
Dysregulation of Synaptic Signaling Genes Is Involved in Biology of Uterine Leiomyoma



Abstract
:1. Introduction
2. Materials and methods
2.1. Subjects
2.2. RNA Sequencing Analysis
2.3. Gene Ontology Analysis
2.4. Extraction of RNA
2.5. Validation Using RT-qPCR
2.6. Correlation Analyses and Machine Learning Prediction Value Estimation
2.7. Integration to Meta-GWAs and In Silico Functional Analysis
3. Results
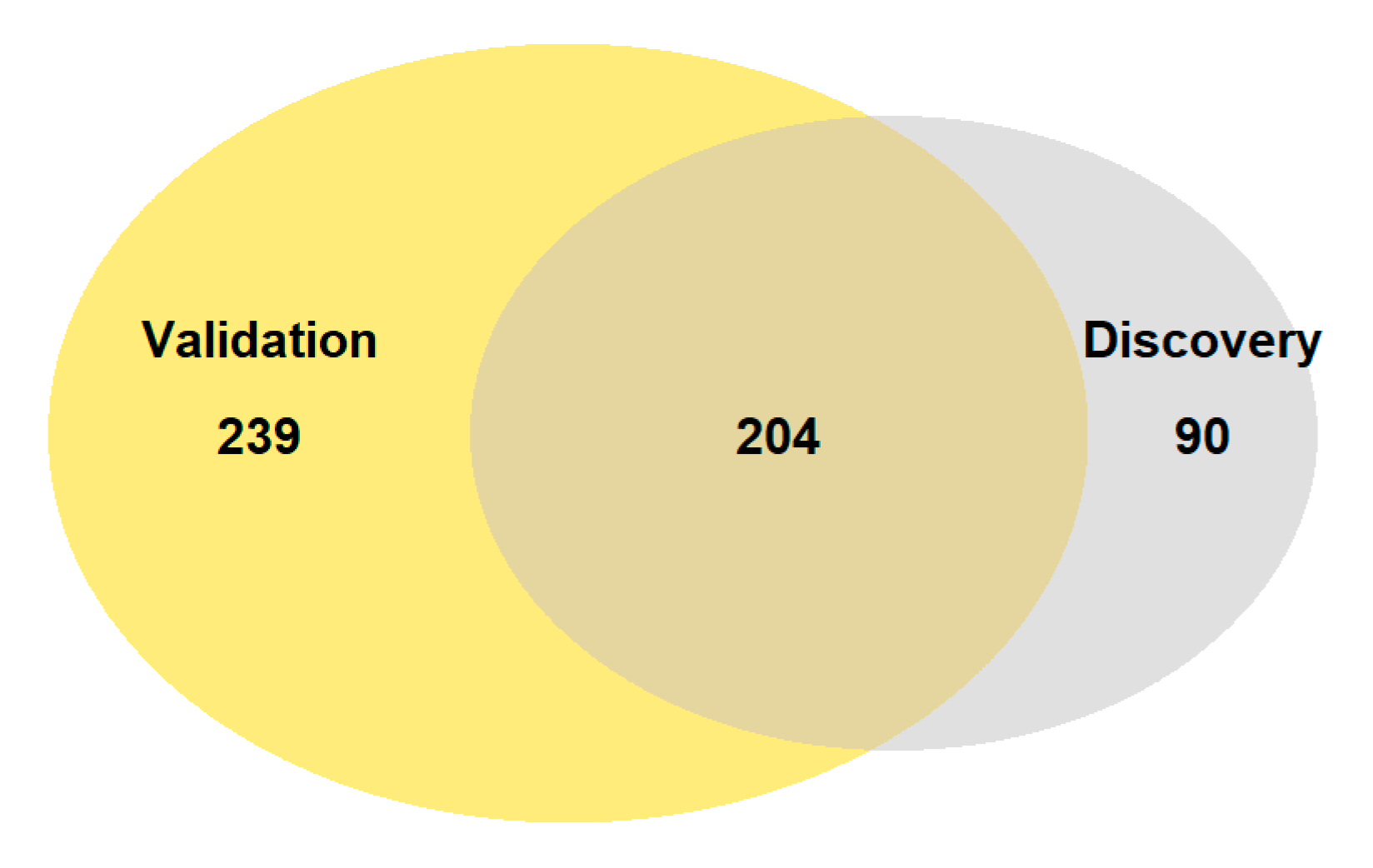
3.1. RNA Sequencing and Differential Expression
3.2. GO Analysis
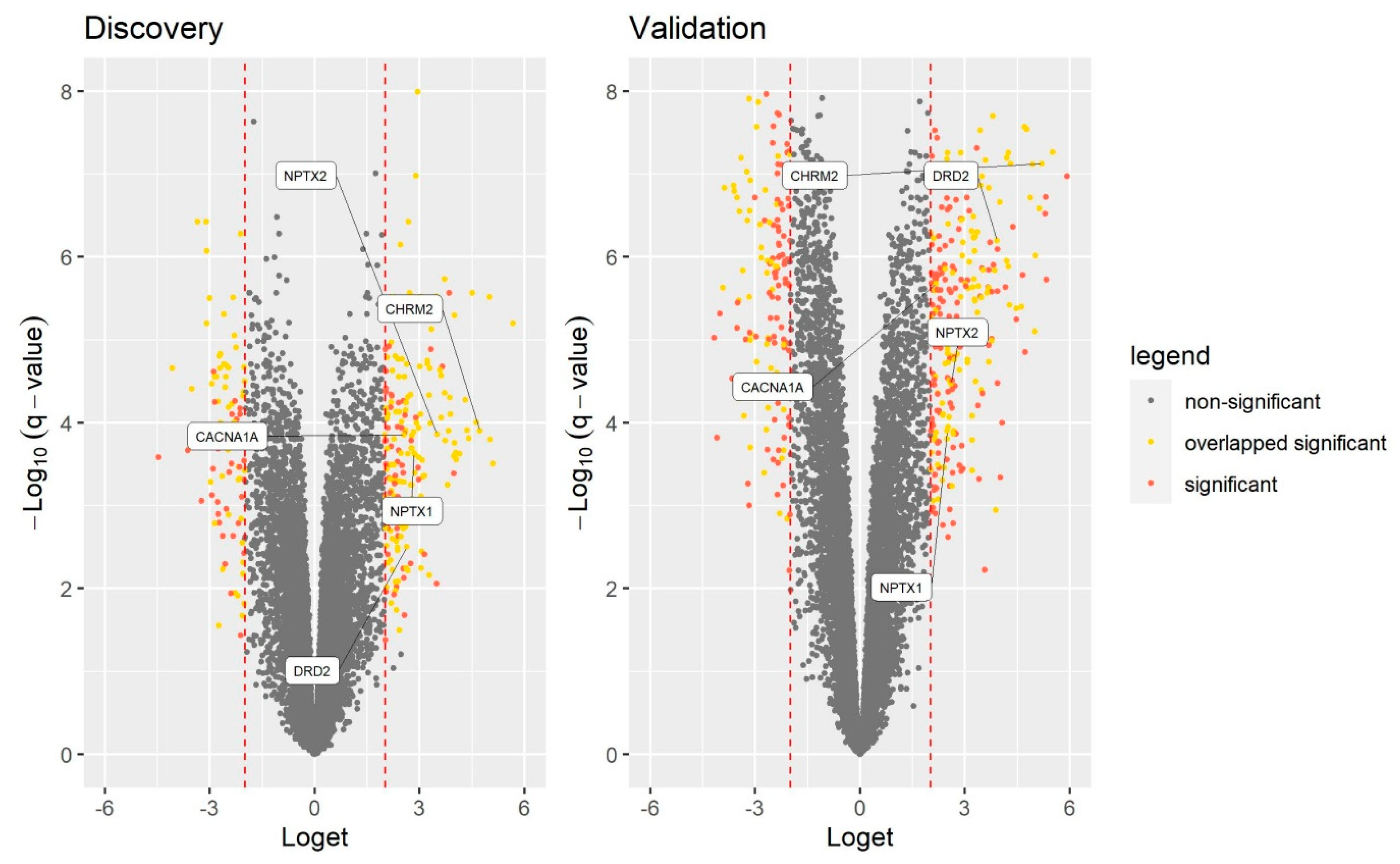
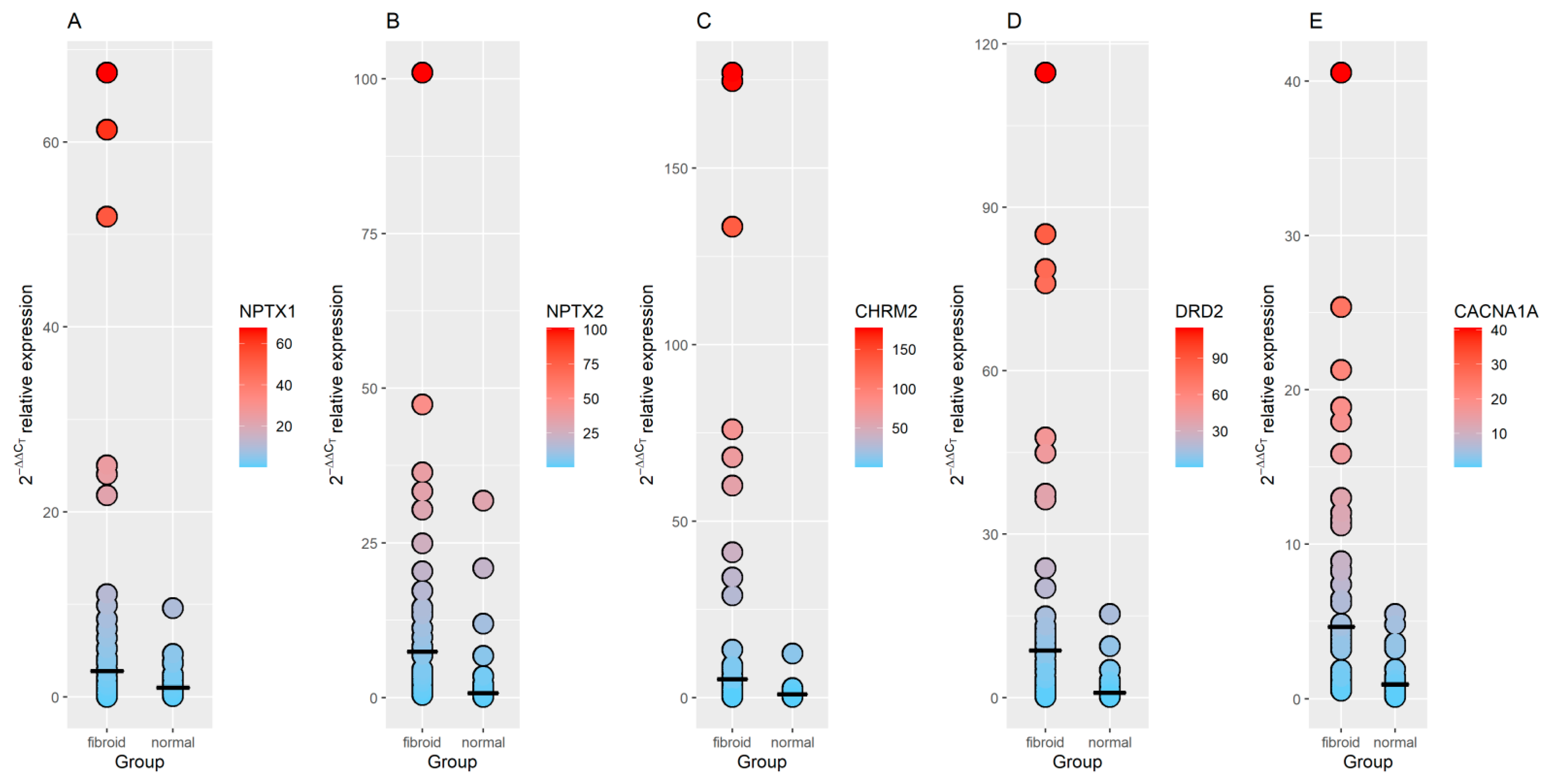
3.3. RT-qPCR Validation of GO Identified Genes
3.4. Between-Gene Regulation of the Identified Genes and Prediction Value Estimation
3.5. Integration into Uterine Leiomyoma Meta-GWAs and Functional Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Stewart, E.A. Clinical practice. Uterine fibroids. N. Engl. J. Med. 2015, 372, 1646–1655. [Google Scholar] [CrossRef]
- Stewart, E.A.; Laughlin-Tommaso, S.K.; Catherino, W.H.; Lalitkumar, S.; Gupta, D.; Vollenhoven, B. Uterine fibroids. Nat. Rev. Dis. Primers 2016, 2, 1–18. [Google Scholar] [CrossRef]
- Bolt, H.M.; Janning, P.; Michna, H.; Degen, G.H. Comparative assessment of endocrine modulators with oestrogenic activity: I. Definition of a hygiene-based margin of safety (HBMOS) for xeno-oestrogens against the background of European developments. Arch. Toxicol. 2001, 74, 649–662. [Google Scholar] [CrossRef]
- Malik, M.; Norian, J.; McCarthy-Keith, D.; Britten, J.; Catherino, W.H. Why Leiomyomas Are Called Fibroids: The Central Role of Extracellular Matrix in Symptomatic Women. Semin. Reprod. Med. 2010, 28, 169–179. [Google Scholar] [CrossRef]
- Islam, S.; Ciavattini, A.; Petraglia, F.; Castellucci, M.; Ciarmela, P. Extracellular matrix in uterine leiomyoma pathogenesis: A potential target for future therapeutics. Hum. Reprod. Update 2018, 24, 59–85. [Google Scholar] [CrossRef] [Green Version]
- Cramer, S.F.; Patel, A. The frequency of uterine leiomyomas. Am. J. Clin. Pathol. 1990, 94, 435–438. [Google Scholar] [CrossRef] [PubMed]
