1. Introduction
Unravelling the detailed molecular determinants by which an enzyme, its substrates or inhibitors follow a precise mechanism of recognition and action is a key step that may drive a better understanding of the behavior of similar molecules and to the design of novel synthetic structures that facilitate biotechnological applicability. Both aims have been addressed and achieved for many proteolytic enzymes and ligands because of their wide distribution in living organisms and their involvement in key functions and distresses, including metallocarboxypeptidases (MCPs or CPs) [
1,
2]. However, although some members of this type of proteases are among the first enzymes for which the basic structure-function features were solved [
3], the great number of genomic and proteomic variants of them, with more than 30 members described nowadays [
4,
5], demand further characterization and the search for new molecular modulators.
The proteinaceous inhibitors of CPs are particularly valuable molecules because of their binding in a substrate-like manner to the enzyme. In several instances this results in an initial recognition and C-terminal cleavage of the inhibitor, followed by the establishment of a very stable complex with the enzyme, which facilitates structural and mechanistic analyses [
5,
6,
7]. This has been the case for few of such proteinaceous inhibitors: those from potato (PCI) [
8], tomato [
9], the intestinal parasite
Ascaris suum (ACI) [
10], the medical leech
Hirudo medicinalis (LCI) [
11], the ticks
Riphicephalus bursa (TCI) [
12] and
Haemaphysalis longicornis (HITCI) [
13] and the mollusk
Nerita versicolor [
14]. Also, the variants from rat and human tissues (Latexin) [
15]. However, the latter case, as well that recently characterized from the marine annelid
Sabellastarte magnifica (SmCI) [
16], differ from the former in being much larger (19–22 kDa vs. 5–8 kDa) and displaying a quite different inhibitory mechanism.
Interestingly, most of the above mentioned examples, displayed a remarkable inhibitory capability, with
Ki values in the nanomolar range (about 1–40 nM), but failed to reach the picomolar level that has been described for certain proteinaceous inhibitors of other proteolytic enzymes, like serine- or cysteine-proteases [
17,
18]. The small size of the C-terminal inhibitory tails of the inhibitors mentioned above as compared to the extended inhibitory regions of serine- or cysteine-protease inhibitors, the number of stabilizing interactions or the entropic constraints needed to anchor a free-moving tail into the enzyme active site may be some of the reasons that could account for the apparent lower performance of CP inhibitors.
Some of these questions might be answered with the detailed analysis of the potent proteinaceous CP inhibitor characterized in depth in this report, isolated from the body of the marine snail Nerita versicolor. It is a small form (with a mass around 5945 Da and 53 residues) stabilized by three disulphides, with the shortest C-tail reported for such a kind of inhibitors (only two residues), but displaying the highest inhibitory power described until now, with Ki in the 1-10 pM and 0.1–0.9 nM ranges for several mammalian carboxypeptidases A and B, respectively. This unusually tight inhibitor is found in several isoforms in the snail and here we report the isolation and identification of four of them, the biochemical and cellular characterization of a main isoform and the development of an efficient recombinant production method in yeast. Also, the location of the inhibitor in the body of the snail and its internalization when added to a given cell culture, among other features. The knowledge of its properties might provide both a further understanding of the limits in the inhibition mechanism and specificities of CPs and, on applied grounds, a basis for a better redesign or the generation of synthetic inhibitors for practical uses.
3. Discussion
This work reports the characterization of the properties, state and in vivo location of a very compact and stable small proteinaceous inhibitor of metalloCPs in the Nerita marine snails that has a powerful inhibitory capacity in the picomolar range, non-previously described for any similar inhibitor of such enzymes.
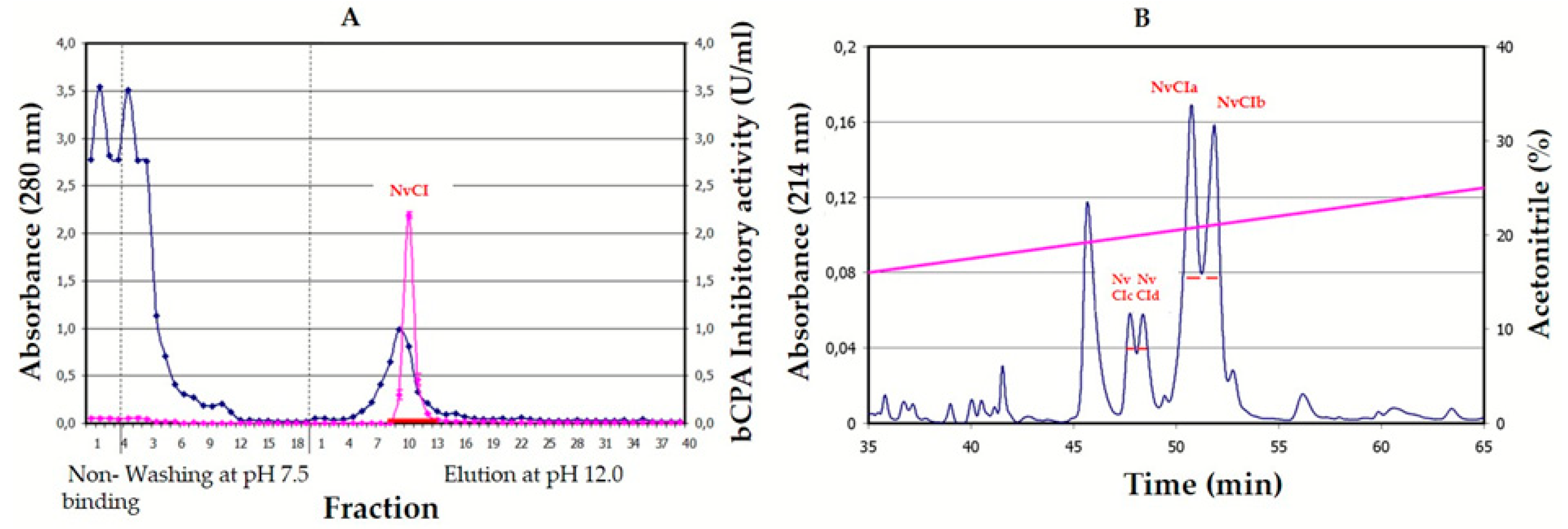
The purification scheme from natural sources, involving a heating pre-treatment for clarification purposes and two chromatographic steps, is very successful in terms of the massive elimination of unwanted material, part of which was very interfering with purification and also because it allows for the dissociation of the inhibitor from complexes (results not shown) and preserves inhibitory power in better terms than other equivalent strategies used to treat crude extracts, like acid or alkaline precipitation, usage of salts or organic solvents. However, the results obtained from competitive titration indicate that only 50.4% of the isolated natural inhibitor was active, pointing to some structural damage that could be related to certain aspects of the purification treatment, like heating or alkaline release from the affinity column. In contrast, the recombinant production of the inhibitor and its purification by successive fractionation steps using anionic and cationic chromatographies yields pure, active and stable material that, although at the cost of a relatively low purification yield that may be improved in future experiments because of the initial very high expression of the recombinant protein.
