
Inflammasome Molecular Insights in Autoimmune Diseases
 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Autoimmune Diseases
1.1. Introduction
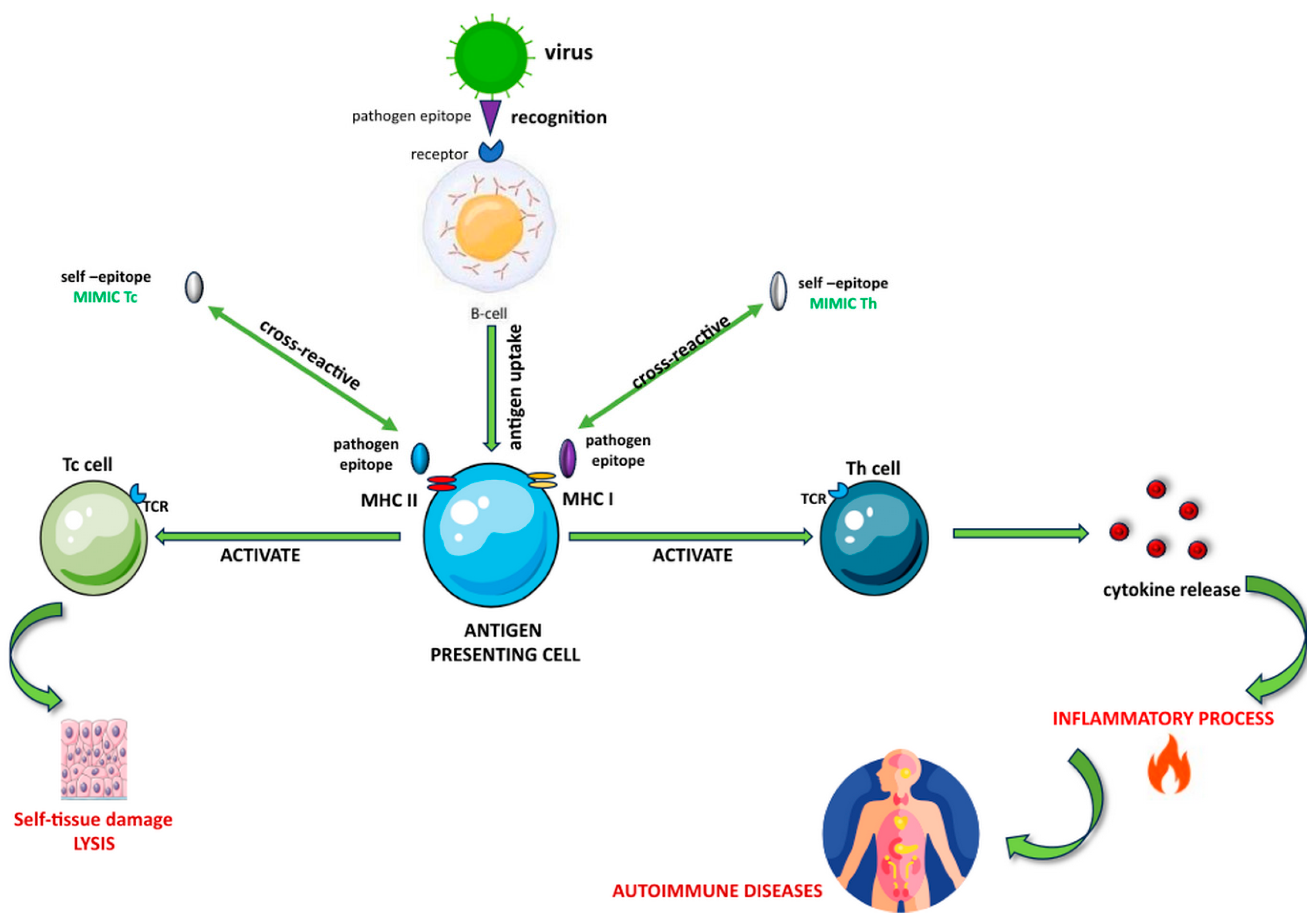
1.2. Molecular Mimicry in Autoimmune Diseases
1.2.1. Molecular Mimicry in Multiple Sclerosis
1.2.2. Molecular Mimicry in Autoimmune Hepatitis
1.2.3. Molecular Mimicry in Autoimmune Thyroid Disease
1.2.4. Molecular Mimicry in Systemic Lupus Erythematosus
1.2.5. Molecular Mimicry in Rheumatoid Arthritis
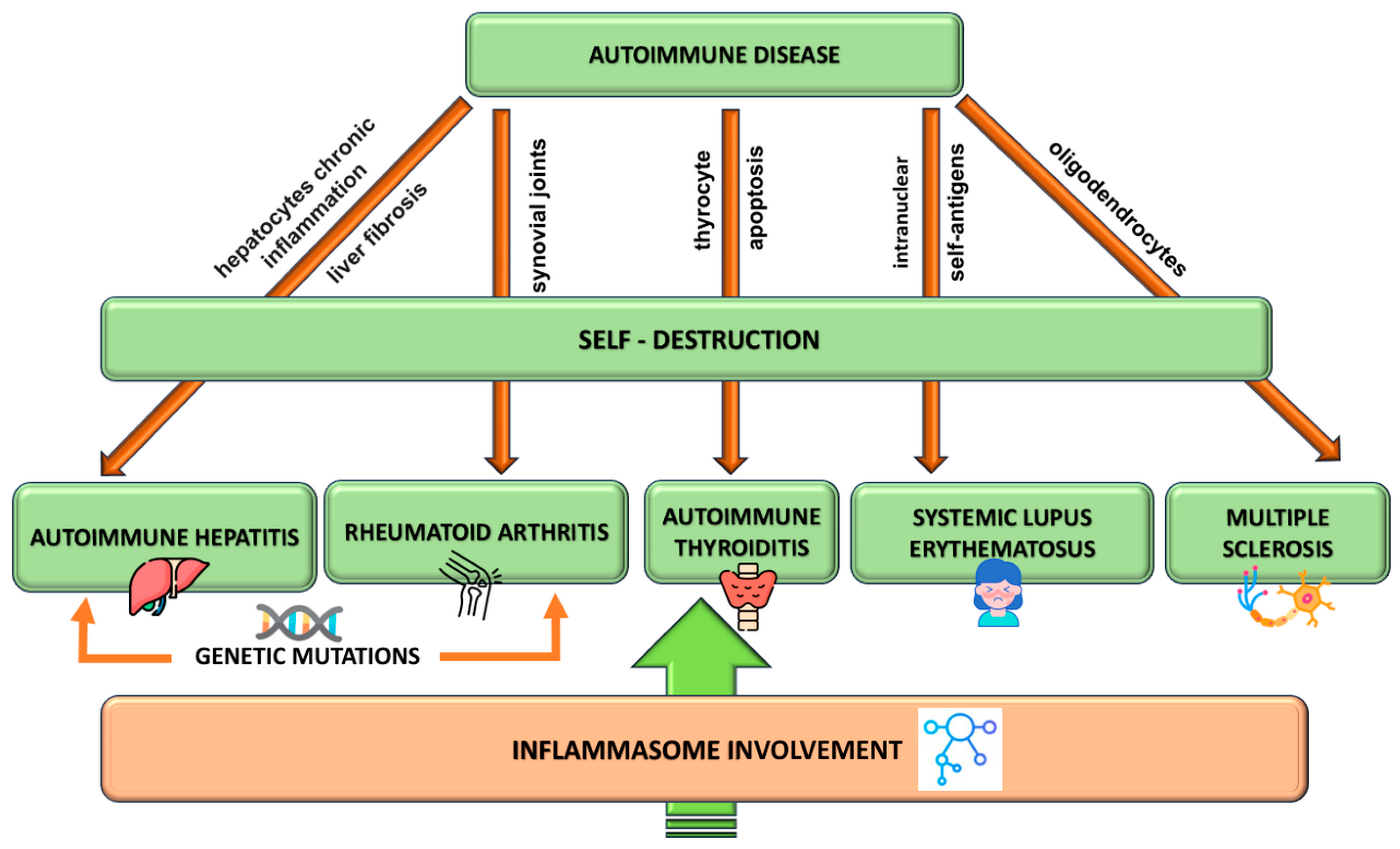
2. Autoimmune Disorders Pathogenesis
2.1. Multiple Sclerosis Pathogenesis
2.2. Autoimmune Hepatitis Pathogenesis
2.3. Autoimmune Thyroid Disease
2.4. Systemic Lupus Erythematosus
2.5. Rheumatoid Arthritis Pathogenesis
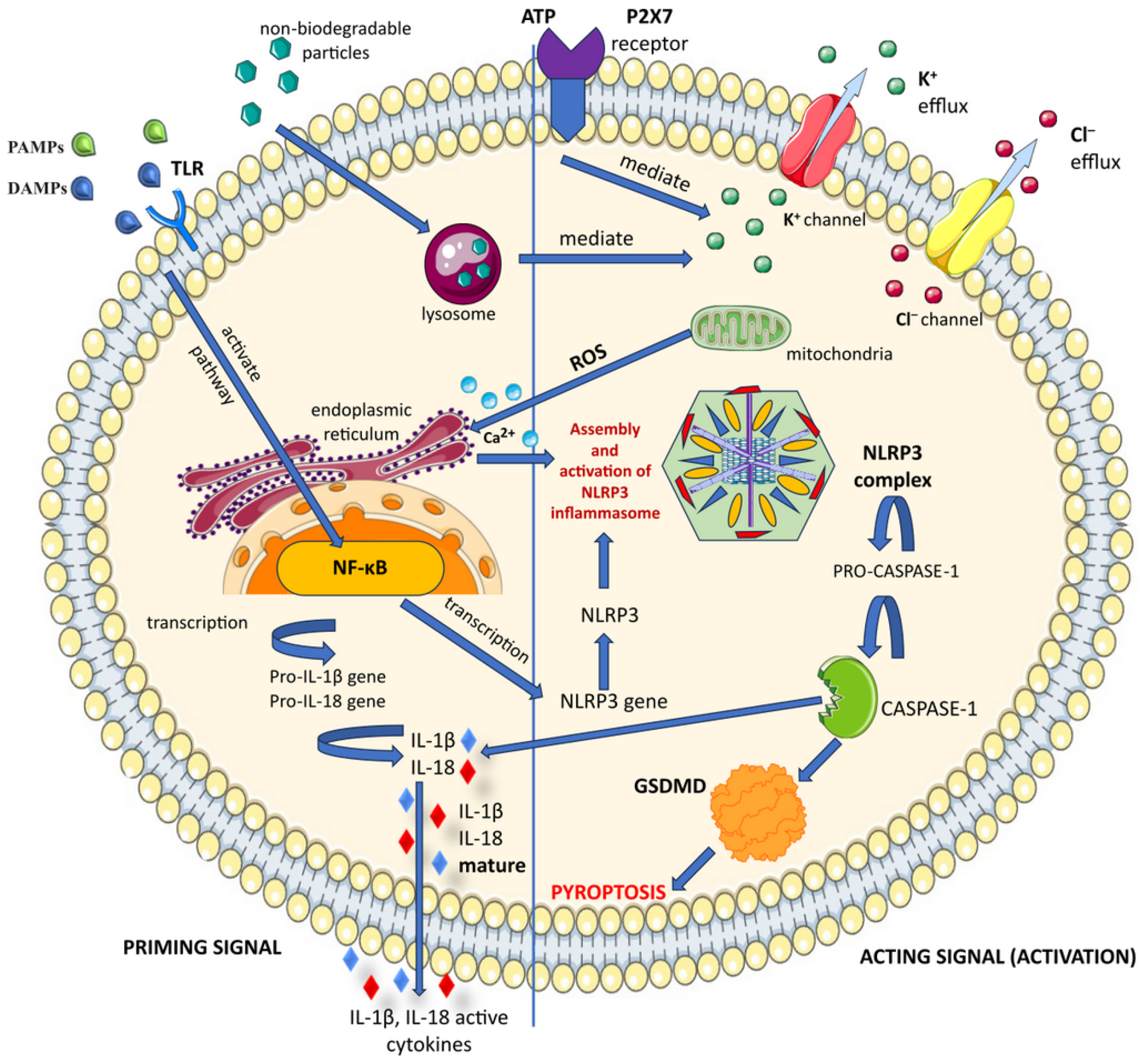
3. Inflammasome Structure and Functioning
3.1. Canonical Inflammasomes
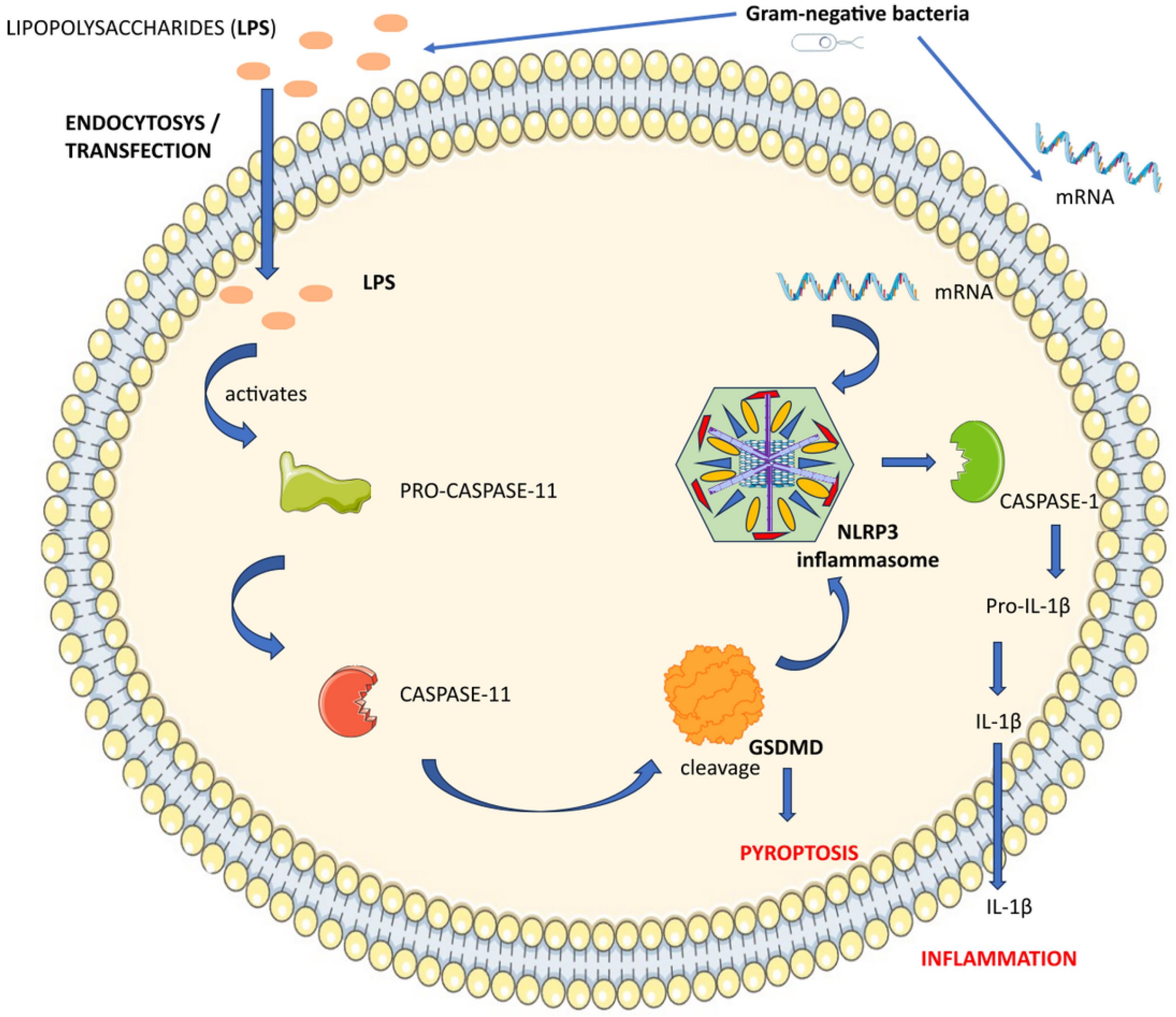
3.2. Non-Canonical Inflammasomes
3.3. Inflammasome Activation
3.4. NLRP3 Canonical Activation
3.5. NLRP3 Non-Canonical Activation
4. Inflammasome and Innate Immune System
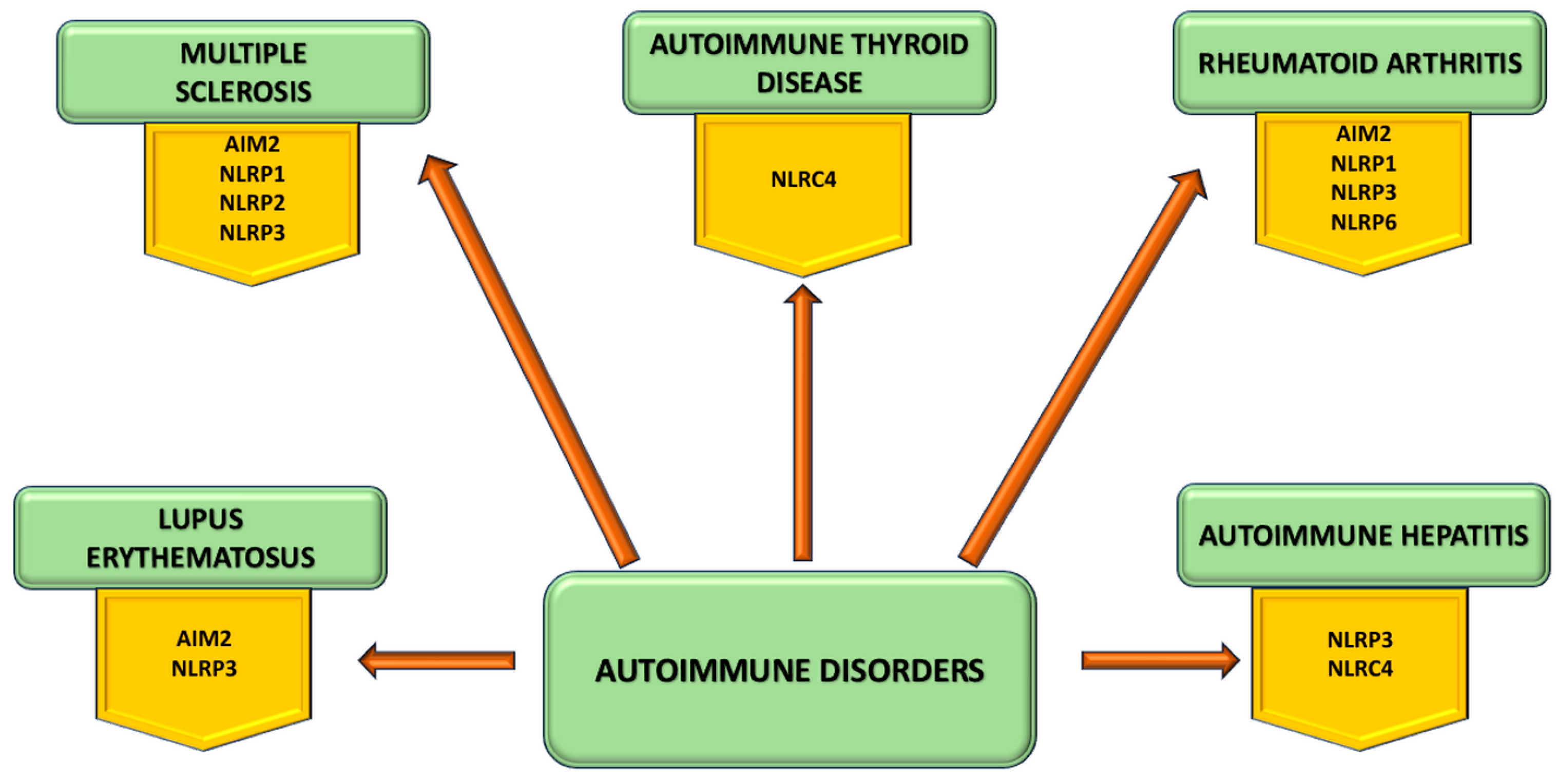
5. Inflammasome Involvement in Autoimmune Disorders
5.1. Inflammasome in Multiple Sclerosis
5.2. Inflammasome in Autoimmune Hepatitis
5.3. Inflammasome in Autoimmune Thyroid Disease
5.4. Inflammasome in Systemic Lupus Erhythematosus
5.5. Inflammasome in Rheumatoid Arthritis
6. Inflammasome as a Therapeutic Target
7. New Biomarkers—Promising Strategies for the Management of Autoimmune Diseases
8. Conclusions
9. Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| 5-HT | 5-hydroxytryptamine |
| 11β-HSD1 | 11β-hydroxysteroid dehydrogenase 1 |
| AID | Autoimmune disease |
| AIH | Autoimmune hepatitis |
| AIM2 | Absent in melanoma 2 |
| AITD | Autoimmune thyroid disease |
| ALR | Absent in melanoma 2-like receptor |
| APC | Antigen-presenting cells |
| ASC | Apoptosis-associated speck-like protein containing a CARD |
| ATP | Adenosine triphosphate |
| BHB | β-Hydroxybutyrate |
| BTK | Bruton tyrosine kinase |
| CARD | Caspase activation and recruitment domain |
| Ch25h | Cholesterol 25-hydroxylase |
| CNS | Central nervous system |
| ConA | Concanavalin A |
| CTL | Cytotoxic T cell |
| DAMP | Damage-associated molecular pattern |
| DNA | Deoxyribonucleic acid |
| EBV | Epstein–Barr virus |
| ER | Endoplasmic reticulum |
| FasL | Fas ligand |
| FLS | Fibroblast-like synoviocytes |
| GD | Graves’ disease |
| GSDMD | Gasdermin D |
| HCV | Hepatitis C virus |
| HT | Hashimoto’s thyroiditis |
| IL | Interleukin |
| IFN-γ | Interferon-gamma |
| IFN-I | Type-I interferon |
| LN | Lupus nephritis |
| LPS | Lipopolysaccharide |
| LRR | Leucine-rich repeat |
| MAC | Membrane attack complex |
| MHC | Major histocompatibility complex |
| MS | Multiple sclerosis |
| Mt | Mitochondrial |
| NACTH/NBD | Nucleotide-binding domain |
| NEK7 | NIMA-related protein kinase 7 |
| NF-κB | Nuclear factor-kappa B |
| NIMA | Never in mitosis gene A |
| NOD | Nucleotide-binding and oligomerization domain |
| NLR | NOD-like receptor |
| NLRC4 | NLR family CARD domain-containing protein 4 |
| NLRP | Nucleotide-binding domain, leucine-rich-containing family, pyrin domain-containing |
| NSAID | Nonsteroidal anti-inflammatory drug |
| P2X7R | P2X purinoceptor 7 |
| PAMP | Pathogen associated molecular pattern |
| PRR | Pattern recognition receptor |
| PYD | Pyrin domain |
| RA | Rheumatoid arthritis |
| RIG-I | Retinoic acid-inducible gene I |
| RLR | RIG-I-like receptor |
| RNA | Ribonucleic acid |
| ROS | Reactive oxygen species |
| SLE | Systemic lupus erythematosus |
| TCR | T cell receptor |
| Th | T helper |
| TLR | Toll-like receptor |
| TNF-α | Tumor necrosis factor-alpha |
| UVB | Ultraviolet radiation type B |
| VDR | Vitamin D receptor |
References
- Ercolini, A.M.; Miller, S.D. The role of infections in autoimmune disease. Clin. Exp. Immunol. 2009, 155, 1–15. [Google Scholar] [CrossRef]
- Sundaresan, B.; Shirafkan, F.; Ripperger, K.; Rattay, K. The Role of Viral Infections in the Onset of Autoimmune Diseases. Viruses 2023, 15, 782. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Y.; Zhang, M.; Jiang, D.; Su, Q.; Shi, J. The role of inflammation in autoimmune disease: A therapeutic target. Front. Immunol. 2023, 14, 1267091. [Google Scholar] [CrossRef]
- Lou, H.; Ling, G.S.; Cao, X. Autoantibodies in systemic lupus erythematosus: From immunopathology to therapeutic target. J. Autoimmun. 2022, 132, 102861. [Google Scholar] [CrossRef]
- Pisetsky, D.S. Pathogenesis of autoimmune disease. Nat. Rev. Nephrol. 2023, 19, 509–524. [Google Scholar] [CrossRef] [PubMed]
- Rojas, M.; Restrepo-Jiménez, P.; Monsalve, D.M.; Pacheco, Y.; Acosta-Ampudia, Y.; Ramírez-Santana, C.; Leung, P.S.C.; Ansari, A.A.; Gershwin, M.E.; Anaya, J.M. Molecular mimicry and autoimmunity. J. Autoimmun. 2018, 95, 100–123. [Google Scholar] [CrossRef]
- Blank, M.; Barzilai, O.; Shoenfeld, Y. Molecular mimicry and auto-immunity. Clin. Rev. Allergy Immunol. 2007, 32, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Fujinami, R.S.; Oldstone, M.B.; Wroblewska, Z.; Frankel, M.E.; Koprowski, H. Molecular mimicry in virus infection: Crossreaction of measles virus phosphoprotein or of herpes simplex virus protein with human intermediate filaments. Proc. Natl. Acad. Sci. USA 1983, 80, 2346–2350. [Google Scholar] [CrossRef]
- Zabriskie, J.B. Rheumatic fever: A model for the pathological consequences of microbial-host mimicry. Clin. Exp. Rheumatol. 1986, 4, 65–73. [Google Scholar]
- Oldstone, M.B. Molecular mimicry and autoimmune disease. Cell 1987, 50, 819–820. [Google Scholar] [CrossRef]