- Marino, J.; Eskenazi, B.; Warner, M.; Samuels, S.; Vercellini, P.P.; Gavoni, N.; Olive, D. Uterine leiomyoma and menstrual cycle characteristics in a population-based cohort study. Hum. Reprod. 2004, 19, 2350–2355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merrill, R.M. Hysterectomy surveillance in the United States, 1997 through 2005. Med. Sci. Monit. 2008, 14, 24–31. [Google Scholar]
- Mehine, M.; Kaasinen, E.; Mäkinen, N.; Katainen, R.; Kämpjärvi, K.; Pitkänen, E.; Heinonen, H.-R.; Bützow, R.; Kilpivaara, O.; Kuosmanen, A.; et al. Characterization of Uterine Leiomyomas by Whole-Genome Sequencing. N. Engl. J. Med. 2013, 369, 43–53. [Google Scholar] [CrossRef] [PubMed]
- Mäkinen, N.; Mehine, M.; Tolvanen, J.; Kaasinen, E.; Li, Y.; Lehtonen, H.J.; Gentile, M.; Yan, J.; Enge, M.; Taipale, M.; et al. MED12, the Mediator Complex Subunit 12 Gene, Is Mutated at High Frequency in Uterine Leiomyomas. Science 2011, 334, 252–255. [Google Scholar] [CrossRef]
- Mehine, M.; Kaasinen, E.; Heinonen, H.-R.; Mäkinen, N.; Kämpjärvi, K.; Sarvilinna, N.; Aavikko, M.; Vähärautio, A.; Pasanen, A.; Bützow, R.; et al. Integrated data analysis reveals uterine leiomyoma subtypes with distinct driver pathways and biomarkers. Proc. Natl. Acad. Sci. USA 2016, 113, 1315–1320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciarmela, P.; Islam, S.; Reis, F.M.; Gray, P.C.; Bloise, E.; Petraglia, F.; Vale, W.; Castellucci, M. Growth factors and myometrium: Biological effects in uterine fibroid and possible clinical implications. Hum. Reprod. Update 2011, 17, 772–790. [Google Scholar] [CrossRef] [Green Version]
- Lee, B.S.; Nowak, R.A. Human leiomyoma smooth muscle cells show increased expression of transforming growth factor-β 3 (TGF β 3) and altered responses to the antiproliferative effects of TGF β. J. Clin. Endocrinol. Metab. 2001, 86, 913–920. [Google Scholar] [CrossRef] [PubMed]
