Early Diagnosis in Prader–Willi Syndrome Reduces Obesity and Associated Co-Morbidities

 , ,
, ,  and
and
Abstract
:1. Introduction
1.1. Clinical Aspects of Prader–Willi Syndrome
1.2. Genetic Aspects of Prader–Willi Syndrome
2. Materials and Methods
Analysis
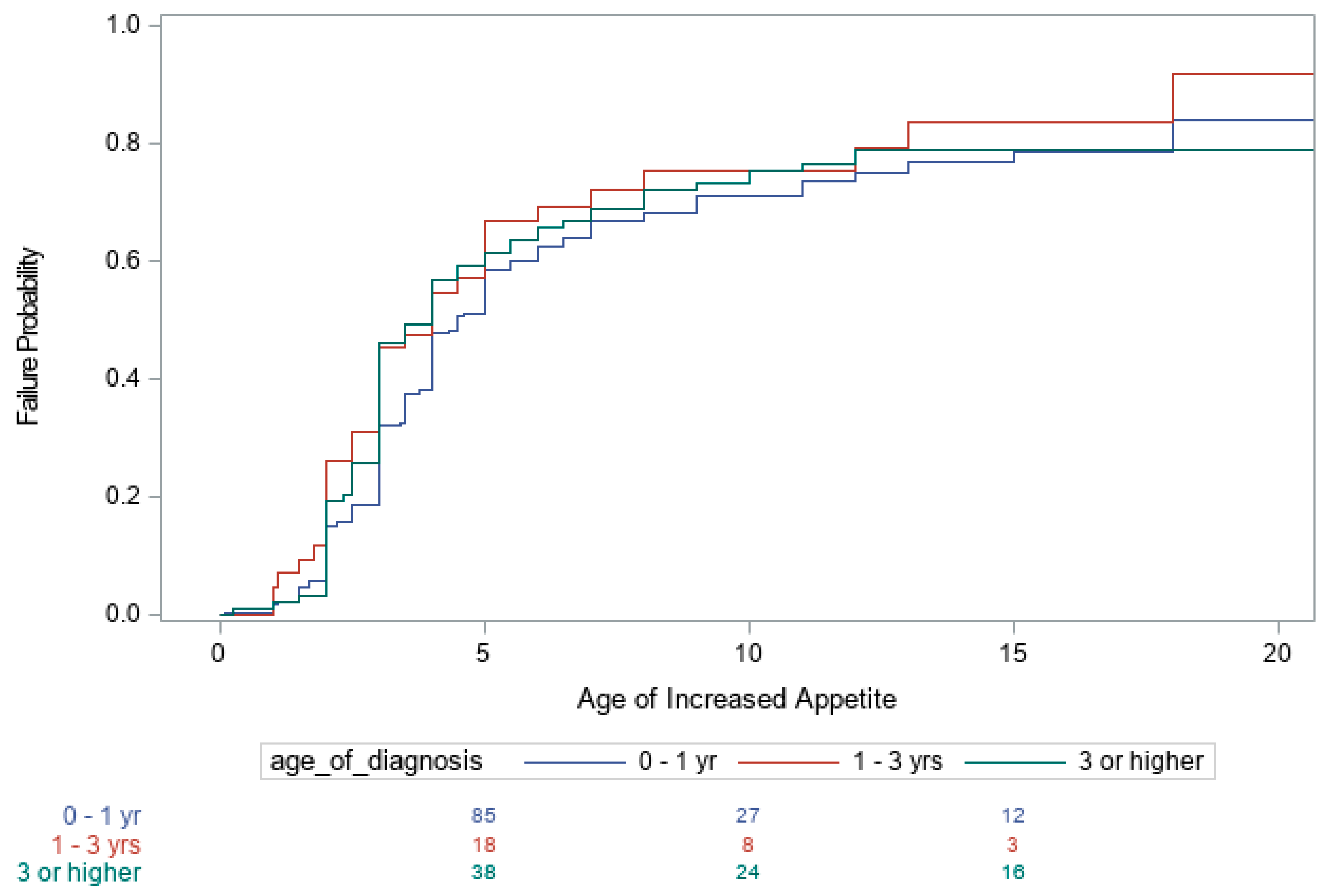
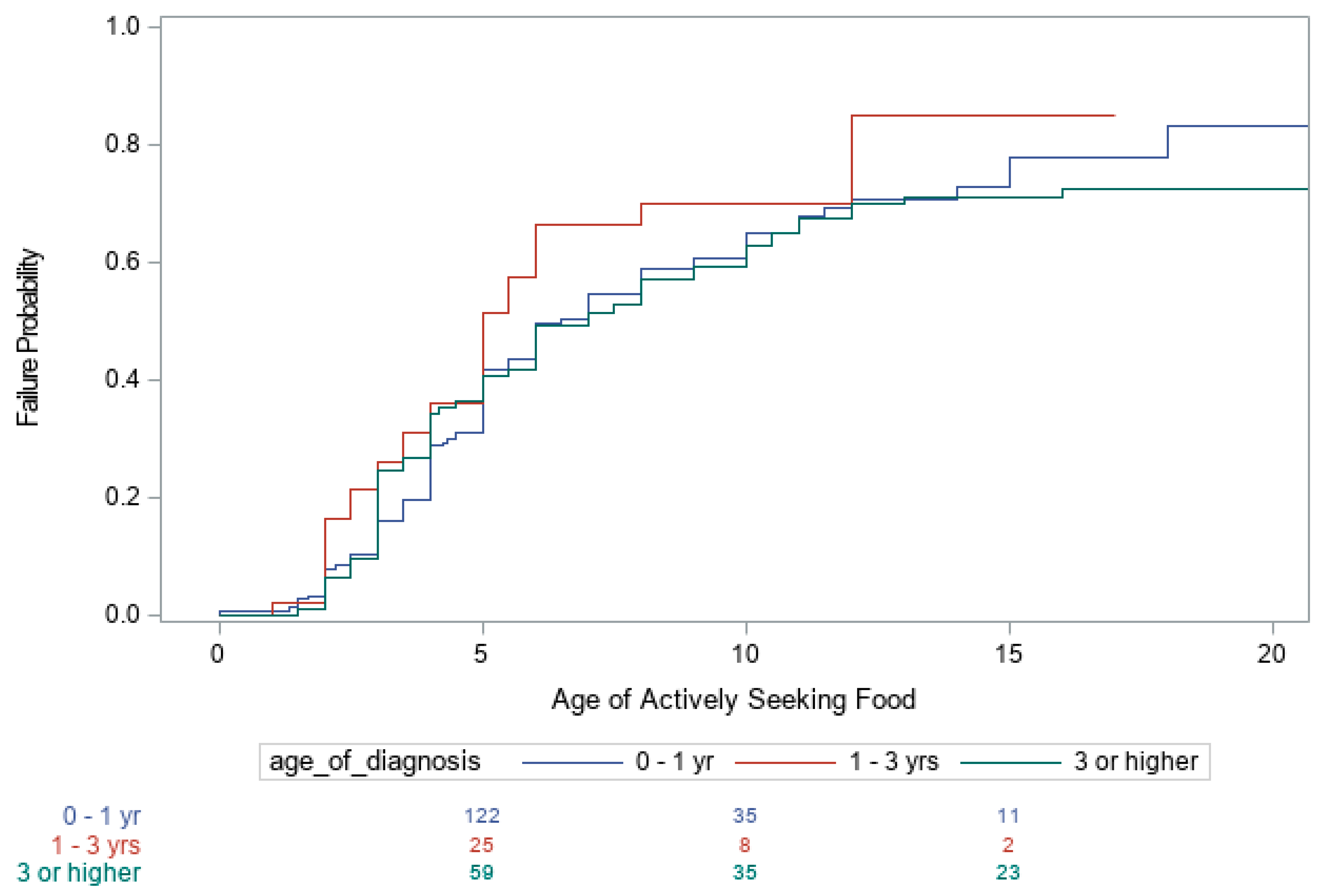
3. Results
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Baker, M.W.; Grossman, W.J.; Laessig, R.H.; Hoffman, G.L.; Brokopp, C.D.; Kurtycz, D.F.; Cogley, M.F.; Litsheim, T.J.; Katcher, M.L.; Routes, J.M. Development of a routine newborn screening protocol for severe combined immunodeficiency. J. Allergy Clin. Immunol. 2009, 124, 522–527. [Google Scholar] [CrossRef] [PubMed]
- Routes, J.M.; Grossman, W.J.; Verbsky, J.; Laessig, R.H.; Hoffman, G.L.; Brokopp, C.D.; Baker, M.W. Statewide newborn screening for severe T-cell lymphopenia. JAMA 2009, 302, 2465–2470. [Google Scholar] [CrossRef] [PubMed]
- Vogel, B.H.; Bonagura, V.; Weinberg, G.A.; Ballow, M.; Isabelle, J.; DiAntonio, L.; Parker, A.; Young, A.; Cunningham-Rundles, C.; Fong, C.T.; et al. Newborn screening for SCID in New York State: Experience from the first two years. J. Clin. Immunol. 2014, 34, 289–303. [Google Scholar] [CrossRef] [PubMed]
- Butler, M.G. Management of obesity in Prader-Willi syndrome. Nat. Clin. Pract. Endocrinol. Metab. 2006, 2, 592–593. [Google Scholar] [CrossRef]