- Olson, J.K.; Croxford, J.L.; Calenoff, M.A.; Dal Canto, M.C.; Miller, S.D. A virus-induced molecular mimicry model of multiple sclerosis. J. Clin. Investig. 2001, 108, 311–318. [Google Scholar] [CrossRef]
- Scherer, M.T.; Ignatowicz, L.; Winslow, G.M.; Kappler, J.W.; Marrack, P. Superantigens: Bacterial and viral proteins that manipulate the immune system. Annu. Rev. Cell Biol. 1993, 9, 101–128. [Google Scholar] [CrossRef]
- Delogu, L.G.; Deidda, S.; Delitala, G.; Manetti, R. Infectious diseases and autoimmunity. J. Infect. Dev. Ctries. 2011, 5, 679–687. [Google Scholar] [CrossRef] [PubMed]
- Benoist, C.; Mathis, D. Autoimmunity provoked by infection: How good is the case for T cell epitope mimicry? Nat. Immunol. 2001, 2, 797–801. [Google Scholar] [CrossRef] [PubMed]
- Wucherpfennig, K.W. Mechanisms for the induction of autoimmunity by infectious agents. J. Clin. Investig. 2001, 108, 1097–1104. [Google Scholar] [CrossRef]
- Acharya, S.; Shukla, S.; Mahajan, S.N.; Diwan, S.K. Molecular mimicry in human diseases--phenomena or epiphenomena? J. Assoc. Physicians India 2010, 58, 163–168. [Google Scholar] [PubMed]
- Cusick, M.F.; Libbey, J.E.; Fujinami, R.S. Molecular mimicry as a mechanism of autoimmune disease. Clin. Rev. Allergy Immunol. 2012, 42, 102–111. [Google Scholar] [CrossRef]
- Miller, S.D.; Vanderlugt, C.L.; Begolka, W.S.; Pao, W.; Yauch, R.L.; Neville, K.L.; Katz-Levy, Y.; Carrizosa, A.; Kim, B.S. Persistent infection with Theiler’s virus leads to CNS autoimmunity via epitope spreading. Nat. Med. 1997, 3, 1133–1136. [Google Scholar] [CrossRef]
- Musette, P.; Bouaziz, J.D. B Cell Modulation Strategies in Autoimmune Diseases: New Concepts. Front. Immunol. 2018, 9, 622. [Google Scholar] [CrossRef]
- Lees, J.R. Targeting antigen presentation in autoimmunity. Cell. Immunol. 2019, 339, 4–9. [Google Scholar] [CrossRef]
- Abou-Raya, A.; Abou-Raya, S. Inflammation: A pivotal link between autoimmune diseases and atherosclerosis. Autoimmun. Rev. 2006, 5, 331–337. [Google Scholar] [CrossRef]
- Geginat, J.; Paroni, M.; Pagani, M.; Galimberti, D.; De Francesco, R.; Scarpini, E.; Abrignani, S. The Enigmatic Role of Viruses in Multiple Sclerosis: Molecular Mimicry or Disturbed Immune Surveillance? Trends Immunol. 2017, 38, 498–512. [Google Scholar] [CrossRef] [PubMed]
- Dendrou, C.A.; Fugger, L.; Friese, M.A. Immunopathology of multiple sclerosis. Nat. Rev. Immunol. 2015, 15, 545–558. [Google Scholar] [CrossRef] [PubMed]
- Vasincu, A.; Rusu, R.N.; Ababei, D.C.; Larion, M.; Bild, W.; Stanciu, G.D.; Solcan, C.; Bild, V. Endocannabinoid Modulation in Neurodegenerative Diseases: In Pursuit of Certainty. Biology 2022, 11, 440. [Google Scholar] [CrossRef] [PubMed]
- Libbey, J.E.; McCoy, L.L.; Fujinami, R.S. Molecular mimicry in multiple sclerosis. Int. Rev. Neurobiol. 2007, 79, 127–147. [Google Scholar] [CrossRef] [PubMed]
- Namaka, M.; Kapoor, S.; Simms, L.; Leong, C.; Grossberndt, A.; Prouta, M.; Frost, E.; Esfahani, F.; Gomori, A.; Mulvey, M.R. Molecular mimicry and multiple sclerosis. Neural Regen. Res. 2011, 6, 1322–1333. [Google Scholar]
- Christen, U.; Hintermann, E. Pathogens and autoimmune hepatitis. Clin. Exp. Immunol. 2019, 195, 35–51. [Google Scholar] [CrossRef]
- Christen, U.; Hintermann, E. Autoantibodies in Autoimmune Hepatitis: Can Epitopes Tell Us about the Etiology of the Disease? Front. Immunol. 2018, 9, 163. [Google Scholar] [CrossRef] [PubMed]
- Muratori, P.; Lenzi, M.; Cassani, F.; Lalanne, C.; Muratori, L. Diagnostic approach to autoimmune hepatitis. Expert Rev. Clin. Immunol. 2017, 13, 769–779. [Google Scholar] [CrossRef]
- Christen, U. Chapter 4—Molecular Mimicry. In Autoantibodies, 3rd ed.; Shoenfeld, Y., Meroni, P.L., Gershwin, M.E., Eds.; Elsevier: San Diego, CA, USA, 2014; pp. 35–42. [Google Scholar] [CrossRef]
- Arndtz, K.; Hirschfield, G.M. The Pathogenesis of Autoimmune Liver Disease. Dig. Dis. 2016, 34, 327–333. [Google Scholar] [CrossRef]
- Liberal, R.; Vergani, D.; Mieli-Vergani, G. Update on Autoimmune Hepatitis. J. Clin. Transl. Hepatol. 2015, 3, 42–52. [Google Scholar] [CrossRef] [PubMed]
- Chiuri, R.M.; Matronola, M.F.; Di Giulio, C.; Comegna, L.; Chiarelli, F.; Blasetti, A. Bartonella henselae infection associated with autoimmune thyroiditis in a child. Horm. Res. Paediatr. 2013, 79, 185–188. [Google Scholar] [CrossRef] [PubMed]
- Tomer, Y.; Villanueva, R. Infection and autoimmune thyroid diseases. In Infection and Autoimmunity; Shoenfeld, Y., Rose, N.R., Eds.; Elsevier: Amsterdam, The Netherlands, 2004; pp. 515–530. [Google Scholar]
- Caza, T.; Oaks, Z.; Perl, A. Interplay of infections, autoimmunity, and immunosuppression in systemic lupus erythematosus. Int. Rev. Immunol. 2014, 33, 330–363. [Google Scholar] [CrossRef]
- Sundar, K.; Jacques, S.; Gottlieb, P.; Villars, R.; Benito, M.E.; Taylor, D.K.; Spatz, L.A. Expression of the Epstein-Barr virus nuclear antigen-1 (EBNA-1) in the mouse can elicit the production of anti-dsDNA and anti-Sm antibodies. J. Autoimmun. 2004, 23, 127–140. [Google Scholar] [CrossRef] [PubMed]
- Tsokos, G.C. Systemic lupus erythematosus. N. Engl. J. Med. 2011, 365, 2110–2121. [Google Scholar] [CrossRef]
- McInnes, I.B.; Schett, G. The pathogenesis of rheumatoid arthritis. N. Engl. J. Med. 2011, 365, 2205–2219. [Google Scholar] [CrossRef] [PubMed]
- Lundberg, K.; Kinloch, A.; Fisher, B.A.; Wegner, N.; Wait, R.; Charles, P.; Mikuls, T.R.; Venables, P.J. Antibodies to citrullinated alpha-enolase peptide 1 are specific for rheumatoid arthritis and cross-react with bacterial enolase. Arthritis Rheum. 2008, 58, 3009–3019. [Google Scholar] [CrossRef] [PubMed]