- Dou, Q.; Zhao, Y.; Tarnuzzer, R.W.; Rong, H.; Williams, R.S.; Schultz, G.S.; Chegini, N. Suppression of transforming growth factor-β (TGF β) and TGF β receptor messenger ribonucleic acid and protein expression in leiomyomata in women receiving gonadotropin-releasing hormone agonist therapy. J. Clin. Endocrinol. Metab. 1996, 81, 3222–3230. [Google Scholar] [PubMed] [Green Version]
- Sozen, I.; Arici, A. Interactions of cytokines, growth factors, and the extracellular matrix in the cellular biology of uterine leiomyomata. Fertil. Steril. 2002, 78, 1–12. [Google Scholar] [CrossRef]
- Moyo, M.B.; Parker, J.B.; Chakravarti, D. Altered chromatin landscape and enhancer engagement underlie transcriptional dysregulation in MED12 mutant uterine leiomyomas. Nat. Commun. 2020, 11, 1–16. [Google Scholar] [CrossRef] [PubMed]
- George, J.; Fan, H.; Johnson, B.; Carpenter, T.J.; Foy, K.K.; Chatterjee, A.; Patterson, A.L.; Koeman, J.; Adams, M.; Madaj, Z.B.; et al. Integrated Epigenome, Exome, and Transcriptome Analyses Reveal Molecular Subtypes and Homeotic Transformation in Uterine Fibroids. Cell Rep. 2019, 29, 4069–4085.e6. [Google Scholar] [CrossRef] [Green Version]
- Gallagher, C.S.; Mäkinen, N.; Harris, H.R.; Rahmioglu, N.; Uimari, O.; Cook, J.P.; Shigesi, N.; Ferreira, T.; Velez-Edwards, D.R.; Edwards, T.L.; et al. Genome-wide association and epidemiological analyses reveal common genetic origins between uterine leiomyomata and endometriosis. Nat. Commun. 2019, 10, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Ponomarenko, I.; Reshetnikov, E.; Polonikov, A.; Verzilina, I.; Sorokina, I.; Yermachenko, A.; Dvornyk, V.; Churnosov, M. Candidate Genes for Age at Menarche Are Associated with Uterine Leiomyoma. Front. Genet. 2021, 11, 512940. [Google Scholar] [CrossRef]
- Hernandez-Pacheco, N.; Gorenjak, M.; Jurgec, S.; Corrales, A.; Jorgensen, A.; Karimi, L.; Vijverberg, S.J.; Berce, V.; Schieck, M.; Acosta-Herrera, M.; et al. Combined analysis of transcriptomic and genetic data for the identification of loci involved in glucocorticosteroid response in asthma. Allergy 2021, 76, 1238–1243. [Google Scholar] [CrossRef]