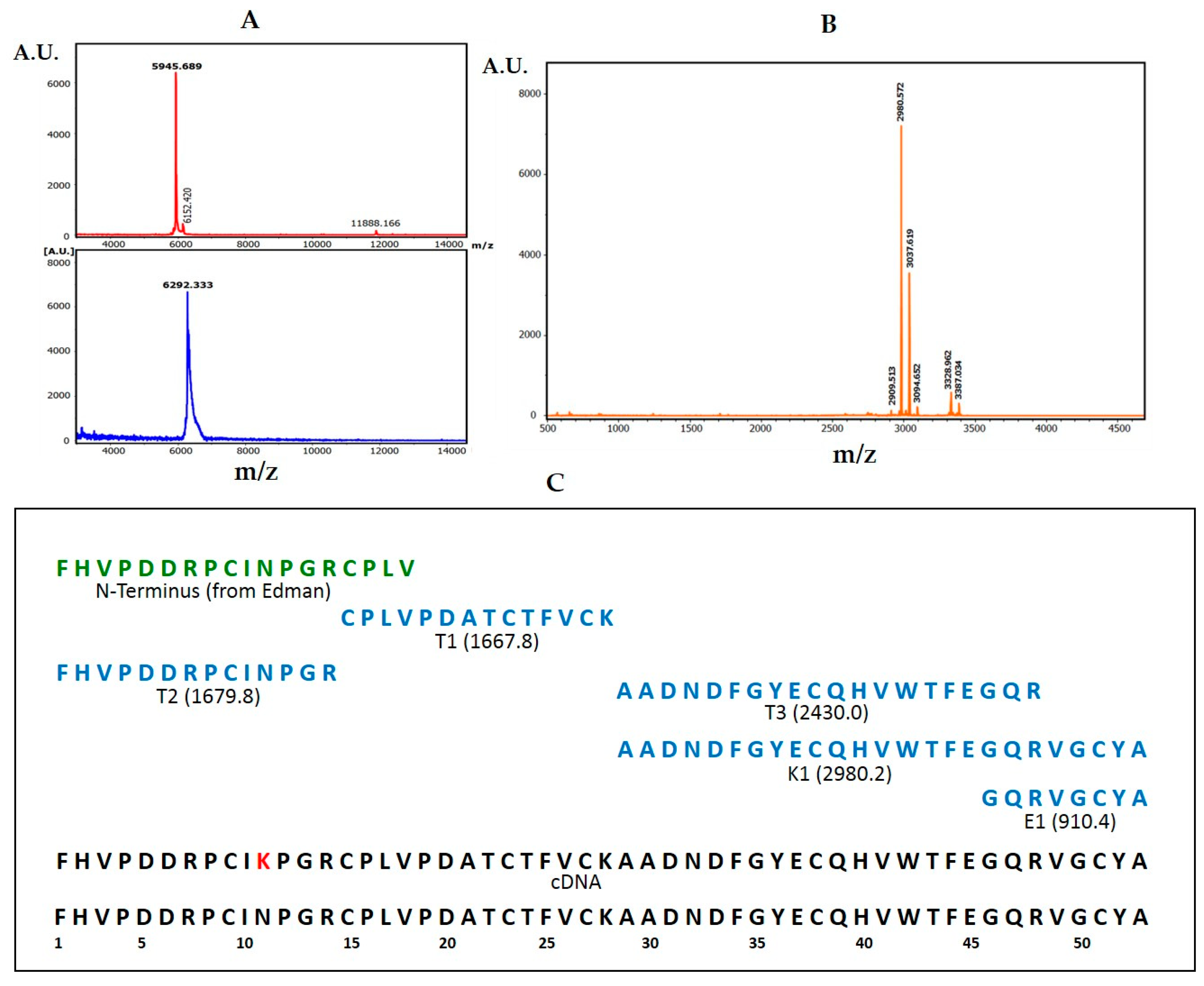
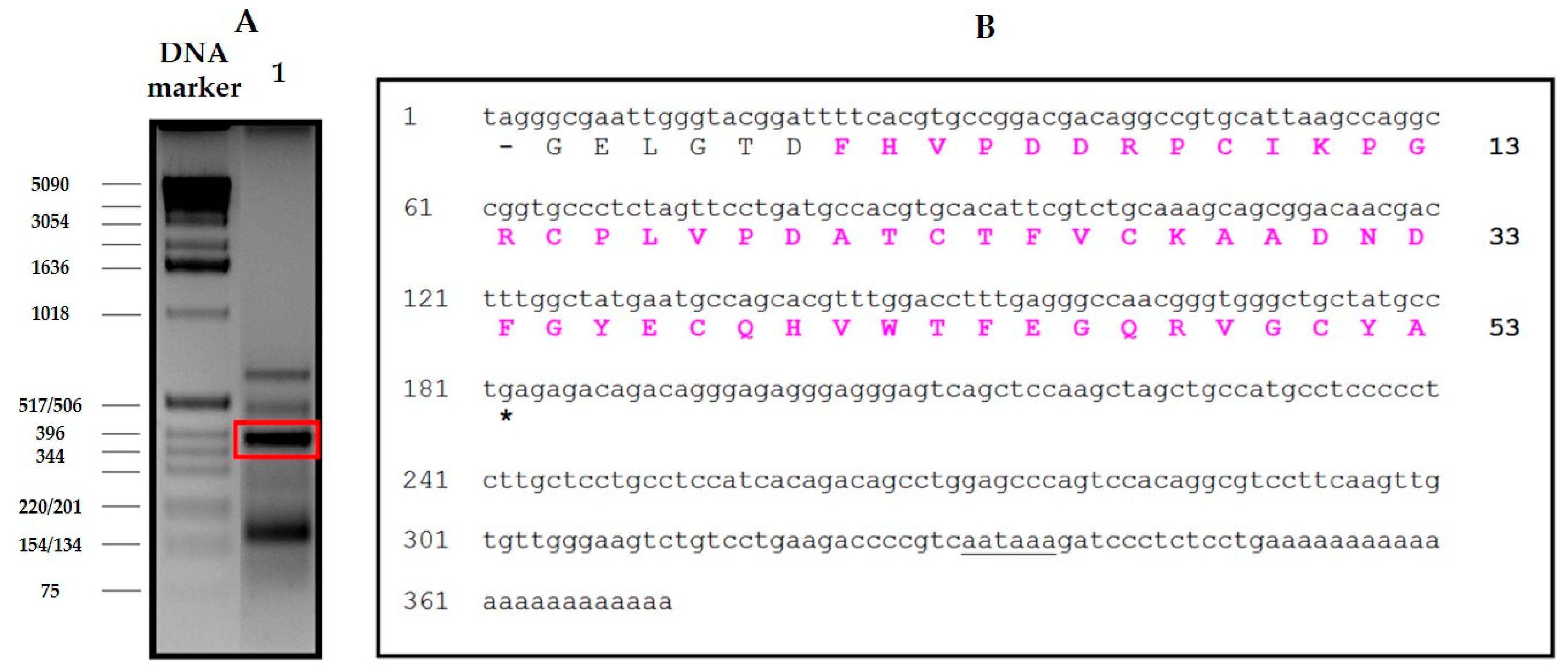
The derivation of the amino acid sequence of NvCI, by both direct approach and through cDNA analysis, showed that the protein consists of 53 residues, with a theoretical mass of 5946 Da. The protein has a high percentage of hydrophobic residues (34%, with two Tyr and one Trp), a slightly higher percentage of acidic than basic residues (13% vs. 11%) which equilibrate in the second major isoform, NvCIb, because of the Asn11Lys substitution, and a notable presence of Pro (9%) and Gly (8%). It lacks Met and contains 6 oxidized Cys, expectedly involved in three intrachain disulphide bonds. When the protein and cDNA sequences are aligned (
Figure 2;
Figure 3), it is evident that the latter has 21 extra nucleotides at the 5′ end, potentially encoding for six additional amino acid residues. In addition, the cDNA sequence of NvCI is also spanning out of the alignment at the 3′ end, after the TGA-stop codon, with 169 untranslated nucleotides plus a polyA tail of 23 residues. The occurrence of additional protein sequences at the N- or C-terminus has been reported for other small protease inhibitors, including those for metalloCPs [
10,
33], and points to its possible role as pro-sequences potentially involved in initial folding steps.