- Rashid, T.; Jayakumar, K.S.; Binder, A.; Ellis, S.; Cunningham, P.; Ebringer, A. Rheumatoid arthritis patients have elevated antibodies to cross-reactive and non cross-reactive antigens from Proteus microbes. Clin. Exp. Rheumatol. 2007, 25, 259–267. [Google Scholar] [PubMed]
- Compston, A.; Coles, A. Multiple sclerosis. Lancet 2008, 372, 1502–1517. [Google Scholar] [CrossRef]
- Ghasemi, N.; Razavi, S.; Nikzad, E. Multiple Sclerosis: Pathogenesis, Symptoms, Diagnoses and Cell-Based Therapy. Cell J. 2017, 19, 1–10. [Google Scholar] [CrossRef]
- Gandhi, R.; Laroni, A.; Weiner, H.L. Role of the innate immune system in the pathogenesis of multiple sclerosis. J. Neuroimmunol. 2010, 221, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Kasper, L.H.; Shoemaker, J. Multiple sclerosis immunology: The healthy immune system vs the MS immune system. Neurology 2010, 74 (Suppl. 1), S2–S8. [Google Scholar] [CrossRef] [PubMed]
- Schoenborn, J.R.; Wilson, C.B. Regulation of interferon-gamma during innate and adaptive immune responses. Adv. Immunol. 2007, 96, 41–101. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, W.; Kolls, J.K.; Zheng, Y. The biological functions of T helper 17 cell effector cytokines in inflammation. Immunity 2008, 28, 454–467. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Zhang, H. The pathogenesis of autoimmune hepatitis. Front. Lab. Med. 2018, 2, 36–39. [Google Scholar] [CrossRef]
- Fan, J.H.; Liu, G.F.; Lv, X.D.; Zeng, R.Z.; Zhan, L.L.; Lv, X.P. Pathogenesis of autoimmune hepatitis. World J. Hepatol. 2021, 13, 879–886. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.I.; Kagihara, J.E.; Tsai, N.C.; Roytman, M.M. Autoimmune Hepatitis in Hawai’i. Hawaii J. Med. Public Health 2015, 74, 270–274. [Google Scholar]
- de Oliveira, L.C.; Goldberg, A.C.; Marin, M.L.; Schneidwind, K.R.; Frade, A.F.; Kalil, J.; Miura, I.K.; Pugliese, R.P.; Danesi, V.L.; Porta, G. Autoimmune Hepatitis in Brazilian Children: IgE and Genetic Polymorphisms in Associated Genes. J. Immunol. Res. 2015, 2015, 679813. [Google Scholar] [CrossRef] [PubMed]
- Czaja, A.J.; Manns, M.P. Advances in the diagnosis, pathogenesis, and management of autoimmune hepatitis. Gastroenterology 2010, 139, 58–72.e4. [Google Scholar] [CrossRef]
- Matías, N.; Fernandez, A.; Manganelli, V.; Colell, A.; Tinari, A.; Malorni, W.; Garcia-Ruiz, C.; Sorice, M.; Fernandez-Checa, J. Cholesterol regulates mitochondrial raft-like domains during TNF/Fas-mediated hepatocellular apoptosis. Chem. Phys. Lipids 2010, 163, S59. [Google Scholar] [CrossRef]
- Tomer, Y. Mechanisms of autoimmune thyroid diseases: From genetics to epigenetics. Annu. Rev. Pathol. 2014, 9, 147–156. [Google Scholar] [CrossRef] [PubMed]
- Rydzewska, M.; Jaromin, M.; Pasierowska, I.E.; Stożek, K.; Bossowski, A. Role of the T and B lymphocytes in pathogenesis of autoimmune thyroid diseases. Thyroid Res. 2018, 11, 2. [Google Scholar] [CrossRef] [PubMed]
- Iwatani, Y.; Watanabe, M. Normal Mechanisms for Self-Tolerance. In Autoimmune Endocrinopathies; Volpé, R., Ed.; Humana Press: Totowa, NJ, USA, 1999; pp. 1–30. [Google Scholar]
- Antonelli, A.; Ferrari, S.M.; Corrado, A.; Di Domenicantonio, A.; Fallahi, P. Autoimmune thyroid disorders. Autoimmun. Rev. 2015, 14, 174–180. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Leví, A.M.; Marazuela, M. Pathogenesis of thyroid autoimmune disease: The role of cellular mechanisms. Endocrinol. Nutr. 2016, 63, 421–429. [Google Scholar] [CrossRef] [PubMed]
- Nanba, T.; Watanabe, M.; Inoue, N.; Iwatani, Y. Increases of the Th1/Th2 cell ratio in severe Hashimoto’s disease and in the proportion of Th17 cells in intractable Graves’ disease. Thyroid 2009, 19, 495–501. [Google Scholar] [CrossRef] [PubMed]
- Bedoya, S.K.; Lam, B.; Lau, K.; Larkin, J., 3rd. Th17 cells in immunity and autoimmunity. Clin. Dev. Immunol. 2013, 2013, 986789. [Google Scholar] [CrossRef] [PubMed]
- Pearce, E.N.; Farwell, A.P.; Braverman, L.E. Thyroiditis. N. Engl. J. Med. 2003, 348, 2646–2655. [Google Scholar] [CrossRef]
- Ameer, M.A.; Chaudhry, H.; Mushtaq, J.; Khan, O.S.; Babar, M.; Hashim, T.; Zeb, S.; Tariq, M.A.; Patlolla, S.R.; Ali, J.; et al. An Overview of Systemic Lupus Erythematosus (SLE) Pathogenesis, Classification, and Management. Cureus 2022, 14, e30330. [Google Scholar] [CrossRef] [PubMed]
- Karrar, S.; Cunninghame Graham, D.S. Abnormal B Cell Development in Systemic Lupus Erythematosus: What the Genetics Tell Us. Arthritis Rheumatol. 2018, 70, 496–507. [Google Scholar] [CrossRef]
- Justiz Vaillant, A.; Goyal, A.; Varacallo, M. Systemic Lupus Erythematosus; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Tayem, M.G.; Shahin, L.; Shook, J.; Kesselman, M.M. A Review of Cardiac Manifestations in Patients With Systemic Lupus Erythematosus and Antiphospholipid Syndrome With Focus on Endocarditis. Cureus 2022, 14, e21698. [Google Scholar] [CrossRef]
- Paroli, M.; Caccavale, R.; Fiorillo, M.T.; Spadea, L.; Gumina, S.; Candela, V.; Paroli, M.P. The Double Game Played by Th17 Cells in Infection: Host Defense and Immunopathology. Pathogens 2022, 11, 1547. [Google Scholar] [CrossRef]
- Scheen, M.; Adedjouma, A.; Esteve, E.; Buob, D.; Abisror, N.; Planche, V.; Fain, O.; Boffa, J.J.; De Seigneux, S.; Mekinian, A.; et al. Kidney disease in antiphospholipid antibody syndrome: Risk factors, pathophysiology and management. Autoimmun. Rev. 2022, 21, 103072. [Google Scholar] [CrossRef]
- Berland, R.; Fernandez, L.; Kari, E.; Han, J.H.; Lomakin, I.; Akira, S.; Wortis, H.H.; Kearney, J.F.; Ucci, A.A.; Imanishi-Kari, T. Toll-like receptor 7-dependent loss of B cell tolerance in pathogenic autoantibody knockin mice. Immunity 2006, 25, 429–440. [Google Scholar] [CrossRef]