- Gorenjak, M.; Zupin, M.; Jezernik, G.; Skok, P.; Potočnik, U. Omics data integration identifies ELOVL7 and MMD gene regions as novel loci for adalimumab response in patients with Crohn’s disease. Sci. Rep. 2021, 11, 1–12. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.; Shi, W. The Subread aligner: Fast, accurate and scalable read mapping by seed-and-vote. Nucleic Acids Res. 2013, 41, e108. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [Green Version]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [Green Version]
- Robinson, M.D.; Oshlack, A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 2010, 11, R25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Law, C.W.; Chen, Y.; Shi, W.; Smyth, G.K. voom: Precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 2014, 15, R29. [Google Scholar] [CrossRef] [Green Version]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Bindea, G.; Mlecnik, B.; Hackl, H.; Charoentong, P.; Tosolini, M.; Kirilovsky, A.; Fridman, W.-H.; Pagès, F.; Trajanoski, Z.; Galon, J. ClueGO: A Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics 2009, 25, 1091–1093. [Google Scholar] [CrossRef] [Green Version]
- Gorenjak, M.; Repnik, K.; Jezernik, G.; Jurgec, S.; Skok, P.; Potočnik, U. Genetic prediction profile for adalimumab response in Slovenian Crohn’s disease patients. Z. Gastroenterol. 2019, 57, 1218–1225. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Liaw, A.; Wiener, M. Classification and Regression by randomForest. R News 2002, 2, 18–22. [Google Scholar]
- Robin, X.; Turck, N.; Hainard, A.; Tiberti, N.; Lisacek, F.; Sanchez, J.-C.; Muller, M.J. pROC: An open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinform. 2011, 12, 77. [Google Scholar] [CrossRef]
- Ward, L.D.; Kellis, M. HaploReg: A resource for exploring chromatin states, conservation, and regulatory motif alterations within sets of genetically linked variants. Nucleic Acids Res. 2012, 40, D930–D934. [Google Scholar] [CrossRef] [Green Version]