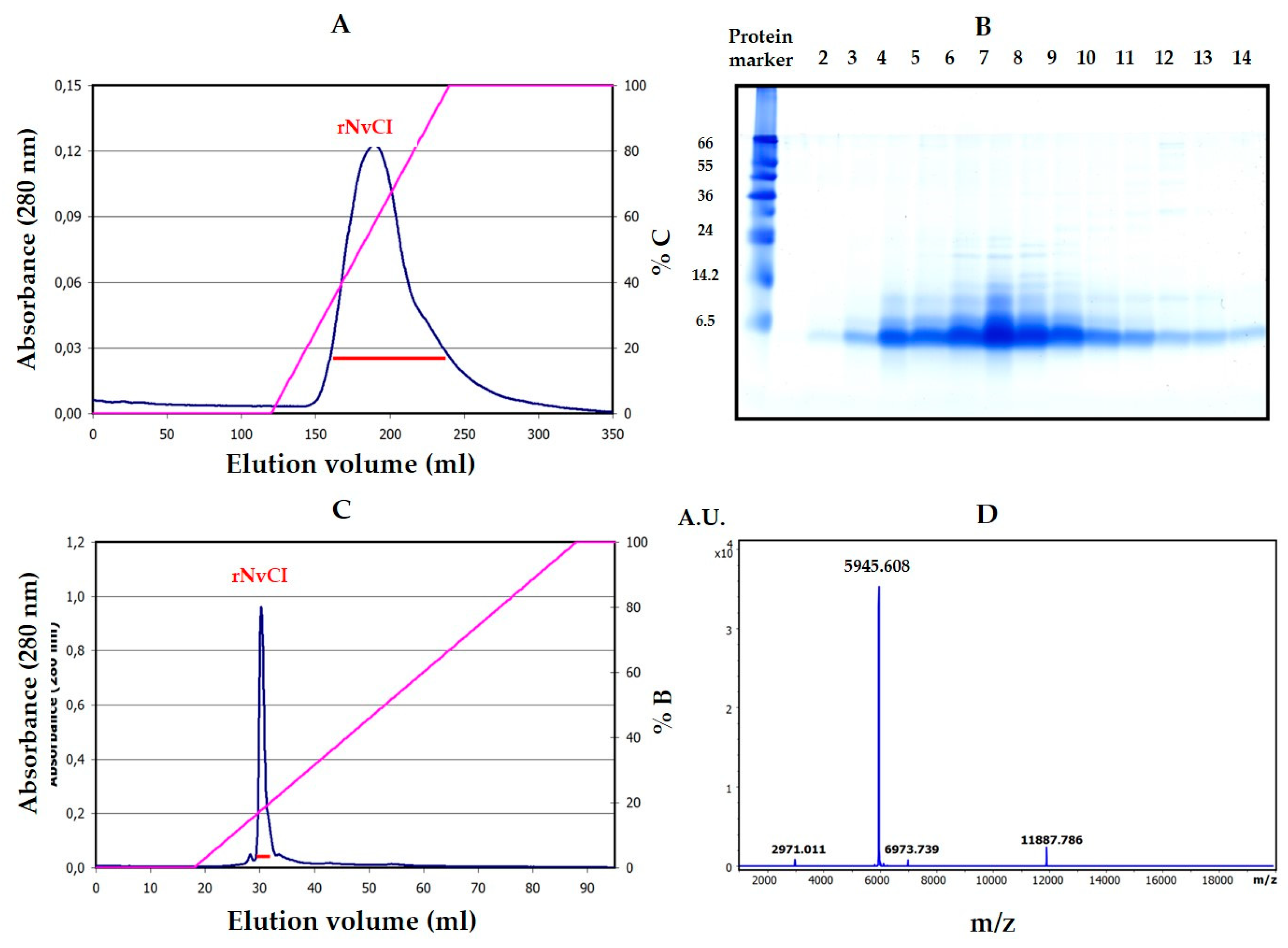
The availability of a recombinant system to produce large quantities of an active form of NvCI identical or equivalent to the natural one was considered a very important point in this research and for future applications of the inhibitor and has been satisfactorily, but only partially, achieved. The system developed in
Pichia pastoris cell cultures is still waiting for improvements in the purification scheme but allows producing hundreds of milligrams of highly active inhibitor (rNvCI) in a few days’ time and with reasonable effort. The produced protein is sequentially identical to the natural main form, NvCIa, as shown by proteomic analysis, and displays the same enzymatic parameters (see
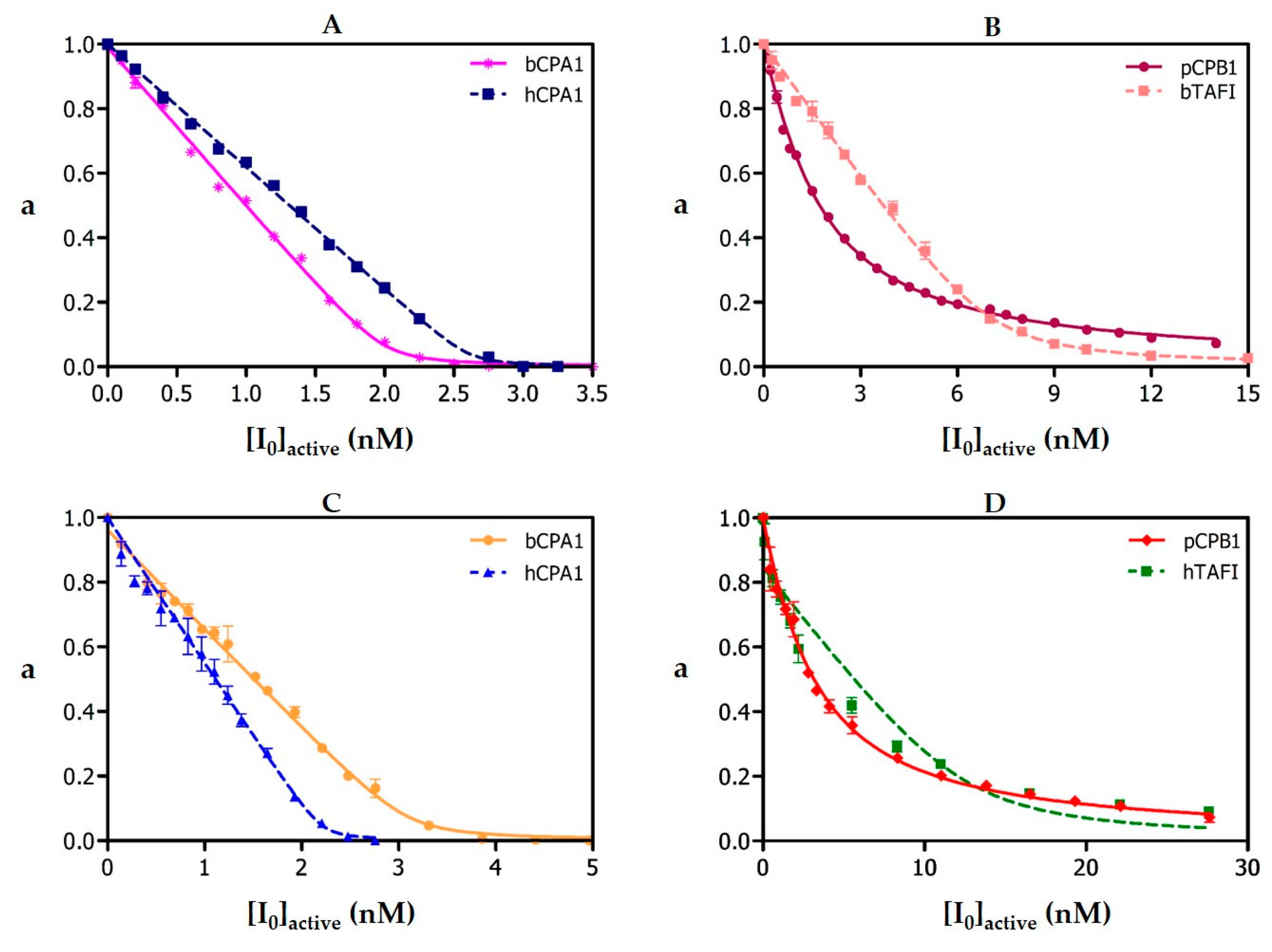
Table 1), but a higher practical activity by competitive titration (95–98% vs. 50.4% for the purified form from the snail). No evidences of glycosylations or other natural or artifactual chemical residue modifications have been observed.
A key point worth to stress from this work is the unusually high inhibitory power shown by NvCI and its structural-functional implications. The number of proteinaceous small inhibitors of metallocarboxypeptidases characterized in depth so far is small, but it seems to be sufficient to delineate their common characteristics: the members of the main subgroup comprising the smallest ones, have their functionally essential region or primary reactive site at the C-terminus, which is normally 3–5 residues long and docks into the protease active site, as has been reported for inhibitors isolated from potato, tomato, leech, intestinal worms and ticks [
5]. On the other hand, the primary interacting site in the largest ones is located in a loop or a region other than the C-terminus, as observed for Latexin [
15]. The here characterized NvCI inhibitor clearly fits with the former case, with the difference that the C-terminal tail is even shorter: only two residues. An alignment of the C-terminus of those inhibitors is shown in
Figure S9. Since the two terminal residues in NvCI (-Tyr-Ala-COOH) are not substantially different from those in the other aligned positions, the clear improvement in its binding strength to metallocarboxypeptidases (from low-medium nanomolar to pico-subnanomolar