- Christensen, S.R.; Shupe, J.; Nickerson, K.; Kashgarian, M.; Flavell, R.A.; Shlomchik, M.J. Toll-like receptor 7 and TLR9 dictate autoantibody specificity and have opposing inflammatory and regulatory roles in a murine model of lupus. Immunity 2006, 25, 417–428. [Google Scholar] [CrossRef] [PubMed]
- Cook, H.T.; Botto, M. Mechanisms of Disease: The complement system and the pathogenesis of systemic lupus erythematosus. Nat. Clin. Pract. Rheumatol. 2006, 2, 330–337. [Google Scholar] [CrossRef]
- Leffler, J.; Bengtsson, A.A.; Blom, A.M. The complement system in systemic lupus erythematosus: An update. Ann. Rheum. Dis. 2014, 73, 1601–1606. [Google Scholar] [CrossRef] [PubMed]
- Branimir, A.; Miroslav, M. Pathogenesis of rheumatoid arthritis. Reumatizam 2014, 61, 19–23. [Google Scholar]
- Klareskog, L.; Rönnelid, J.; Lundberg, K.; Padyukov, L.; Alfredsson, L. Immunity to citrullinated proteins in rheumatoid arthritis. Annu. Rev. Immunol. 2008, 26, 651–675. [Google Scholar] [CrossRef]
- Choy, E. Understanding the dynamics: Pathways involved in the pathogenesis of rheumatoid arthritis. Rheumatology 2012, 51 (Suppl. 5), v3–v11. [Google Scholar] [CrossRef] [PubMed]
- Smolen, J.S.; Steiner, G. Therapeutic strategies for rheumatoid arthritis. Nat. Rev. Drug Discov. 2003, 2, 473–488. [Google Scholar] [CrossRef]
- Smolen, J.S.; Aletaha, D.; Koeller, M.; Weisman, M.H.; Emery, P. New therapies for treatment of rheumatoid arthritis. Lancet 2007, 370, 1861–1874. [Google Scholar] [CrossRef] [PubMed]
- McInnes, I.B.; Schett, G. Cytokines in the pathogenesis of rheumatoid arthritis. Nat. Rev. Immunol. 2007, 7, 429–442. [Google Scholar] [CrossRef] [PubMed]
- Firestein, G.S. Evolving concepts of rheumatoid arthritis. Nature 2003, 423, 356–361. [Google Scholar] [CrossRef] [PubMed]
- Rantapää-Dahlqvist, S.; de Jong, B.A.; Berglin, E.; Hallmans, G.; Wadell, G.; Stenlund, H.; Sundin, U.; van Venrooij, W.J. Antibodies against cyclic citrullinated peptide and IgA rheumatoid factor predict the development of rheumatoid arthritis. Arthritis Rheum. 2003, 48, 2741–2749. [Google Scholar] [CrossRef] [PubMed]
- Pesu, M.; Laurence, A.; Kishore, N.; Zwillich, S.H.; Chan, G.; O’Shea, J.J. Therapeutic targeting of Janus kinases. Immunol. Rev. 2008, 223, 132–142. [Google Scholar] [CrossRef] [PubMed]
- Schroder, K.; Tschopp, J. The inflammasomes. Cell 2010, 140, 821–832. [Google Scholar] [CrossRef] [PubMed]
- Downs, K.P.; Nguyen, H.; Dorfleutner, A.; Stehlik, C. An overview of the non-canonical inflammasome. Mol. Aspects Med. 2020, 76, 100924. [Google Scholar] [CrossRef]
- Ahmad, B.; Achek, A.; Farooq, M.; Choi, S. Accelerated NLRP3 inflammasome-inhibitory peptide design using a recurrent neural network model and molecular dynamics simulations. Comput. Struct. Biotechnol. J. 2023, 21, 4825–4835. [Google Scholar] [CrossRef]
- Cheburu, C.N.; Stoica, B.; Neamţu, A.; Vasile, C. Biocompatibility testing of chitosan hydrogels. Rev. Med. Chir. Soc. Med. Nat. Iasi 2011, 115, 864–870. [Google Scholar]
- Neamtu, A.; Tamba, B.; Patras, X. Molecular dynamics simulations of chondroitin sulfate in explicit solvent: Point charge water models compared. Cellul. Chem. Technol. 2013, 47, 191–202. [Google Scholar]
- Orlowsky, E.; Stabler, T.; Montell, E.; Vergés, J.; Kraus, V. Monosodium urate crystal induced macrophage inflammation is attenuated by chondroitin sulphate: Pre-clinical model for gout prophylaxis? BMC Musculoskelet. Disord. 2014, 15, 318. [Google Scholar] [CrossRef]
- Sheng, N.; Xing, F.; Zhang, Q.-Y.; Tan, J.; Nie, R.; Huang, K.; Li, H.-X.; Jiang, Y.-L.; Tan, B.; Xiang, Z.; et al. A pleiotropic SIS-based hydrogel with immunomodulation via NLRP3 inflammasome inhibition for diabetic bone regeneration. Chem. Eng. J. 2024, 480, 147985. [Google Scholar] [CrossRef]
- Xiao, L.; Magupalli, V.G.; Wu, H. Cryo-EM structures of the active NLRP3 inflammasome disc. Nature 2023, 613, 595–600. [Google Scholar] [CrossRef] [PubMed]
- Sharma, D.; Kanneganti, T.D. The cell biology of inflammasomes: Mechanisms of inflammasome activation and regulation. J. Cell Biol. 2016, 213, 617–629. [Google Scholar] [CrossRef] [PubMed]
- Broz, P.; Dixit, V.M. Inflammasomes: Mechanism of assembly, regulation and signalling. Nat. Rev. Immunol. 2016, 16, 407–420. [Google Scholar] [CrossRef] [PubMed]
- Lamkanfi, M.; Dixit, V.M. Mechanisms and functions of inflammasomes. Cell 2014, 157, 1013–1022. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Zhao, Y.; Shao, F. Non-canonical activation of inflammatory caspases by cytosolic LPS in innate immunity. Curr. Opin. Immunol. 2015, 32, 78–83. [Google Scholar] [CrossRef] [PubMed]
- Kayagaki, N.; Warming, S.; Lamkanfi, M.; Vande Walle, L.; Louie, S.; Dong, J.; Newton, K.; Qu, Y.; Liu, J.; Heldens, S.; et al. Non-canonical inflammasome activation targets caspase-11. Nature 2011, 479, 117–121. [Google Scholar] [CrossRef] [PubMed]
- Diamond, C.E.; Khameneh, H.J.; Brough, D.; Mortellaro, A. Novel perspectives on non-canonical inflammasome activation. Immunotargets Ther. 2015, 4, 131–141. [Google Scholar] [CrossRef]
- Mangan, M.S.J.; Olhava, E.J.; Roush, W.R.; Seidel, H.M.; Glick, G.D.; Latz, E. Targeting the NLRP3 inflammasome in inflammatory diseases. Nat. Rev. Drug Discov. 2018, 17, 588–606. [Google Scholar] [CrossRef]
- Shaw, P.J.; McDermott, M.F.; Kanneganti, T.D. Inflammasomes and autoimmunity. Trends Mol. Med. 2011, 17, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, O.; Akira, S. Pattern recognition receptors and inflammation. Cell 2010, 140, 805–820. [Google Scholar] [CrossRef]
- Yi, Y.S. Role of inflammasomes in inflammatory autoimmune rheumatic diseases. Korean J. Physiol. Pharmacol. 2018, 22, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Kanneganti, T.D.; Lamkanfi, M.; Núñez, G. Intracellular NOD-like receptors in host defense and disease. Immunity 2007, 27, 549–559. [Google Scholar] [CrossRef] [PubMed]