- Consortium, G. Erratum: Genetic effects on gene expression across human tissues. Nature 2018, 553, 530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pruim, R.J.; Welch, R.P.; Sanna, S.; Teslovich, T.M.; Chines, P.S.; Gliedt, T.P.; Boehnke, M.; Abecasis, G.; Willer, C.J. LocusZoom: Regional visualization of genome-wide association scan results. Bioinformatics 2010, 26, 2336–2337. [Google Scholar] [CrossRef] [Green Version]
- Boles, N.C.; Hirsch, S.E.; Le, S.; Corneo, B.; Najm, F.; Minotti, A.P.; Wang, Q.; Lotz, S.; Tesar, P.; Fasano, C.A. NPTX1 Regulates Neural Lineage Specification from Human Pluripotent Stem Cells. Cell Rep. 2014, 6, 724–736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeo, C.-Y.; Whitman, M. Nodal Signals to Smads through Cripto-Dependent and Cripto-Independent Mechanisms. Mol. Cell 2001, 7, 949–957. [Google Scholar] [CrossRef]
- Paul, E.; Burns, G.; Carpenter, T.; Grey, J.; Fazleabas, A.; Teixeira, J. Transcriptome Analyses of Myometrium from Fibroid Patients Reveals Phenotypic Differences Compared to Non-Diseased Myometrium. Int. J. Mol. Sci. 2021, 22, 3618. [Google Scholar] [CrossRef]
- Zhou, C.; Qin, Y.; Xie, Z.; Zhang, J.; Yang, M.; Li, S.; Chen, R. NPTX1 is a novel epigenetic regulation gene and associated with prognosis in lung cancer. Biochem. Biophys. Res. Commun. 2015, 458, 381–386. [Google Scholar] [CrossRef]
- Libiger, O.; Shaw, L.M.; Watson, M.H.; Nairn, A.C.; Umaña, K.L.; Biarnes, M.C.; Canet-Avilés, R.M.; Jack, C.R., Jr.; Breton, Y.A.; Cortes, L. Longitudinal CSF proteomics identifies NPTX2 as a prognostic biomarker of Alzheimer’s disease. Alzheimers Dement. 2021. [Google Scholar] [CrossRef]
- Xu, G.; Fan, L.; Zhao, S.; OuYang, C. Neuronal pentraxin II (NPTX2) hypermethylation promotes cell proliferation but inhibits cell cycle arrest and apoptosis in gastric cancer cells by suppressing the p53 signaling pathway. Bioengineered 2021, 12, 1311–1323. [Google Scholar] [CrossRef]
- Hsieh, Y.-Y.; Chang, C.-C.; Bau, D.-T.; Tsai, F.-J.; Tsai, C.-H.; Chen, C.-P. The p21 codon 31∗C- and DRD2 codon 313∗T-related genotypes/alleles, but not XRCC1 codon 399, hOGG1 codon 326, and DRD1-48 polymorphisms, are correlated with the presence of leiomyoma. Fertil. Steril. 2009, 91, 869–877. [Google Scholar] [CrossRef]