- Butler, M.G.; Sturich, J.; Lee, J.; Myers, S.E.; Whitman, B.Y.; Gold, J.A.; Kimonis, V.; Scheimann, A.; Terrazas, N.; Driscoll, D.J. Growth standards of infants with Prader-Willi syndrome. Pediatrics 2011, 127, 687–695. [Google Scholar] [CrossRef] [PubMed]
- Cassidy, S.B.; Schwartz, S.; Miller, J.L.; Driscoll, D.J. Prader-Willi syndrome. Genet. Med. 2012, 14, 10–26. [Google Scholar] [CrossRef] [Green Version]
- Angulo, M.A.; Butler, M.G.; Cataletto, M.E. Prader-Willi syndrome: A review of clinical, genetic, and endocrine findings. J. Endocrinol. Investig. 2015, 38, 1249–1263. [Google Scholar] [CrossRef]
- Butler, M.G. Prader-Willi syndrome: Current understanding of cause and diagnosis. Am. J. Med. Genet. 1990, 35, 319–332. [Google Scholar] [CrossRef] [Green Version]
- Butler, M.G.; Manzardo, A.M.; Heinemann, J.; Loker, C.; Loker, J. Causes of death in Prader-Willi syndrome: Prader-Willi Syndrome Association (USA) 40-year mortality survey. Genet. Med. 2017, 19, 635–642. [Google Scholar] [CrossRef]
- Butler, M.G. Single Gene and Syndromic Causes of Obesity: Illustrative Examples. Prog. Mol. Biol. Transl. Sci. 2016, 140, 1–45. [Google Scholar] [CrossRef]
- Stevenson, D.A.; Heinemann, J.; Angulo, M.; Butler, M.G.; Loker, J.; Rupe, N.; Kendell, P.; Cassidy, S.B.; Scheimann, A. Gastric rupture and necrosis in Prader-Willi syndrome. J. Pediatr. Gastroenterol. Nutr. 2007, 45, 272–274. [Google Scholar] [CrossRef] [PubMed]