- Lamkanfi, M.; Dixit, V.M. Inflammasomes and their roles in health and disease. Annu. Rev. Cell Dev. Biol. 2012, 28, 137–161. [Google Scholar] [CrossRef] [PubMed]
- Strowig, T.; Henao-Mejia, J.; Elinav, E.; Flavell, R. Inflammasomes in health and disease. Nature 2012, 481, 278–286. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Callaway, J.B.; Ting, J.P. Inflammasomes: Mechanism of action, role in disease, and therapeutics. Nat. Med. 2015, 21, 677–687. [Google Scholar] [CrossRef] [PubMed]
- Latz, E.; Xiao, T.S.; Stutz, A. Activation and regulation of the inflammasomes. Nat. Rev. Immunol. 2013, 13, 397–411. [Google Scholar] [CrossRef]
- de Torre-Minguela, C.; Mesa Del Castillo, P.; Pelegrín, P. The NLRP3 and Pyrin Inflammasomes: Implications in the Pathophysiology of Autoinflammatory Diseases. Front. Immunol. 2017, 8, 43. [Google Scholar] [CrossRef]
- Zhang, Y.; Yang, W.; Li, W.; Zhao, Y. NLRP3 Inflammasome: Checkpoint Connecting Innate and Adaptive Immunity in Autoimmune Diseases. Front. Immunol. 2021, 12, 732933. [Google Scholar] [CrossRef]
- Lang, T.; Lee, J.P.W.; Elgass, K.; Pinar, A.A.; Tate, M.D.; Aitken, E.H.; Fan, H.; Creed, S.J.; Deen, N.S.; Traore, D.A.K.; et al. Macrophage migration inhibitory factor is required for NLRP3 inflammasome activation. Nat. Commun. 2018, 9, 2223. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Zeng, M.Y.; Yang, D.; Motro, B.; Núñez, G. NEK7 is an essential mediator of NLRP3 activation downstream of potassium efflux. Nature 2016, 530, 354–357. [Google Scholar] [CrossRef] [PubMed]
- Ito, M.; Shichita, T.; Okada, M.; Komine, R.; Noguchi, Y.; Yoshimura, A.; Morita, R. Bruton’s tyrosine kinase is essential for NLRP3 inflammasome activation and contributes to ischaemic brain injury. Nat. Commun. 2015, 6, 7360. [Google Scholar] [CrossRef] [PubMed]
- Shao, B.Z.; Xu, Z.Q.; Han, B.Z.; Su, D.F.; Liu, C. NLRP3 inflammasome and its inhibitors: A review. Front. Pharmacol. 2015, 6, 262. [Google Scholar] [CrossRef] [PubMed]
- Bauernfeind, F.G.; Horvath, G.; Stutz, A.; Alnemri, E.S.; MacDonald, K.; Speert, D.; Fernandes-Alnemri, T.; Wu, J.; Monks, B.G.; Fitzgerald, K.A.; et al. Cutting edge: NF-kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J. Immunol. 2009, 183, 787–791. [Google Scholar] [CrossRef] [PubMed]
- Guan, Y.; Gu, Y.; Li, H.; Liang, B.; Han, C.; Zhang, Y.; Liu, Q.; Wei, W.; Ma, Y. NLRP3 inflammasome activation mechanism and its role in autoimmune liver disease. Acta Biochim. Biophys. Sin. 2022, 54, 1577–1586. [Google Scholar] [CrossRef] [PubMed]
- Cookson, B.T.; Brennan, M.A. Pro-inflammatory programmed cell death. Trends Microbiol. 2001, 9, 113–114. [Google Scholar] [CrossRef] [PubMed]
- He, W.T.; Wan, H.; Hu, L.; Chen, P.; Wang, X.; Huang, Z.; Yang, Z.H.; Zhong, C.Q.; Han, J. Gasdermin D is an executor of pyroptosis and required for interleukin-1β secretion. Cell Res. 2015, 25, 1285–1298. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Zhao, Y.; Wang, K.; Shi, X.; Wang, Y.; Huang, H.; Zhuang, Y.; Cai, T.; Wang, F.; Shao, F. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 2015, 526, 660–665. [Google Scholar] [CrossRef]
- Gong, T.; Yang, Y.; Jin, T.; Jiang, W.; Zhou, R. Orchestration of NLRP3 Inflammasome Activation by Ion Fluxes. Trends Immunol. 2018, 39, 393–406. [Google Scholar] [CrossRef]
- Ozaki, E.; Campbell, M.; Doyle, S.L. Targeting the NLRP3 inflammasome in chronic inflammatory diseases: Current perspectives. J. Inflamm. Res. 2015, 8, 15–27. [Google Scholar] [CrossRef] [PubMed]
- Soriano-Teruel, P.M.; García-Laínez, G.; Marco-Salvador, M.; Pardo, J.; Arias, M.; DeFord, C.; Merfort, I.; Vicent, M.J.; Pelegrín, P.; Sancho, M.; et al. Identification of an ASC oligomerization inhibitor for the treatment of inflammatory diseases. Cell Death Dis. 2021, 12, 1155. [Google Scholar] [CrossRef] [PubMed]
- Kayagaki, N.; Stowe, I.B.; Lee, B.L.; O’Rourke, K.; Anderson, K.; Warming, S.; Cuellar, T.; Haley, B.; Roose-Girma, M.; Phung, Q.T.; et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 2015, 526, 666–671. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Zhao, Y.; Wang, Y.; Gao, W.; Ding, J.; Li, P.; Hu, L.; Shao, F. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature 2014, 514, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Junqueira, C.; Crespo, Â.; Ranjbar, S.; de Lacerda, L.B.; Lewandrowski, M.; Ingber, J.; Parry, B.; Ravid, S.; Clark, S.; Schrimpf, M.R.; et al. FcγR-mediated SARS-CoV-2 infection of monocytes activates inflammation. Nature 2022, 606, 576–584. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Ye, X.; Escames, G.; Lei, W.; Zhang, X.; Li, M.; Jing, T.; Yao, Y.; Qiu, Z.; Wang, Z.; et al. The NLRP3 inflammasome: Contributions to inflammation-related diseases. Cell Mol. Biol. Lett. 2023, 28, 51. [Google Scholar] [CrossRef] [PubMed]
- Moretti, J.; Jia, B.; Hutchins, Z.; Roy, S.; Yip, H.; Wu, J.; Shan, M.; Jaffrey, S.R.; Coers, J.; Blander, J.M. Caspase-11 interaction with NLRP3 potentiates the noncanonical activation of the NLRP3 inflammasome. Nat. Immunol. 2022, 23, 705–717. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.; Ou, Z.; Zhang, W.; Zhu, X.; Li, P.; Gong, J. Cathepsin B regulates non-canonical NLRP3 inflammasome pathway by modulating activation of caspase-11 in Kupffer cells. Cell Prolif. 2018, 51, e12487. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Li, X.; Luo, Z.; Li, M.; Liu, H.; Zhu, Z.; Wang, J.; Lu, P.; Wang, L.; Yang, C.; et al. Purinergic receptor P2X7 contributes to abdominal aortic aneurysm development via modulating macrophage pyroptosis and inflammation. Transl. Res. 2023, 258, 72–85. [Google Scholar] [CrossRef]