- Zhang, L.; Wen, Y.; Zhang, Q.; Chen, Y.; Wang, J.; Shi, K.; Du, L.; Bao, X. CACNA1A Gene Variants in Eight Chinese Patients With a Wide Range of Phenotypes. Front. Pediatr. 2020, 8, 577544. [Google Scholar] [CrossRef]
- Kurokawa, T.; Nakagawa, T.; Matsusaka, K.; Fukuyo, M.; Mima, M.; Misawa, K.; Rahmutulla, B.; Ikeda, J.-I.; Hanazawa, T.; Okamoto, Y.; et al. Establishment of epigenetic markers to predict irradiation efficacy against oropharyngeal cancer. Cancer Sci. 2020, 111, 1407–1416. [Google Scholar] [CrossRef]
- Ho, C.-M.; Huang, C.-J.; Huang, C.-Y.; Wu, Y.-Y.; Chang, S.-F.; Cheng, W.-F. Promoter Methylation status of HIN-1 associated with outcomes of ovarian clear cell adenocarcinoma. Mol. Cancer 2012, 11, 53. [Google Scholar] [CrossRef] [Green Version]
- Branham, M.T.; Marzese, D.; Laurito, S.R.; Gago, F.E.; Orozco, J.I.; Tello, O.M.; Vargas-Roig, L.M.; Roqué, M. Methylation profile of triple-negative breast carcinomas. Oncogenesis 2012, 1, e17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aleksandrovych, V.; Kurnik-Łucka, M.; Bereza, T.; Białas, M.; Pasternak, A.; Cretoiu, D.; Walocha, J.A.; Gil, K. The Autonomic Innervation and Uterine Telocyte Interplay in Leiomyoma Formation. Cell Transplant. 2019, 28, 619–629. [Google Scholar] [CrossRef]
- Barski, A.; Cuddapah, S.; Cui, K.; Roh, T.-Y.; Schones, D.E.; Wang, Z.; Wei, G.; Chepelev, I.; Zhao, K. High-Resolution Profiling of Histone Methylations in the Human Genome. Cell 2007, 129, 823–837. [Google Scholar] [CrossRef] [Green Version]
- Park, J.-A.; Kim, A.-J.; Kang, Y.; Jung, Y.-J.; Kim, H.K.; Kim, K.-C. Deacetylation and methylation at histone H3 lysine 9 (H3K9) coordinate chromosome condensation during cell cycle progression. Mol. Cells 2011, 31, 343–349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karmodiya, K.; Krebs, A.R.; Oulad-Abdelghani, M.; Kimura, H.; Tora, L. H3K9 and H3K14 acetylation co-occur at many gene regulatory elements, while H3K14ac marks a subset of inactive inducible promoters in mouse embryonic stem cells. BMC Genom. 2012, 13, 424. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Data | Values |