Ki, three orders of magnitude) must be related to a much better fit with the enzyme at the primary reactive site and/or to additional effects in the secondary interacting sites. Interestingly, in several previously reported cases, the inhibitor loses one or two C-terminal residues when binding to the protease, after which both become strongly attached, capturing the scissed residue mid-between [
7,
11] and acting, therefore, as “pseudo-substrates”. This is not the case for NvCI.
Some details of the binding and action mechanism of NvCI on metallocarboxypeptidases started to emerge in a work of our group on the NvCI-hCPA4 complex, previously reported because of the easy crystallization and X-ray analysis of the complex [
14], when many characteristics of the whole system were still under preliminary study. In short, it was hypothesized that whilst in the other small proteinaceous inhibitors of metallocarboxypeptidases the primary reactive site only covers the S1 and S2 subsites of the enzyme, in the case of the NvCI-hCPA4 complex the inhibitor also covers the S3 subsite through a double hydrogen bond between residues Cys51 (involved in a disulfide bond) and Tyr52, and residue Glu163 of the enzyme. This would be based on a different orientation of Cys51 which, in turn, would give rise to a richer hydrogen bonding network between NvCI and hCPA4. Another factor could be the larger interface found between both counterparts as compared to other cases. It may seem surprising that such small changes can give rise to three orders of magnitude change in the inhibition constants in the extreme cases. Further experimentation is required for a deeper interpretation. In the present work, we have shown that such extraordinary binding power and low
Ki values previously found for hCPA4 are matched or even beaten in complexes of NvCI with bCPA and hCPA1 in which the Glu163 position is kept. It also suffers a weakening in complexes with enzymes that maintain Glu163 but differ in C-terminal specificity (hCPB and hTAFI) and dramatically decrease in the complex with hCPA2 due to the interplay of two factors: the substitution Glu163Asp and a selectivity pocket designed to host residues bigger than Val. Such advance in knowledge could facilitate its future engineering, i.e., towards more specific or convenient forms.