- Stevenson, D.A.; Heinemann, J.; Angulo, M.; Butler, M.G.; Loker, J.; Rupe, N.; Kendell, P.; Clericuzio, C.L.; Scheimann, A.O. Deaths due to choking in Prader-Willi syndrome. Am. J. Med. Genet. Part A 2007, 143, 484–487. [Google Scholar] [CrossRef] [PubMed]
- Shoffstall, A.J.; Gaebler, J.A.; Kreher, N.C.; Niecko, T.; Douglas, D.; Strong, T.V.; Miller, J.L.; Stafford, D.E.; Butler, M.G. The High Direct Medical Costs of Prader-Willi Syndrome. J. Pediatr. 2016, 175, 137–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindgren, A.C.; Lindberg, A. Growth hormone treatment completely normalizes adult height and improves body composition in Prader-Willi syndrome: Experience from KIGS (Pfizer International Growth Database). Horm. Res. 2008, 70, 182–187. [Google Scholar] [CrossRef] [PubMed]
- Nishi, Y.; Tanaka, T. Growth Hormone Treatment and Adverse Events. Pediatr. Endocrinol. Rev. 2017, 14 (Suppl. 1), 235–239. [Google Scholar] [CrossRef]
- Grugni, G.; Marzullo, P. Diagnosis and treatment of GH deficiency in Prader-Willi syndrome. Best Pract. Res. Clin. Endocrinol. Metab. 2016, 30, 785–794. [Google Scholar] [CrossRef]
- Irizarry, K.A.; Miller, M.; Freemark, M.; Haqq, A.M. Prader Willi Syndrome: Genetics, Metabolomics, Hormonal Function, and New Approaches to Therapy. Adv. Pediatr. 2016, 63, 47–77. [Google Scholar] [CrossRef]
- Butler, M.G.; Lee, J.; Cox, D.M.; Manzardo, A.M.; Gold, J.A.; Miller, J.L.; Roof, E.; Dykens, E.; Kimonis, V.; Driscoll, D.J. Growth Charts for Prader-Willi Syndrome During Growth Hormone Treatment. Clin. Pediatr. 2016, 55, 957–974. [Google Scholar] [CrossRef] [Green Version]