|---|---|
| Age mean (95% CI) | 43.3 (40.6–45.4) |
| Positive familial history (%) | 17.7 |
| Menarche mean (95% CI) | 12.6 (12.1–14.1) |
| Pregnancy mean (95% CI) | 3 (2.5–3.5) |
| Parity mean (95% CI) | 1.9 (1.6–2.2) |
| Miscarriage mean (95% CI) | 0.6 (0.2–1.0) |
| Oral contraceptives (%) | 60 |
| Progestin therapy (%) | 16 |
| BMI mean (95% CI) | 26.3 (25.0–28.5) |
| GENE | ACCESSION | FW 5′ to 3′ | RV 5′ to 3′ |
|---|---|---|---|
| NPTX1 | NM_002522.4 | GTGGCAGTGGCGAGAACT | GGTCCCAGATGTTGAAGTGG |
| NPTX2 | NM_002523.3 | CAGGACGGAGAGAAGCTG | AGTGGCATCAAACCTACCC |
| CHRM2 | NM_001006630.2 | CTATCAACCCTGCCTGCTAT | ACCTTGTAGCGCCTATGTTC |
| DRD2 | NM_000795.4 | TCCCAGCAGAAGGAGAAGAA | TGTTCAGGATGTGTGTGATGAA |
| CACNA1A | NM_000068.4 | TTGTGGTGTTCCCCTTCTTC | ACATGCGGTACTGGAAGCTC |
| GO ID | GO TERM | ONTOLOGY | BONFERRONI p | GENES |
|---|---|---|---|---|
| GO:0098644 | complex of collagen trimers | Cellular component | 0.004 | COL11A1, COL2A1, COL4A4 |
| GO:0009308 | amine metabolic process | Biological process | 0.004 | ATCAY, HDC, INMT, TDO2 |
| GO:0098960 | postsynaptic neurotransmitter receptor activity | Biological process | 0.006 | CHRM2, DRD2, NPTX1, NPTX2 |
| GO:0032835 | glomerulus development | Biological process | 0.015 | BMP7, CD24, COL4A4 |
| GO:0001822 | kidney development | Biological process | 0.020 | BMP7, CD24, COL4A4, OSR1, STRA6 |
| GO:0099565 | chemical synaptic transmission, postsynaptic | Biological process | 0.025 | CHRM2, DRD2, NPTX1, NPTX2 |
| GO:0072001 | renal system development | Biological process | 0.025 | BMP7, CD24, COL4A4, OSR1, STRA6 |
| GO:0001655 | urogenital system development | Biological process | 0.027 | BMP7, CD24, COL4A4, OSR1, STRA6 |
| GO:0098916 | anterograde trans-synaptic signaling | Biological process | 0.027 | CACNA1A, CHRM2, DRD2, NPTX1, NPTX2 |
| GO:0007268 | chemical synaptic transmission | Biological process | 0.027 | CACNA1A, CHRM2, DRD2, NPTX1, NPTX2 |
| GO:0099537 | trans-synaptic signaling | Biological process | 0.030 | CACNA1A, CHRM2, DRD2, NPTX1, NPTX2 |