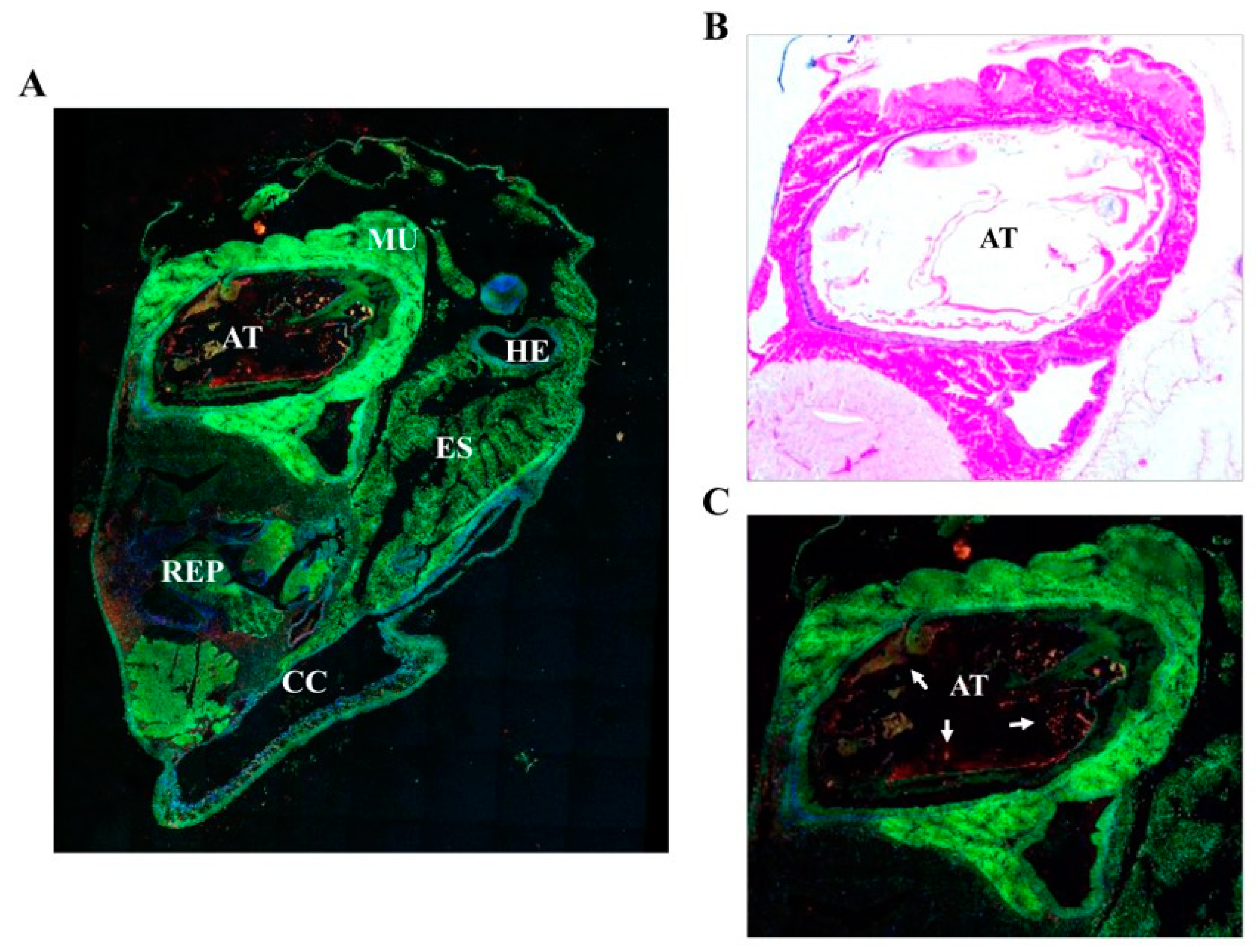
The main in vivo location of NvCI in the surface and the regions of the animal facing the open entrance of the shell and gut suggests a potential functional role of NvCI in the protection of the snail against possible damaging actions from the environment. In fact, this hypothesis would fit with the increasingly convincing evidences that biological defense and protection against predators or invading species is one of the main properties that explain the occurrence of such protease inhibitors in marine invertebrates [
34], as well as in animals and plants, in general [
35,
36]. Specific activity studies and analysis of the biological niche would be required to substantiate this view for NvCI. It would be interesting to investigate whether this hypothesis can be extended to other marine invertebrates in which the occurrence of proteinaceous inhibitors of M14 MCPs is known. This would be the case of the well based work on the inhibitor in
Sabellastarte magnifica [
16], or the just initially unveiled occurrence of equivalent inhibitors in other eight marine invertebrates encompassing Mollusca, Cnidaria, Annelida and Chordata phyla, recently reported by our group [
32].
A remaining question is to what extend NvCI could have biotechnological and/or biomedical potential uses as a drug or lead compound given its exceptional capability to inhibit metallocarboxypeptidases and the growing evidences on the involvement of those enzymes in important biological mechanisms and diseases [
5,
37]. In favor of such uses are its lack of toxicity when added to cellular assays, its stability in the presence of human blood plasma or serum and the fairly abundant recombinant production of NvCI shown in this work. This indication is also congruent with the frequently reported short-term stability, innocuousness and bioavailability of related proteinaceous inhibitors when tested in cellulo and in vivo in mice and humans [
38]. At this respect, it is worth remembering that a well-established circulatory carboxypeptidase, plasma Carboxypeptidase B or TAFIa [
39], is a key factor in the stabilization of blood clots and an important target for fibrinolytic therapeutic strategies based on inhibitors [
40]. Also, that mastocytosis is a growing conjoint of diseases [
41] that gives rise, among others, to massive extracellular release of carboxypeptidase A3 from degranulated mastocytes, constituting a potential therapeutic target for carboxypeptidase inhibitors [
42]. Both enzymes are strongly inhibited by NvCI, as shown in
Table 1. Overall, the very small, compact and stable NvCI proteinaceous inhibitor, which is potent and specific for the carboxypeptidase M14A subfamily, can be of practical use, either directly, or after redesign towards increased specificity or as minimized forms.
4. Materials and Methods
4.1. Purification of Natural NvCI
Nerita versicolor snails were collected in the tropical sea near La Habana (Cuba) and validated by the Cuban Oceanographic Institute. The body of the snails were removed from the shell, washed with seawater, homogenized in a home blender, tissue-filtered, clarified by heating at 60 °C for 30min, centrifuged at 6000 rpm, freeze-dried and kept at −20 °C. After solubilization in the column equilibration buffer (500mM NaCl, 20mM Tris-HCl, pH 7.5), the solution was centrifuged again and loaded on a CPA-glyoxal Sepharose
® CL-4B column (1.6 × 9.9 cm, containing 3.5mg of CPA/ml gel) [
31]. Non-retained molecules were removed by washing the column with equilibration buffer and elution was performed with 10 mM NaOH, pH 12.0. The whole process was carried out at room temperature. The elution profile was followed at 280nm in a Pharmacia FPLC equipment (GE Amersham, UK) and peaks were collected in tubes containing a neutralizing buffer. The fraction containing NvCI was then applied to a Jupiter 0.39 × 15cm HPLC-C4 column (Phenomenex, Torrance, CA, USA), equilibrated and washed with 0.1% TFA in water (solution A), and elution was performed with 0.1%
v/v TFA in acetonitrile (solution B) using the following gradient: 10% of solution B during 15 min followed by a linear gradient from 10 to 40% over 100 min and a linear gradient from 40 to 98% over 1 min. The flow rate was 0.5 ml/min at room temperature. After evaporation of the organic solvent, the fractions containing NvCI, as verified by MALDI-MS and inhibition analysis, were freeze-dried and kept at −80 °C. Total protein concentration was measured in the crude extracts using the bicinchoninic acid (BCA) method [
43].
4.2. Primary Structure Determination
The NvCIa isoform was dissolved in 50mM TrisHCl (pH 8.0) and 2% SDS, or 6M guanidinium chloride, heated at 95 °C for 5 min, centrifuged at 6000 rpm for 20 min and the supernatant transferred to a clean tube. The sample was reduced with 20 mM DTT at 56 °C for 30 min, alkylated with iodoacetamide (20 mM, at 25 °C, for 30 min, in the dark), desalted and concentrated by ZipTipC4 pipette tips (Millipore, Burlington, MA, USA). MALDI-TOF-MS analysis using DHAP as a matrix were made before and after reduction-alkylation to derive the number of free- or disulfide-linked cysteines in the molecule. Subsequently, an aliquot of the reduced-alkylated protein was subjected to eighteen cycles of automated EDMAN degradation. Additional aliquots were treated with Lys-C endoproteinase trypsin and Glu-C endoproteinase at a 40/1 w/w ratios and for 1h at 37 °C and the digest were desalted using Zip TipC4 pipette tips and analyzed by MALDI.TOF-MS using α-CHCA as a matrix to generate a peptide mass fingerprint (PMF). The peptide fragments were subsequently subjected to CID fragmentation within the spectrometer, using the LIFT method and experimental details previously reported [
32]. Peptide alignment and protein sequence analyses from the fragmentation spectra were performed using the Bruker Daltonics software and Mascot search engine, also following the mentioned previous report, with incorporation of the Edman degradation data and the help of the parallel analysis of the NvCI cDNA. Comparison of the PMF and fragmentation data derived from each NvCI isoform (a, b, c and d) allowed to derive the corresponding sequences and differences.