- Heksch, R.; Kamboj, M.; Anglin, K.; Obrynba, K. Review of Prader-Willi syndrome: The endocrine approach. Transl. Pediatr. 2017, 6, 274–285. [Google Scholar] [CrossRef]
- Dykens, E.M.; Roof, E.; Hunt-Hawkins, H. Cognitive and adaptive advantages of growth hormone treatment in children with Prader-Willi syndrome. J. Child. Psychol. Psychiatry 2017, 58, 64–74. [Google Scholar] [CrossRef]
- Butler, M.G.; Matthews, N.A.; Patel, N.; Surampalli, A.; Gold, J.A.; Khare, M.; Thompson, T.; Cassidy, S.B.; Kimonis, V.E. Impact of genetic subtypes of Prader-Willi syndrome with growth hormone therapy on intelligence and body mass index. Am. J. Med. Genet. Part A 2019, 179, 1826–1835. [Google Scholar] [CrossRef] [PubMed]
- Butler, M.G.; Hartin, S.N.; Hossain, W.A.; Manzardo, A.M.; Kimonis, V.; Dykens, E.; Gold, J.A.; Kim, S.J.; Weisensel, N.; Tamura, R.; et al. Molecular genetic classification in Prader-Willi syndrome: A multisite cohort study. J. Med. Genet. 2019, 56, 149–153. [Google Scholar] [CrossRef] [PubMed]
- Manzardo, A.M.; Heinemann, J.; McManus, B.; Loker, C.; Loker, J.; Butler, M.G. Venous Thromboembolism in Prader-Willi Syndrome: A Questionnaire Survey. Genes 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Driscoll, D.J.; Miller, J.L.; Schwartz, S.; Cassidy, S.B. Prader- Willi Syndrome. Available online: https://www.ncbi.nlm.nih.gov/books/NBK1330/ (accessed on 28 September 2019).