| GO:0030594 | neurotransmitter receptor activity | Molecular function | 0.031 | CHRM2, DRD2, GRIA2, NPTX1, NPTX2 |
| GO:0044106 | cellular amine metabolic process | Biological process | 0.033 | ATCAY, HDC, TDO2 |
| GO:0099536 | synaptic signaling | Biological process | 0.034 | CACNA1A, CHRM2, DRD2, NPTX1, NPTX2 |
| GO:0010469 | regulation of signaling receptor activity | Biological process | 0.034 | CRHBP, NPTX1, NPTX2 |
| Discovery | Validation | RT-qPCR | ||||
|---|---|---|---|---|---|---|
| GENE | LOGET | q VALUE | LOGET | q VALUE | FC | p VALUE |
| NPTX1 | 2.84 | 2.3 × 10−4 | 2.49 | 1.3 × 10−4 | 2.55 | 0.013 |
| NPTX2 | 3.50 | 1.4 × 10−4 | 2.60 | 3.4 × 10−5 | 7.34 | 0.015 |
| DRD2 | 2.63 | 3.2 × 10−3 | 3.91 | 6.4 × 10−7 | 6.08 | 0.016 |
| CHRM2 | 4.72 | 1.3 × 10−4 | 5.19 | 7.5 × 10−8 | 5.77 | 0.018 |
| CACNA1A | 2.55 | 1.4 × 10−4 | 2.18 | 2.1 × 10−6 | 4.68 | 0.024 |
| GENE | SNP | CONS | REF | ALT | EFFECT SIZE | ST ERROR | p VALUE | N |
|---|---|---|---|---|---|---|---|---|
| NPTX1 | rs9906819 | 3’ downstream | A | C | 0.036 | 0.011 | 0.0013 | 244324 |
| NPTX2 | rs817758 | 5’ upstream | T | C | 0.085 | 0.029 | 0.0028 | 226299 |
| CHRM2 | rs77571733 | 3’ downstream | A | G | −0.172 | 0.049 | 0.0004 | 238961 |
| DRD2 | rs139711611 | 5’ upstream | A | C | −0.151 | 0.053 | 0.0044 | 238961 |
| CACNA1A | rs112605945 | intron variant | T | C | 0.078 | 0.026 | 0.0021 | 244324 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krsteski, J.; Gorenjak, M.; But, I.; Pakiž, M.; Potočnik, U. Dysregulation of Synaptic Signaling Genes Is Involved in Biology of Uterine Leiomyoma. Genes 2021, 12, 1179. https://doi.org/10.3390/genes12081179
Krsteski J, Gorenjak M, But I, Pakiž M, Potočnik U. Dysregulation of Synaptic Signaling Genes Is Involved in Biology of Uterine Leiomyoma. Genes. 2021; 12(8):1179. https://doi.org/10.3390/genes12081179
Chicago/Turabian StyleKrsteski, Jovan, Mario Gorenjak, Igor But, Maja Pakiž, and Uroš Potočnik. 2021. "Dysregulation of Synaptic Signaling Genes Is Involved in Biology of Uterine Leiomyoma" Genes 12, no. 8: 1179. https://doi.org/10.3390/genes12081179