- Medzhitov, R.; Janeway, C., Jr. Innate immunity. N. Engl. J. Med. 2000, 343, 338–344. [Google Scholar] [CrossRef]
- Mogensen, T.H. Pathogen recognition and inflammatory signaling in innate immune defenses. Clin. Microbiol. Rev. 2009, 22, 240–273. [Google Scholar] [CrossRef] [PubMed]
- Chou, W.C.; Jha, S.; Linhoff, M.W.; Ting, J.P. The NLR gene family: From discovery to present day. Nat. Rev. Immunol. 2023, 23, 635–654. [Google Scholar] [CrossRef]
- Olson, J.K.; Miller, S.D. The innate immune response affects the development of the autoimmune response in Theiler’s virus-induced demyelinating disease. J. Immunol. 2009, 182, 5712–5722. [Google Scholar] [CrossRef] [PubMed]
- Firestein, G.S.; Zvaifler, N.J. How important are T cells in chronic rheumatoid synovitis?: II. T cell-independent mechanisms from beginning to end. Arthritis Rheum. 2002, 46, 298–308. [Google Scholar] [CrossRef] [PubMed]
- Man, S.M.; Karki, R.; Kanneganti, T.D. Molecular mechanisms and functions of pyroptosis, inflammatory caspases and inflammasomes in infectious diseases. Immunol. Rev. 2017, 277, 61–75. [Google Scholar] [CrossRef] [PubMed]
- Kaur, M.; Singh, M.; Silakari, O. Inhibitors of switch kinase ‘spleen tyrosine kinase’ in inflammation and immune-mediated disorders: A review. Eur. J. Med. Chem. 2013, 67, 434–446. [Google Scholar] [CrossRef] [PubMed]
- de Zoete, M.R.; Palm, N.W.; Zhu, S.; Flavell, R.A. Inflammasomes. Cold Spring Harb. Perspect. Biol. 2014, 6, a016287. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Guo, J.; Bi, L. Role of the NLRP3 inflammasome in autoimmune diseases. Biomed. Pharmacother. 2020, 130, 110542. [Google Scholar] [CrossRef] [PubMed]
- Ma, P.; Zha, S.; Shen, X.; Zhao, Y.; Li, L.; Yang, L.; Lei, M.; Liu, W. NFAT5 mediates hypertonic stress-induced atherosclerosis via activating NLRP3 inflammasome in endothelium. Cell Commun. Signal 2019, 17, 102. [Google Scholar] [CrossRef] [PubMed]
- Evavold, C.L.; Kagan, J.C. How Inflammasomes Inform Adaptive Immunity. J. Mol. Biol. 2018, 430, 217–237. [Google Scholar] [CrossRef]
- Swanson, K.V.; Deng, M.; Ting, J.P. The NLRP3 inflammasome: Molecular activation and regulation to therapeutics. Nat. Rev. Immunol. 2019, 19, 477–489. [Google Scholar] [CrossRef] [PubMed]
- Vanaja, S.K.; Rathinam, V.A.; Fitzgerald, K.A. Mechanisms of inflammasome activation: Recent advances and novel insights. Trends Cell Biol. 2015, 25, 308–315. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Yu, H.; Bu, Z.; Wen, L.; Yan, L.; Feng, J. Focus on the Role of the NLRP3 Inflammasome in Multiple Sclerosis: Pathogenesis, Diagnosis, and Therapeutics. Front. Mol. Neurosci. 2022, 15, 894298. [Google Scholar] [CrossRef] [PubMed]
- Imani, D.; Azimi, A.; Salehi, Z.; Rezaei, N.; Emamnejad, R.; Sadr, M.; Izad, M. Association of nod-like receptor protein-3 single nucleotide gene polymorphisms and expression with the susceptibility to relapsing-remitting multiple sclerosis. Int. J. Immunogenet. 2018, 45, 329–336. [Google Scholar] [CrossRef] [PubMed]
- Keane, R.W.; Dietrich, W.D.; de Rivero Vaccari, J.P. Inflammasome Proteins As Biomarkers of Multiple Sclerosis. Front. Neurol. 2018, 9, 135. [Google Scholar] [CrossRef] [PubMed]
- Voet, S.; Mc Guire, C.; Hagemeyer, N.; Martens, A.; Schroeder, A.; Wieghofer, P.; Daems, C.; Staszewski, O.; Vande Walle, L.; Jordao, M.J.C.; et al. A20 critically controls microglia activation and inhibits inflammasome-dependent neuroinflammation. Nat. Commun. 2018, 9, 2036. [Google Scholar] [CrossRef] [PubMed]
- McKenzie, B.A.; Mamik, M.K.; Saito, L.B.; Boghozian, R.; Monaco, M.C.; Major, E.O.; Lu, J.Q.; Branton, W.G.; Power, C. Caspase-1 inhibition prevents glial inflammasome activation and pyroptosis in models of multiple sclerosis. Proc. Natl. Acad. Sci. USA 2018, 115, E6065–E6074. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, J.I.; Cayrol, R.; Prat, A. Disruption of central nervous system barriers in multiple sclerosis. Biochim. Biophys. Acta 2011, 1812, 252–264. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Benveniste, E.N. Immune function of astrocytes. Glia 2001, 36, 180–190. [Google Scholar] [CrossRef]
- Correale, J.; Farez, M.F. The Role of Astrocytes in Multiple Sclerosis Progression. Front. Neurol. 2015, 6, 180. [Google Scholar] [CrossRef]
- Mallucci, G.; Peruzzotti-Jametti, L.; Bernstock, J.D.; Pluchino, S. The role of immune cells, glia and neurons in white and gray matter pathology in multiple sclerosis. Prog. Neurobiol. 2015, 127–128, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Hou, L.; Yang, J.; Li, S.; Huang, R.; Zhang, D.; Zhao, J.; Wang, Q. Glibenclamide attenuates 2,5-hexanedione-induced neurotoxicity in the spinal cord of rats through mitigation of NLRP3 inflammasome activation, neuroinflammation and oxidative stress. Toxicol. Lett. 2020, 331, 152–158. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Chen, X.; Tao, J.; Shi, A.; Zhang, J.; Yu, P. Exosomes Regulate NLRP3 Inflammasome in Diseases. Front. Cell Dev. Biol. 2021, 9, 802509. [Google Scholar] [CrossRef] [PubMed]
- Reubold, T.F.; Hahne, G.; Wohlgemuth, S.; Eschenburg, S. Crystal structure of the leucine-rich repeat domain of the NOD-like receptor NLRP1: Implications for binding of muramyl dipeptide. FEBS Lett. 2014, 588, 3327–3332. [Google Scholar] [CrossRef]
- Maver, A.; Lavtar, P.; Ristić, S.; Stopinšek, S.; Simčič, S.; Hočevar, K.; Sepčić, J.; Drulović, J.; Pekmezović, T.; Novaković, I.; et al. Identification of rare genetic variation of NLRP1 gene in familial multiple sclerosis. Sci. Rep. 2017, 7, 3715. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Xing, F.; Qu, M.; Yang, Z.; Liu, Y.; Yao, Y.; Xing, N. NLRP2 in health and disease. Immunology 2024, 171, 170–180. [Google Scholar] [CrossRef] [PubMed]