- Henkhaus, R.S.; Kim, S.J.; Kimonis, V.E.; Gold, J.A.; Dykens, E.M.; Driscoll, D.J.; Butler, M.G. Methylation-specific multiplex ligation-dependent probe amplification and identification of deletion genetic subtypes in Prader-Willi syndrome. Genet. Test. Mol. Biomark. 2012, 16, 178–186. [Google Scholar] [CrossRef]
- Hartin, S.N.; Hossain, W.A.; Francis, D.; Godler, D.E.; Barkataki, S.; Butler, M.G. Analysis of the Prader-Willi syndrome imprinting center using droplet digital PCR and next-generation whole-exome sequencing. Mol. Genet. Genomic Med. 2019, 7, e00575. [Google Scholar] [CrossRef] [PubMed]
- Bittel, D.C.; Kibiryeva, N.; Butler, M.G. Methylation-specific multiplex ligation-dependent probe amplification analysis of subjects with chromosome 15 abnormalities. Genet. Test. 2007, 11, 467–475. [Google Scholar] [CrossRef] [PubMed]
- Bar, C.; Diene, G.; Molinas, C.; Bieth, E.; Casper, C.; Tauber, M. Early diagnosis and care is achieved but should be improved in infants with Prader-Willi syndrome. Orphanet J. Rare Dis. 2017, 12, 118. [Google Scholar] [CrossRef] [PubMed]
- Inaba, Y.; Schwartz, C.E.; Bui, Q.M.; Li, X.; Skinner, C.; Field, M.; Wotton, T.; Hagerman, R.J.; Francis, D.; Amor, D.J.; et al. Early detection of fragile X syndrome: Applications of a novel approach for improved quantitative methylation analysis in venous blood and newborn blood spots. Clin. Chem. 2014, 60, 963–973. [Google Scholar] [CrossRef]
- Godler, D.E.; Inaba, Y.; Schwartz, C.E.; Bui, Q.M.; Shi, E.Z.; Li, X.; Herlihy, A.S.; Skinner, C.; Hagerman, R.J.; Francis, D.; et al. Detection of skewed X-chromosome inactivation in Fragile X syndrome and X chromosome aneuploidy using quantitative melt analysis. Expert Rev. Mol. Med. 2015, 17, e13. [Google Scholar] [CrossRef] [Green Version]
- Butler, M.G.; Lee, J.; Manzardo, A.M.; Gold, J.A.; Miller, J.L.; Kimonis, V.; Driscoll, D.J. Growth charts for non-growth hormone treated Prader-Willi syndrome. Pediatrics 2015, 135, e126–e135. [Google Scholar] [CrossRef]
- Butler, M.G.; Sturich, J.; Myers, S.E.; Gold, J.A.; Kimonis, V.; Driscoll, D.J. Is gestation in Prader-Willi syndrome affected by the genetic subtype? J. Assist. Reprod. Genet. 2009, 26, 461–466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gold, J.A.; Ruth, C.; Osann, K.; Flodman, P.; McManus, B.; Lee, H.S.; Donkervoort, S.; Khare, M.; Roof, E.; Dykens, E.; et al. Frequency of Prader-Willi syndrome in births conceived via assisted reproductive technology. Genet. Med. 2014, 16, 164–169. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.L.; Lynn, C.H.; Driscoll, D.C.; Goldstone, A.P.; Gold, J.A.; Kimonis, V.; Dykens, E.; Butler, M.G.; Shuster, J.J.; Driscoll, D.J. Nutritional phases in Prader-Willi syndrome. Am. J. Med. Genet. Part A 2011, 155, 1040–1049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butler, M.G.; Kimonis, V.; Dykens, E.; Gold, J.A.; Miller, J.; Tamura, R.; Driscoll, D.J. Prader-Willi syndrome and early-onset morbid obesity NIH rare disease consortium: A review of natural history study. Am. J. Med. Genet. Part A 2018, 176, 368–375. [Google Scholar] [CrossRef] [PubMed]
- Manzardo, A.M.; Loker, J.; Heinemann, J.; Loker, C.; Butler, M.G. Survival trends from the Prader-Willi Syndrome Association (USA) 40-year mortality survey. Genet. Med. 2018, 20, 24–30. [Google Scholar] [CrossRef] [PubMed]
- Mahmoud, R.; Singh, P.; Weiss, L.; Lakatos, A.; Oakes, M.; Hossain, W.; Butler, M.G.; Kimonis, V. Newborn screening for Prader-Willi syndrome is feasible: Early diagnosis for better outcomes. Am. J. Med. Genet. Part A 2019, 179, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Angulo, M.A.; Castro-Magana, M.; Lamerson, M.; Arguello, R.; Accacha, S.; Khan, A. Final adult height in children with Prader-Willi syndrome with and without human growth hormone treatment. Am. J. Med. Genet. Part A 2007, 143, 1456–1461. [Google Scholar] [CrossRef]
