL 3-phosphoglycerate kinase (EC2.7.2.3), and 5 mmol/L 3-phosphoglyceric acid as start reagent. 3-Phosphoglycerate kinase (PGK; EC2.7.2.3)—0.15 mmol/L NADH, 1 mmol/L ATP, 8 U/mL glyceraldehyde-3-phosphate dehydrogenase (EC1.2.1.12), and 5 mmol/L 3-phosphoglyceric acid as start reagent. Phosphoglycerate mutase (PGAM; EC5.4.2.1)—0.15 mmol/L NADH, 1 mmol/L ADP, 2.5 mmol/L 2,3-diphospho-glyceric acid, 5 U/mL enolase (EC4.2.1.11), 50 U/mL pyruvate kinase (EC2.7.1.40), 60 U/mL L-lactate dehydrogenase (EC1.1.1.27), and 5 mmol/L 3-phosphoglyceric acid as start reagent. Enolase (ENO; EC4.2.1.11)—0.15 mmol/L NADH, 1 mmol/L ADP, 50 U/mL pyruvate kinase (EC2.7.1.40), 15 U/mL L-lactate dehydrogenase (EC1.1.1.27), and 1 mmol/L 2-phosphoglyceric acid as start reagent. Pyruvate kinase (PK)—0.15 mmol/L NADH, 1 mmol/L ADP, 1 mmol/L fructose 1,6-bisphosphate, 60 U/mL L-lactate dehydrogenase (EC1.1.1.27) and 2 mmol/L phosphoenolpyruvate as start reagent. Lactate dehydrogenase (LDH)—0.15 mmol/L NADH, and 1 mmol/L pyruvate as start reagent. Glucose-6-phosphate dehydrogenase (G6PDH)—0.4 mmol/L NADP+, and 5 mmol/L glucose 6-phosphate as start reagent.
2.9. ATP Assay
Primary rat HSCs were seeded in black 96-well plates and after 24 h of attachment, cells were treated. After 72 h, cells were lysed according to the manufacturer’s instructions (Cell-Triter GLO, Promega, Madison, WI, USA) and luminescence was recorded in a Synergy H4 (BioTek) plate reader at RT.
2.10. Immunofluorescence Microscopy
Primary rat HSCs were seeded directly after isolation in 12-well plates containing 18 mm glass coverslips. Cells on coverslips were washed, fixed (4% PFA, 10 min) and permeabilized (0.1% Triton X-100, 10 min) prior non-specific blocking (2% BSA, 30 min). After blocking, cell on coverslips were incubated for 1 h at RT with the primary antibodies goat polyclonal collagen-I (Southern Biotech, 1310-01, 1/200) and mouse monoclonal αSMA (Sigma Aldrich (Munich, Germany), A5228, 1/100), washed three times with blocking solution and incubated with secondary antibodies goat anti-rabbit Alexa Fluor 488 or rabbit anti-mouse Alexa Fluor 488 (Thermo Fisher Scientific, 30 min, at RT). After secondary antibody incubation, coverslips were washed three times and mounted with Vectashield Antifade Mounting Medium with DAPI (Vector Laboratories, Gdynia, Poland). Coverslips were air-dried, sealed using nail polish, stored at 4 °C and covered from light until further use. Images were obtained in a Zeiss 410 inverted laser scan microscope (Leica Microsystems, Wetzlar, Germany) with 16× or 40× magnification objectives using immersion oil and processed using ImageJ software (public domain, developed at the National Institutes of Health).
2.11. Statistical Analysis
All data are presented as mean ± standard deviation. Significance of differences between groups was tested by one-way ANOVA and t-tests. Calculations were made using the software GraphPad Prism 5. Results were considered statistically different with p value < 0.05.
4. Discussion
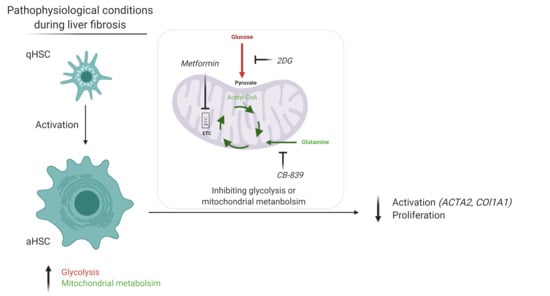
In this study, we show that HSC transdifferentiation is characterized by simultaneous induction of glycolysis and mitochondrial oxidative phosphorylation (OXPHOS) and is accompanied by extensive mitochondrial fusion to meet the high energy demands associated with induced cell proliferation, migration, contraction, migration and ECM production. The HSC transdifferentiation-associated induction of OXPHOS (~5-fold) was stronger than glycolysis (~3-fold). HSC activation was suppressed by targeting glycolysis or mitochondrial metabolism, either glutaminolysis or OXPHOS, separately, but most pronounced by inhibiting OXPHOS. Human activated HSCs appear to be more sensitive to metabolic inhibitors compared to activated rat HSCs. Thus, metabolic shifts associated with HSC transdifferentiation reveal novel and potent targets for the treatment of liver fibrosis.