- Burm, S.M.; Zuiderwijk-Sick, E.A.; EJ‘t Jong, A.E.; van der Putten, C.; Veth, J.; Kondova, I.; Bajramovic, J.J. Inflammasome-induced IL-1β secretion in microglia is characterized by delayed kinetics and is only partially dependent on inflammatory caspases. J. Neurosci. 2015, 35, 678–687. [Google Scholar] [CrossRef] [PubMed]
- Minkiewicz, J.; de Rivero Vaccari, J.P.; Keane, R.W. Human astrocytes express a novel NLRP2 inflammasome. Glia 2013, 61, 1113–1121. [Google Scholar] [CrossRef] [PubMed]
- Heneka, M.T.; McManus, R.M.; Latz, E. Inflammasome signalling in brain function and neurodegenerative disease. Nat. Rev. Neurosci. 2018, 19, 610–621. [Google Scholar] [CrossRef]
- Govindarajan, V.; de Rivero Vaccari, J.P.; Keane, R.W. Role of inflammasomes in multiple sclerosis and their potential as therapeutic targets. J. Neuroinflamm. 2020, 17, 260. [Google Scholar] [CrossRef]
- Noroozi, S.; Meimand, H.A.E.; Arababadi, M.K.; Nakhaee, N.; Asadikaram, G. The Effects of IFN-β 1a on the Expression of Inflammasomes and Apoptosis-Associated Speck-Like Proteins in Multiple Sclerosis Patients. Mol. Neurobiol. 2017, 54, 3031–3037. [Google Scholar] [CrossRef]
- Banerjee, I.; Behl, B.; Mendonca, M.; Shrivastava, G.; Russo, A.J.; Menoret, A.; Ghosh, A.; Vella, A.T.; Vanaja, S.K.; Sarkar, S.N.; et al. Gasdermin D Restrains Type I Interferon Response to Cytosolic DNA by Disrupting Ionic Homeostasis. Immunity 2018, 49, 413–426.e5. [Google Scholar] [CrossRef]
- Villani, R.; Serviddio, G.; Avolio, C.; Cassano, T.; D’Amico, E. Autoimmune liver disease and multiple sclerosis: State of the art and future perspectives. Clin. Exp. Med. 2023, 23, 3321–3338. [Google Scholar] [CrossRef] [PubMed]
- Fazeli, P.; Kalani, M.; Hosseini, M. T memory stem cell characteristics in autoimmune diseases and their promising therapeutic values. Front. Immunol. 2023, 14, 1204231. [Google Scholar] [CrossRef]
- Luan, J.; Zhang, X.; Wang, S.; Li, Y.; Fan, J.; Chen, W.; Zai, W.; Wang, S.; Wang, Y.; Chen, M.; et al. NOD-Like Receptor Protein 3 Inflammasome-Dependent IL-1β Accelerated ConA-Induced Hepatitis. Front. Immunol. 2018, 9, 758. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Wang, G.; Ansari, G.A.S.; Khan, M.F. Trichloroethene metabolite dichloroacetyl chloride induces apoptosis and compromises phagocytosis in Kupffer Cells: Activation of inflammasome and MAPKs. PLoS ONE 2018, 13, e0210200. [Google Scholar] [CrossRef]
- Wang, H.; Wang, G.; Liang, Y.; Du, X.; Boor, P.J.; Sun, J.; Khan, M.F. Redox regulation of hepatic NLRP3 inflammasome activation and immune dysregulation in trichloroethene-mediated autoimmunity. Free Radic. Biol. Med. 2019, 143, 223–231. [Google Scholar] [CrossRef]
- de Jesus, A.A.; Canna, S.W.; Liu, Y.; Goldbach-Mansky, R. Molecular mechanisms in genetically defined autoinflammatory diseases: Disorders of amplified danger signaling. Annu. Rev. Immunol. 2015, 33, 823–874. [Google Scholar] [CrossRef]
- Wen, J.; Xuan, B.; Liu, Y.; Wang, L.; He, L.; Meng, X.; Zhou, T.; Wang, Y. Updating the NLRC4 Inflammasome: From Bacterial Infections to Autoimmunity and Cancer. Front. Immunol. 2021, 12, 702527. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Sun, Z.; Xie, J.; Ji, W.; Cui, Y.; Ai, Z.; Liang, G. Inflammasome and pyroptosis in autoimmune liver diseases. Front. Immunol. 2023, 14, 1150879. [Google Scholar] [CrossRef]
- Guo, Q.; Wu, Y.; Hou, Y.; Liu, Y.; Liu, T.; Zhang, H.; Fan, C.; Guan, H.; Li, Y.; Shan, Z.; et al. Cytokine Secretion and Pyroptosis of Thyroid Follicular Cells Mediated by Enhanced NLRP3, NLRP1, NLRC4, and AIM2 Inflammasomes Are Associated with Autoimmune Thyroiditis. Front. Immunol. 2018, 9, 1197. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Mao, C.; Dong, L.; Kang, P.; Ding, C.; Zheng, T.; Wang, X.; Xiao, Y. Excessive Iodine Promotes Pyroptosis of Thyroid Follicular Epithelial Cells in Hashimoto’s Thyroiditis Through the ROS-NF-κB-NLRP3 Pathway. Front. Endocrinol. 2019, 10, 778. [Google Scholar] [CrossRef] [PubMed]
- Aotsuka, S.; Nakamura, K.; Nakano, T.; Kawakami, M.; Goto, M.; Okawa-Takatsuji, M.; Kinoshita, M.; Yokohari, R. Production of intracellular and extracellular interleukin-1 alpha and interleukin-1 beta by peripheral blood monocytes from patients with connective tissue diseases. Ann. Rheum. Dis. 1991, 50, 27–31. [Google Scholar] [CrossRef] [PubMed]
- Boswell, J.M.; Yui, M.A.; Burt, D.W.; Kelley, V.E. Increased tumor necrosis factor and IL-1 beta gene expression in the kidneys of mice with lupus nephritis. J. Immunol. 1988, 141, 3050–3054. [Google Scholar] [CrossRef] [PubMed]
- Kahlenberg, J.M.; Kang, I. Advances in Disease Mechanisms and Translational Technologies: Clinicopathologic Significance of Inflammasome Activation in Autoimmune Diseases. Arthritis Rheumatol. 2020, 72, 386–395. [Google Scholar] [CrossRef]
- Kahlenberg, J.M.; Carmona-Rivera, C.; Smith, C.K.; Kaplan, M.J. Neutrophil extracellular trap-associated protein activation of the NLRP3 inflammasome is enhanced in lupus macrophages. J. Immunol. 2013, 190, 1217–1226. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Tao, X.; Tao, J. Strategies of Targeting Inflammasome in the Treatment of Systemic Lupus Erythematosus. Front. Immunol. 2022, 13, 894847. [Google Scholar] [CrossRef]
- Guarda, G.; Braun, M.; Staehli, F.; Tardivel, A.; Mattmann, C.; Förster, I.; Farlik, M.; Decker, T.; Du Pasquier, R.A.; Romero, P.; et al. Type I interferon inhibits interleukin-1 production and inflammasome activation. Immunity 2011, 34, 213–223. [Google Scholar] [CrossRef]