- Carrel, A.L.; Myers, S.E.; Whitman, B.Y.; Allen, D.B. Sustained benefits of growth hormone on body composition, fat utilization, physical strength and agility, and growth in Prader-Willi syndrome are dose-dependent. J. Pediatr. Endocrinol. Metab. 2001, 14, 1097–1105. [Google Scholar] [CrossRef]
- Butler, M.G.; Smith, B.K.; Lee, J.; Gibson, C.; Schmoll, C.; Moore, W.V.; Donnelly, J.E. Effects of growth hormone treatment in adults with Prader-Willi syndrome. Growth Horm. IGF Res. 2013, 23, 81–87. [Google Scholar] [CrossRef]
- Carrel, A.L.; Myers, S.E.; Whitman, B.Y.; Eickhoff, J.; Allen, D.B. Long-term growth hormone therapy changes the natural history of body composition and motor function in children with prader-willi syndrome. J. Clin. Endocrinol. Metab. 2010, 95, 1131–1136. [Google Scholar] [CrossRef]
- Mogul, H.R.; Lee, P.D.; Whitman, B.Y.; Zipf, W.B.; Frey, M.; Myers, S.; Cahan, M.; Pinyerd, B.; Southren, A.L. Growth hormone treatment of adults with Prader-Willi syndrome and growth hormone deficiency improves lean body mass, fractional body fat, and serum triiodothyronine without glucose impairment: Results from the United States multicenter trial. J. Clin. Endocrinol. Metab. 2008, 93, 1238–1245. [Google Scholar] [CrossRef] [PubMed]
- Hoybye, C. Five-years growth hormone (GH) treatment in adults with Prader-Willi syndrome. Acta Paediatr. 2007, 96, 410–413. [Google Scholar] [CrossRef] [PubMed]
- Myers, S.E.; Whitman, B.Y.; Carrel, A.L.; Moerchen, V.; Bekx, M.T.; Allen, D.B. Two years of growth hormone therapy in young children with Prader-Willi syndrome: Physical and neurodevelopmental benefits. Am. J. Med. Genet. Part A 2007, 143, 443–448. [Google Scholar] [CrossRef] [PubMed]
- Butler, M.G.; Roberts, J.; Hayes, J.; Tan, X.; Manzardo, A.M. Growth hormone receptor (GHR) gene polymorphism and Prader-Willi syndrome. Am. J. Med. Genet. Part A 2013, 161, 1647–1653. [Google Scholar] [CrossRef]
- Butler, M.G.; Manzardo, A.M.; Forster, J.L. Prader-Willi Syndrome: Clinical Genetics and Diagnostic Aspects with Treatment Approaches. Curr. Pediatr. Rev. 2016, 12, 136–166. [Google Scholar] [CrossRef]
- Khare, M.; Gold, J.A.; Wencel, M.; Billimek, J.; Surampalli, A.; Duarte, B.; Pontello, A.; Galassetti, P.; Cassidy, S.; Kimonis, V.E. Effect of genetic subtypes and growth hormone treatment on bone mineral density in Prader-Willi syndrome. J. Pediatr. Endocrinol. Metab. 2014, 27, 511–518. [Google Scholar] [CrossRef]
- Galassetti, P.; Saetrum Opgaard, O.; Cassidy, S.B.; Pontello, A. Nutrient intake and body composition variables in Prader-Willi syndrome—Effect of growth hormone supplementation and genetic subtype. J. Pediatr. Endocrinol. Metab. 2007, 20, 491–500. [Google Scholar] [CrossRef]
- Whitman, B.Y.; Myers, S.; Carrel, A.; Allen, D. The behavioral impact of growth hormone treatment for children and adolescents with Prader-Willi syndrome: A 2-year, controlled study. Pediatrics 2002, 109, e35. [Google Scholar] [CrossRef]
- Goldstone, A.P.; Holland, A.J.; Hauffa, B.P.; Hokken-Koelega, A.C.; Tauber, M. Recommendations for the diagnosis and management of Prader-Willi syndrome. J. Clin. Endocrinol. Metab. 2008, 93, 4183–4197. [Google Scholar] [CrossRef]
- Butler, M.G.; Miller, J.L.; Forster, J.L. Prader-Willi Syndrome—Clinical Genetics, Diagnosis and Treatment Approaches: An Update. Curr. Pediatr. Rev. 2019. [Google Scholar] [CrossRef]
- Lossie, A.C.; Whitney, M.M.; Amidon, D.; Dong, H.J.; Chen, P.; Theriaque, D.; Hutson, A.; Nicholls, R.D.; Zori, R.T.; Williams, C.A.; et al. Distinct phenotypes distinguish the molecular classes of Angelman syndrome. J. Med. Genet. 2001, 38, 834–845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dagli, A.I.; Mueller, J.; Williams, C.A. Angelman Syndrome; Adam, M.P., Ardinger, H.H., Pagon, R.A., Eds.; [Updated 2017 Dec 21]. GeneReviews® [Internet]; University of Washington: Seattle, WA, USA, 1993–2019. Available online: https://www.ncbi.nlm.nih.gov/books/NBK1144/ (accessed on 28 September 2019).