Total RNA was isolated from the marine snail body using the Nucleospin kit (Macherey-Nagel, Düren, Germany), and poly(A) + RNA was purified using the Nucleotrap kit (Macherey-Nagel, Düren, Germany) both according to manufacturer’s instructions. The first strand of NvCI cDNA was synthesized using the adaptor oligonucleotide R0R1polydT (5´CCGGAATTCACTGCAGGGTACCCAATACGACTCACTATAGGGCTTTTTTTTTTTTTTTTT-3´) and avian myeloblastosis virus reverse transcriptase according to the supplier’s protocols. For cloning the NvCI cDNA, four semi-degenerated oligonucleotides were designed based on its N-terminal sequence, P1_NvCI1-8: 5’-TTYCAYGTSCCNGAYGAYCGNCC-3’, P2_NvCI1-8: 5’-TTYCAYGTSCCNGAYGAYAGRCC-3’, P3_NvCI1-8: 5’-TTYCAYGTWCCNGAYGAYCGNCC-3’ and P4_NvCI1-8: 5’- TTYCAYGTWCCNGAYGAYAGRCC -3’. Where, Y = C/T, R = A/G, S = C/G, W = A/T, N = A/C/G/T.
In order to isolate the specific gene product of the RT-PCR, a PCR step was subsequently performed using the specific primers for NvCI and the R0 primer: 5´-CCGGAATTCACTGCAGGGT-3´. PCR was conducted as 35 cycles each at 95 °C for 30s, annealing at 60 °C for 30 s, and extension at 72 °C for 90 s. PCR products were separated by electrophoresis on 2% agarose gels. Selected and purified PCR product was cloned into pBE vector to generate the pBE-NvCI construct (corresponding to NvCI amino acid residues from 1 to 53).
4.3. Recombinant Production and Biophysical Analysis of rNvCI
4.3.1. Cloning, Expression and Purification in Pichia Pastoris
The plasmid construct for recombinant NvCI was based on a synthetic codon-optimized sequence (Geneart, Regensburg, GE), fused in frame to the Saccharomyces cerevisiae prepro α-factor signal under the AOX1 gene promoter. Zeocin hyper-resistant P. pastoris transformants (at high antibiotic concentration) were selected to generate enrichment in recombinant strains with multiple copies of the integrated vector. Recombinant production of NvCI was carried out in a 3 L autoclavable bioreactor (Applikon Biotechnology B.V., Delft, Netherlands), methanol autocontrolled, monitored by cell density, cell weight, protein concentration (by the BCA method) as well as NvCI concentration and integrity (by HPLC, MALDI-MS and activity) along the three and a half days of operation (36 h cell growth until 129g/L cell wet weight, followed by 48 h of induction by 2–3 g methanol per liter of culture, at 25 °C and pH 4.0. According to protein and NvCI analyses, 550 mg/L of recombinant NvCI were obtained in the centrifuged culture broth and about 330 mg/L were produced as a final purification yield at the end of the process.
The purification scheme comprised two steps: (1) A weak cation exchange chromatography of the rNvCI fermentation supernatant on a Waters Accell™ Plus CM column (1.6 cm × 20.0 cm) using buffers A (20 mM sodium citrate, pH 3.0), B (20 mM Tris-HCl pH 7.0) and C (20 mM Tris-HCl pH 7.0, containing 1 M NaCl). The column was equilibrated at 1 ml/min with buffer A over 3 column volumes (CV). Sample loading was performed at 1 ml/min in buffer A. Non-retained molecules were removed by washing the column at 1 ml/min with buffer A and 3 CV. Elution was performed starting with 100% buffer B, at 2 ml/min and 4 CV, followed by a linear gradient from 0 to 100% C at 2 ml/min with 4 CV, and 100% C at 2 ml/min and 1 CV. The whole process was carried out at room temperature. (2) A weak anion exchange chromatography of the elution peak from the previous chromatography on a TSK-GEL™DEAE-5PW column (7.5 cm × 7.5 mm) using buffers A (20 mM Tris-HCl pH 8.5) and B (B: 20 mM Tris-HCl pH 8.5, containing 1 M NaCl). The column was equilibrated with buffer A over 5 CV. Sample loading was performed in buffer A. Non-retained molecules were removed by washing the column with buffer A over 5 CV. Elution was performed using a linear gradient from 0% to 100% B over 20 CV followed by 100% B over 10 CV. The flow rate was 1 ml/min at room temperature. The purified sample was analyzed by MALDI-TOF MS on a MTP 384 target plate polished steel T F (Bruker Daltonics), followed by deposition of 1 μL of DHAP as a matrix. The purified sample was subsequently reduced and S-carbamidomethylated using the procedures described in [
32].