The first question this study sought to determine was the actual contribution of glycolysis and oxidative phosphorylation towards HSC activation by comparing freshly isolated versus culture-activated primary HSCs. Several reports have shown that HSCs depend on glycolysis and glutaminolysis to transdifferentiate towards active HSCs [
7,
8]. These reports analyzed protein and mRNA expression of key enzymes in glycolysis and glutaminolysis during rat HSC activation [
7,
8]. However, actual glycolytic rat and/or mitochondrial respiration were not quantified. Interestingly, others have shown that plastic-cultured r-aHSCs and LX-2 cells have higher mitochondrial and glycolytic metabolism compared to matrigel-cultured r-aHSCs and LX-2 cells [
9]. Here, we show that the glycolytic rate is increased approximately 3-fold in culture-activated rHSCs versus freshly isolated r-qHSCs. Surprisingly though, the mitochondrial oxygen consumption rate increased even more, by about 5-fold. This is the first time that the simultaneous activation of glycolysis and mitochondrial metabolism in culture-activated versus freshly isolated HSCs is reported. Interestingly, the induction of mitochondrial OXPHOS was accompanied by extensive mitochondrial fusion, without changes in mitochondrial mass. Mitochondria play a key role in regulating many different processes like calcium homeostasis, generation and control of reactive oxygen species, regulation of programmed cell death and ATP production [
21]. The mitochondrial morphology is highly linked to its functions. Mitochondrial fragmentation occurs in response to nutrient excess and cellular dysfunction and mitochondrial fusion is associated with increased cell bioenergetic demands [
19]. This suggests that the increase in mitochondrial fusion is a direct response to the increasing energetic demand during HSC activation. Surprisingly, the observed increase in mitochondrial respiration (5-fold) and mitochondrial fusion was not accompanied by elevated levels of mitochondrial ETC complex proteins. This might be explained by the fact that the mitochondrial ETC proteins form super complexes in response to high energy demands [
22] to create a more efficient ETC machinery without upregulating total protein levels. It needs to be noted though that the normalization in the two methods, e.g., mitochondrial respiration per cell number and ETC proteins per total cellular protein, is different. HSCs grow enormously in size, and thus protein content, during activation in vitro, and the absolute amount of mitochondrial ETC protein increase along with it. Still, mitochondrial DNA copy number did not increase during HSC transdifferentiation. Thus, it will be interesting to analyze whether ETC super complexes indeed are formed in activating HSCs to accommodate the high energy demands for cell proliferation, migration, contraction and ECM production. Interestingly, mitochondrial fusion was also observed in human primary culture-activated HSCs and in LX-2 cells, suggesting it is a well-conserved process. Unfortunately, we were not able to compare mitochondrial morphology and activity in isolated human qHSCs versus aHSCs, as the isolation and purification protocol of human HSCs from resected liver tissue includes a culturing phase where plastic-adherent HSCs are enriched over other contaminating hepatic cell types (such as hepatocytes and endothelial cells). The human HSCs rapidly activate in this culturing step, precluding a direct comparison of qHSCs versus aHSCs.
We observed a strong downregulation of PGC1-α and VDAC during the activation of HSCs, suggesting a decrease in mitochondrial biogenesis and mass. PGC1-α is a co-activator of PPARγ, which is a transcription factor that is markedly downregulated during HSC activation. However, the role of PGC1-α in mitochondrial biogenesis is mostly driven by its role as a co-activator of nuclear respiratory factor 2 (NRF2) [
23]. Given the fact that PPARγ is virtually absent in aHSCs, the reduced levels of PGC1-α may therefore remain sufficient to activate NRF2 and maintain mitochondrial copy number. The accompanying reduction in VDAC protein levels during HSC activation might be due to a differential stability and/or turnover of this protein compared to total mitochondrial turnover.