{kind=link}
{kind=link}
{kind=link}
| Category | Frequency (%) |
|---|---|
| Age of Diagnosis (yrs.) | |
| Mean = 3.1 | |
| Median = 0.3 | |
| SD = 6.7 | |
| Min =0.0 | |
| Max = 48.0 | |
| Age of Diagnosis Category | |
| <1 yr. | 217 (62%) |
| ≥1 yr. and <3 yrs. | 42 (12%) |
| ≥3 yrs. | 93 (26%) |
| Gender | |
| Female | 194 (55%) |
| Male | 158 (45%) |
| Ethicity | |
| White | 328 (93%) |
| Non-White | 24 (7%) |
| Prader–Willi Molecular Class | |
| Deletion | 216 (61%) |
| Imprinting Defect | 11 (3%) |
| Uniparental Disomy | 125 (36%) |
| First Becoming Heavy | Increased Appetite | Actively Seeking Food | |
|---|---|---|---|
| Effect | Hazard Ratio (95% CI) p value | Hazard Ratio (95% CI) p value | Hazard Ratio (95% CI) p value |
| Gender (ref = female) | 0.99 (0.73, 1.33) 0.990 | 1.13 (0.88, 1.46) 0.332 | 1.09 (0.83, 1.44) 0.525 |
| PWS Molecular Class | |||
| Deletion vs. UPD | 0.90 (0.66, 1.23) 0.499 | 1.31 (1.00, 1.72) 0.054 | 1.14 (0.85, 1.52) 0.393 |
| ID vs. UPD | 1.45 (0.69, 3.07) 0.326 | 0.72 (0.33, 1.58) 0.415 | 1.05 (0.50, 2.20) 0.893 |
| Ethnicity (ref = white) | 0.46 (0.28, 0.78) 0.004 | 0.76 (0.48, 1.21) 0.179 | 0.73 (0.44, 1.21) 0.224 |
| Age of Diagnosis | |||
| <1 vs. 1–3 | 0.67 (0.43, 1.04) 0.077 | 0.70 (0.47, 1.03) 0.067 | 0.72 (0.48, 1.09) 0.125 |
| <1 vs. >3 yrs. | 0.48 (0.35, 0.66) < 0.001 | 0.90 (0.67, 1.20) 0.456 | 1.05 (0.77, 1.43) 0.754 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kimonis, V.E.; Tamura, R.; Gold, J.-A.; Patel, N.; Surampalli, A.; Manazir, J.; Miller, J.L.; Roof, E.; Dykens, E.; Butler, M.G.; et al. Early Diagnosis in Prader–Willi Syndrome Reduces Obesity and Associated Co-Morbidities. Genes 2019, 10, 898. https://doi.org/10.3390/genes10110898
Kimonis VE, Tamura R, Gold J-A, Patel N, Surampalli A, Manazir J, Miller JL, Roof E, Dykens E, Butler MG, et al. Early Diagnosis in Prader–Willi Syndrome Reduces Obesity and Associated Co-Morbidities. Genes. 2019; 10(11):898. https://doi.org/10.3390/genes10110898
Chicago/Turabian StyleKimonis, Virginia E., Roy Tamura, June-Anne Gold, Nidhi Patel, Abhilasha Surampalli, Javeria Manazir, Jennifer L. Miller, Elizabeth Roof, Elisabeth Dykens, Merlin G. Butler, and et al. 2019. "Early Diagnosis in Prader–Willi Syndrome Reduces Obesity and Associated Co-Morbidities" Genes 10, no. 11: 898. https://doi.org/10.3390/genes10110898