4.3.2. Biophysical Analyses
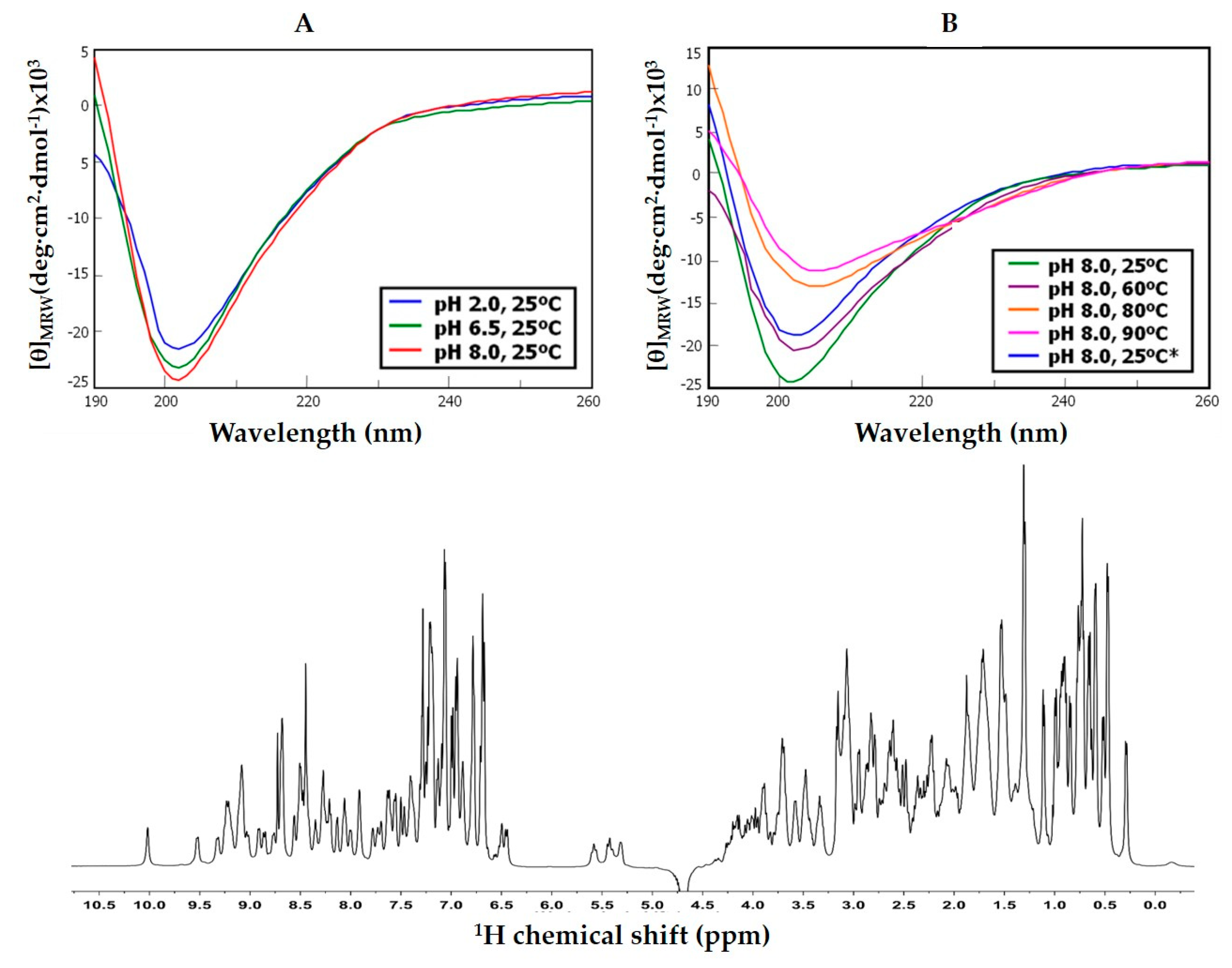
The folding state and stability of NvCI was investigated on the natural and recombinant forms by CD. The proteins, either in 0.1% TFA (v/v) or 20mM sodium phosphate (pH 2.0, 6.5 or 8.0) were analyzed at 0.05–0.06 mg/ml, from 25 to 90 °C in a Jasco J-710 spectropolarimeter (Pfugnstadt, Germany) in the 190–320nm range. Additional analyses were made under folding conditions (20mM sodium phosphate, pH 8.0) in the absence and presence of reducing reagent (1 or 20 mM DTT), a redox-pair (1mM Cys + 0.5mM cystine), and either absence or presence of denaturant (8 M urea). Complementary 1H-NMR experiments were performed in a 500MHz NMR (Bruker Advance) on rNvCI in 20 mM sodium phosphate (pH 2.0 and 6.5), in 90% 1H2O + 10% D2O, at 298 K. DQF-COSY, NOESY, TOCSY and HSQC spectra were obtained in a TCI cryoprobe of 5mm and used for provisional backbone assignment.
4.4. Enzymatic Analysis of the Inhibitory Activity
Inhibitory activities against CPA- and CPB-type forms (bovine and human variants of CPA1, CPA2, CPA4, CPB1, and TAFI) were assayed for all extracts, subject to the experimental conditions that enzyme (E), substrate (S) and inhibitor (I) concentrations as well as the [I]/[E] ratio and preincubation time followed the requirements for tight-binding inhibitor assays [
44]. Similar approaches were followed to measure the activities of proteases of different catalytic types, like pepsin, papain, trypsin and subtilisin [
45,
46,
47,
48]. All assays were performed in triplicate at 25 °C in a multiplexed manner. For 96-well assays an iEMS spectrophotometer reader/dispenser FM (Labsystems, Finland) was used with a final reaction volume of 250 μL. For cuvette assays, a Cary 400 Bio spectrophotometer (Varian Inc., Palo Alto, CA, USA) spectrophotometer was used with a final reaction volume of 1 ml. The reactions were followed at 5 s intervals for 5 min and recorded as initial velocities. In the protease inhibition assays, mixtures of activity buffer, biological sample and enzyme were preincubated at 25 °C for 10min, before substrate addition. The assay conditions were as follows: for CPA-like enzymes, 7.0nM CPA, 0.1mM AAFP substrate in 20mM Tris-HCl pH 7.5, 500mMNaCl, 1%
v/v dimethyl sulfoxide (DMSO), 0.05%
w/v BRIJ-35 was used [
49]. For CPB-like enzymes, CPB 3.0nM CPB, 0.1mM AAFA substrate and activity buffer of 20 mM Tris-HCl pH [
50].
The
Ki values of natural and recombinant NvCI against bCPA1, hCPA1, hCPA2, rat CPA3, hCPA4, hCPB1, bTAFI, hTAFI, hCPD and hCPZ were determined by measuring the enzymatic residual activity (
Vi/
V0 = a) at different inhibitor concentrations and using a fixed enzyme and substrate concentrations as described above, where v
i and v
0 are the fraction of enzymatic activity in the presence and absence of inhibitor, respectively, in terms of initial velocities. The determination of
Ki values was carried out on equilibrium conditions ([E
0]/Ki ≤ 10), using a previously determined preincubation time of 15 min, required to establish enzyme-inhibitor equilibrium. Working temperature was 37 °C. The best estimates of
Ki values were obtained by fitting the experimental data to the equation for tight-binding inhibitors [
19] by non-linear regression using the GraphPad Prisma 5 software (GraphPad Software, Inc., San Diego, CA, USA) at
p < 0.05. The real
Ki values against each of these A/B-type MCPs were calculated according to the equation described by Morrison and Copeland [
19,
20].
4.5. Histology and Immunofluorescence Experiments
Adult Nerita versicolor specimens were obtained from the coast nearby La Habana, Cuba, and left several days only in sea water and without food intake. Whole bodies were extracted from the shell and operculum and fixed in 10% formalin, embedded in paraffin, and cut into 5-µm serial sections. A set of histological sections were stained with hematoxylin and eosin following standard protocols. For immunolocalization experiments, sections were stained with fluorescent-labelled antibodies raised against rNvCI. Such antibodies were produced in house by the SCAC (Servei de Cultius Cel·lulars, producció d´Anticossos i Citometria) at the UAB. The antibody did not cross react with other proteins presents in the Nerita versicolor extract, as tested by Western blot analysis. All incubations were performed at room temperature in a humid chamber. After deparaffinization and rehydration, the sections were immersed in 3% H2O2 in distilled water for 35 min to block endogenous peroxidase activity and then washed three times with distilled H2O. The slides were placed in 10 mM sodium citrate, pH 6.0, and incubated in a water bath at 98 °C for 20 min. Next, the preparations were washed 3 times with PBS, pH 7.4, and blocked with normal goat serum (30%) in PBS for 1 h at room temperature. The sections were then incubated overnight with primary antibody (dilution 1:50) in normal goat serum (30%) in PBS for 1 h at room temperature. The sections were then incubated overnight with primary antibody (dilution 1:50) in normal goat serum (30%) in PBS at 4 °C. After incubation, sections were washed three time with buffer and incubated with an Alexa647 labelled secondary antibody (goat anti-rabbit antibody, dilution 1:500) for 30 min. After washing, sections were incubated with DAPI for 15 min at room temperature and washed with PBS buffer. Finally, the sections were mounted with Prolong Gold mounting medium (Molecular Probes, Eugene, OR, USA). As a negative control, preimmune serum instead of a primary antibody was used. Fluorescence imaging was acquired in a Leica TCS SP5 (Wetzlar, Germany) confocal fluorescence microscope.