To unravel new metabolic targets to treat liver fibrosis, we inhibited glycolysis (by 2DG), as well as two mitochondrial metabolic pathways, i.e., glutaminolysis (by CB-839) and OXPHOS (by rotenone and metformin). Inhibiting glycolysis suppressed cell proliferation and expression of fibrogenic markers in fully activated human HSCs. Surprisingly, though, this was not observed for fully activated rat HSCs. The latter result seems to contradict an earlier report [
7] that observed significant anti-proliferative and anti-fibrogenic effects of 2DG in rat HSCs [
7]. In that study, 2DG treatments were started 3 days after rat primary HSC isolation followed by an exposure to 2DG for an additional 3 days. Three day-cultured rat HSCs are, however, not fully activated yet. In contrast, we started 2DG treatment 7 days after isolation, a stage where maximum activation is achieved and reversal, rather that prevention, of the cell proliferation and fibrogenic phenotype of HSCs can be established. This may suggest that inhibiting glycolysis during the activation indeed suppresses fibrogenesis, but once full HSC activation is reached, 2DG is not really effective in reversing the fibrogenic phenotype of rat HSCs. Interestingly, 2DG treatment in r-aHSCs decreased cellular ATP levels, however, this was not sufficient to suppress cell proliferation and activation markers in r-aHSCs. Still, 2DG did suppress cell proliferation and expression of fibrogenic markers in fully activated human HSCs which indicates that this may be species dependent. Unfortunately, inhibitors of glycolysis that have potent “therapeutic” effects in in vitro experiments studying cancer cell proliferation have not yet been successfully implemented for the treatment of patients [
24]. 2DG itself reached phase I/II clinical trials for anti-cancer treatment after encouraging animal experiments, but was discontinued for phase 3 clinical trials because no significant effects on tumor growth was observed in patients (
https://clinicaltrials.gov/ct2/show/NCT00633087). Furthermore, it has been recently described that targeting hexokinase 2, one of the rate-limiting enzymes in glycolysis, with the natural compound costunolide, was highly efficient in reducing HSC activation in vitro and preventing liver damage in vivo [
25,
26]. This suggests that inhibiting glycolysis, with better compounds, could be a potential strategy to treat liver fibrosis.
Mitochondrial metabolism is upregulated 5-fold in aHSCs versus qHSCs, so we targeted mitochondrial metabolism by inhibiting glutaminase with CB-839, complex I of the ETC with metformin (in human HSCs) and rotenone (in rat HSCs). CB-839 is a novel glutaminase inhibitor that is currently being evaluated in several clinical trials to test its anti-carcinogenic properties in patients, as it effectively kills cancer cells in in vitro and in vivo models [
27,
28,
29]. Similar as observed for 2DG, CB-839 had different effects in fully activated human and rat HSCs; it significantly suppressed proliferation and expression of fibrogenic markers in human HSCs, while minimal to no effects were observed for rat HSCs. The results using human HSCs are promising, though, and may further support the notion that human HSCs rely more on different energy-generating metabolic processes than rat HSCs. CB-839 was developed to specifically inhibit the glutaminase C (GAC) splice variant of the human glutaminase (GLS1) gene [
28], so species differences of this specific splicing variant could also affect the pharmacodynamics of this drug. Interestingly, it has been shown that culturing primary human and rat HSCs in media lacking glutamine in the presence of glucose significantly delays proliferation and activation [
8]. The expression of glutaminase is enhanced in chronically injured human livers, providing further support that targeting glutaminolysis with glutaminase inhibitors (such as BPAN and CB-839) may become a successful approach for treating liver fibrosis.
The most important and novel finding of our work was that targeting complex I of the mitochondrial ETC with either metformin (in human HSCs) or rotenone (in rat HSCs) most potently suppressed the proliferation and expression of fibrogenic markers in HSCs. Furthermore, rotenone reduced cellular ATP levels in r-aHSCs, suggesting these cells rely on mitochondrial-derived ATP to proliferate and maintain the activated status. This uncovers mitochondrial OXPHOS as a novel therapeutic target for the treatment of liver fibrosis. Metformin has been in clinical use for the treatment of metabolic disease, particularly type 2 diabetes, for decades already. Interestingly, metformin reduced the mortality rate by 57% in diabetic patients with cirrhotic liver disease [
30]. Though this does not provide proof that this is related to an anti-fibrotic effect of metformin, other studies also support this therapeutic action of metformin. For instance, metformin suppressed
COL1A1 expression and induced lipogenic genes in human lung fibroblasts in vitro [
31]. Moreover, it reversed already established lung fibrosis in mice [
32]. Thus, metformin, and inhibitors of mitochondrial OXPHOS in general, may have therapeutic potential in the treatment of liver fibrosis in chronic liver diseases.
In summary, we show that HSC activation is associated with simultaneous induction of glycolysis and mitochondrial metabolism to support the high energy demand associated with a fibrogenic phenotype. This uncovers novel targets for developing effective therapies to halt and/or reverse liver fibrosis, specifically targeting mitochondrial glutaminolysis or inhibiting the complex I of the ETC.



 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}