4.6. Cytotoxicity Evaluation of NvCI
Cytotoxicity effect of NvCI was tested in a cell culture system using the human hepatocyte cell line HepG2 (American Type Culture Collection (ATCC)). The cells were grown in Dulbecco’s modified Eagle’s medium (DMEM) medium supplemented with 10% (v/v) heat inactivated fetal bovine serum, 2 mM glutamine (Life Technologies Inc.), in a highly humidified atmosphere of 90% air with 10% CO
2 at 37 °C. Growth inhibitory effect was measured by the microculture tetrazolium [2,3-bis-(2-methoxy-4-nitro-5-sulfophenyl)-2H-tetrazolium-5-carboxanilide, XTT] assay [
51]. Following the addition of different inhibitor concentrations to quadruplicate wells, plates were incubated at 37 °C for 72 h. Aliquots of 20 µl of XTT solution were then added to each well. After 3 h, the color formed was quantitated by a spectrophotometric plate reader (PerkinElmer Victor3 V) at a wavelength of 490 nm. Cell viability was expressed as a percentage of the control level.
4.7. Cellular Uptake of NvCI
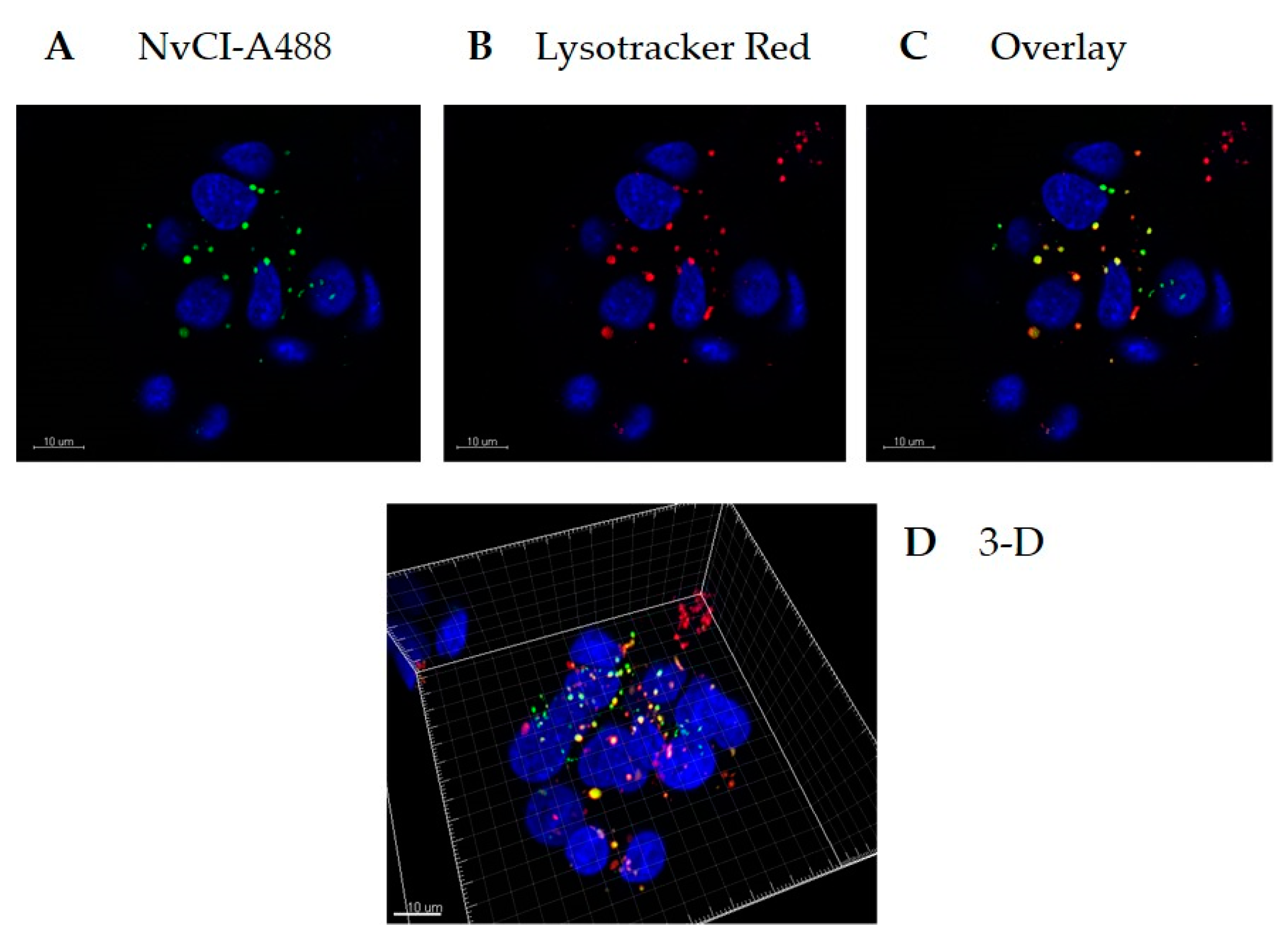
Recombinant NvCI was fluorescently labelled with an Alexa Fluor 488 Protein Labeling Kit (Molecular Probes, Eugene, OR, USA). HepG2 cells were seeded in a 35 mm glass-bottom culture dish (MatTek Corporation, Ashland, MA, USA) at a density of 4 × 105 cells/well and incubated for 24 h. The cells were incubated with 20 µg/ml Alexa 488-NvCI for 4 h at 37 °C. Prior to imaging, lysosomes and nuclei were stained with Lysotracker Red and Hoechst (Invitrogen, Carlsbard, CA, USA), respectively, for 90 or 10 min.
4.8. Molecular and Functional Stability of PCI in Blood Plasma and Serum
rNvCI was dissolved in human plasma and serum samples (provided by Sigma-Aldrich, Saint Louis, MO, USA), at concentrations ranging from 0.001 to 0.5 mg/mL, the mixture was incubated at 37 °C, 100 μL aliquots were withdrawn at 1, 3 and 9 h, and mixed with 100 μL of 40 mM Tris-HCl, 1MNaCl, pH 7.5. The aliquots were analyzed by affinity proteomics on human CPA-microaffinity spin columns (100 μL each, from ThermoFisher, Waltham, MA, USA), followed by RP-HPLC on a C4 column and MALDI-TOF.MS, by a procedure and conditions for biological extracts described previously [
32]. The captured NvCI was released by acid treatment with 110 μL of 0.5% v/v TFA, and 95 μL were applied on a RP Jupiter 5u C4 300A 250 × 10 mm (Phenomenex) HPLC column, with detection at 214 and 280 nm. A calibration graph for NvCI was derived in the 0.001-0.5 range, as shown in Fig.S7. The remaining 10 μL were desalted in ZipTip C4 (Millipore, Burlington, MA, USA) and analyzed by MALDI-TOF MS in a Bruker Xtreme (Billerica, MA, USA) spectrometer.

 ,
, 











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}