- Yu, X.; Du, Y.; Cai, C.; Cai, B.; Zhu, M.; Xing, C.; Tan, P.; Lin, M.; Wu, J.; Li, J.; et al. Inflammasome activation negatively regulates MyD88-IRF7 type I IFN signaling and anti-malaria immunity. Nat. Commun. 2018, 9, 4964. [Google Scholar] [CrossRef]
- Broderick, L.; De Nardo, D.; Franklin, B.S.; Hoffman, H.M.; Latz, E. The inflammasomes and autoinflammatory syndromes. Annu. Rev. Pathol. 2015, 10, 395–424. [Google Scholar] [CrossRef]
- Zhang, W.; Cai, Y.; Xu, W.; Yin, Z.; Gao, X.; Xiong, S. AIM2 facilitates the apoptotic DNA-induced systemic lupus erythematosus via arbitrating macrophage functional maturation. J. Clin. Immunol. 2013, 33, 925–937. [Google Scholar] [CrossRef] [PubMed]
- Choulaki, C.; Papadaki, G.; Repa, A.; Kampouraki, E.; Kambas, K.; Ritis, K.; Bertsias, G.; Boumpas, D.T.; Sidiropoulos, P. Enhanced activity of NLRP3 inflammasome in peripheral blood cells of patients with active rheumatoid arthritis. Arthritis Res. Ther. 2015, 17, 257. [Google Scholar] [CrossRef]
- Sui, J.; Li, H.; Fang, Y.; Liu, Y.; Li, M.; Zhong, B.; Yang, F.; Zou, Q.; Wu, Y. NLRP1 gene polymorphism influences gene transcription and is a risk factor for rheumatoid arthritis in han chinese. Arthritis Rheum. 2012, 64, 647–654. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Guo, N.; Ma, Y.; Ning, B.; Wang, Y.; Kou, L. Inhibition of P2X4 suppresses joint inflammation and damage in collagen-induced arthritis. Inflammation 2014, 37, 146–153. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.Y.; Li, K.T.; Yang, H.X.; Yang, B.; Lu, X.; Zhao, L.D.; Fei, Y.Y.; Chen, H.; Wang, L.; Li, J.; et al. Complement C1q synergizes with PTX3 in promoting NLRP3 inflammasome over-activation and pyroptosis in rheumatoid arthritis. J. Autoimmun. 2020, 106, 102336. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Gu, Y.; Zeng, X.; Wang, J. NLRP3 inflammasome regulates Th17 differentiation in rheumatoid arthritis. Clin. Immunol. 2018, 197, 154–160. [Google Scholar] [CrossRef] [PubMed]
- Mouasni, S.; Gonzalez, V.; Schmitt, A.; Bennana, E.; Guillonneau, F.; Mistou, S.; Avouac, J.; Ea, H.K.; Devauchelle, V.; Gottenberg, J.E.; et al. The classical NLRP3 inflammasome controls FADD unconventional secretion through microvesicle shedding. Cell Death Dis. 2019, 10, 190. [Google Scholar] [CrossRef] [PubMed]
- Grenier, J.M.; Wang, L.; Manji, G.A.; Huang, W.J.; Al-Garawi, A.; Kelly, R.; Carlson, A.; Merriam, S.; Lora, J.M.; Briskin, M.; et al. Functional screening of five PYPAF family members identifies PYPAF5 as a novel regulator of NF-kappaB and caspase-1. FEBS Lett. 2002, 530, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Ghimire, L.; Paudel, S.; Jin, L.; Jeyaseelan, S. The NLRP6 inflammasome in health and disease. Mucosal Immunol. 2020, 13, 388–398. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Luo, Z. NLRP6 facilitates the interaction between TAB2/3 and TRIM38 in rheumatoid arthritis fibroblast-like synoviocytes. FEBS Lett. 2017, 591, 1141–1149. [Google Scholar] [CrossRef]
- Jiang, Q.; Wang, X.; Huang, E.; Wang, Q.; Wen, C.; Yang, G.; Lu, L.; Cui, D. Inflammasome and Its Therapeutic Targeting in Rheumatoid Arthritis. Front. Immunol. 2021, 12, 816839. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Chang, D.K.; Goel, A.; Boland, C.R.; Bugbee, W.; Boyle, D.L.; Firestein, G.S. Microsatellite instability and suppressed DNA repair enzyme expression in rheumatoid arthritis. J. Immunol. 2003, 170, 2214–2220. [Google Scholar] [CrossRef]
- Moodley, D.; Mody, G.; Patel, N.; Chuturgoon, A.A. Mitochondrial depolarisation and oxidative stress in rheumatoid arthritis patients. Clin. Biochem. 2008, 41, 1396–1401. [Google Scholar] [CrossRef] [PubMed]
- Dinarello, C.A.; Novick, D.; Kim, S.; Kaplanski, G. Interleukin-18 and IL-18 binding protein. Front. Immunol. 2013, 4, 289. [Google Scholar] [CrossRef] [PubMed]
- Jesus, A.A.; Goldbach-Mansky, R. IL-1 blockade in autoinflammatory syndromes. Annu. Rev. Med. 2014, 65, 223–244. [Google Scholar] [CrossRef]
- Lamkanfi, M.; Mueller, J.L.; Vitari, A.C.; Misaghi, S.; Fedorova, A.; Deshayes, K.; Lee, W.P.; Hoffman, H.M.; Dixit, V.M. Glyburide inhibits the Cryopyrin/Nalp3 inflammasome. J. Cell Biol. 2009, 187, 61–70. [Google Scholar] [CrossRef]
- Zahid, A.; Li, B.; Kombe, A.J.K.; Jin, T.; Tao, J. Pharmacological Inhibitors of the NLRP3 Inflammasome. Front. Immunol. 2019, 10, 2538. [Google Scholar] [CrossRef]
- Reboldi, A.; Dang, E.V.; McDonald, J.G.; Liang, G.; Russell, D.W.; Cyster, J.G. Inflammation. 25-Hydroxycholesterol suppresses interleukin-1-driven inflammation downstream of type I interferon. Science 2014, 345, 679–684. [Google Scholar] [CrossRef] [PubMed]
- Coll, R.C.; Robertson, A.A.; Chae, J.J.; Higgins, S.C.; Muñoz-Planillo, R.; Inserra, M.C.; Vetter, I.; Dungan, L.S.; Monks, B.G.; Stutz, A.; et al. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat. Med. 2015, 21, 248–255. [Google Scholar] [CrossRef]
- Zhang, X.; Xu, A.; Lv, J.; Zhang, Q.; Ran, Y.; Wei, C.; Wu, J. Development of small molecule inhibitors targeting NLRP3 inflammasome pathway for inflammatory diseases. Eur. J. Med. Chem. 2020, 185, 111822. [Google Scholar] [CrossRef]
- Cao, S.; Weaver, C.M. Bioactives in the Food Supply: Effects on CVD Health. Curr. Atheroscler. Rep. 2022, 24, 655–661. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Wang, Y.; Chen, G. New Potentiality of Bioactive Substances: Regulating the NLRP3 Inflammasome in Autoimmune Diseases. Nutrients 2023, 15, 4584. [Google Scholar] [CrossRef] [PubMed]
- Daryabor, G.; Gholijani, N.; Kahmini, F.R. A review of the critical role of vitamin D axis on the immune system. Exp. Mol. Pathol. 2023, 132–133, 104866. [Google Scholar] [CrossRef]
- Rao, Z.; Chen, X.; Wu, J.; Xiao, M.; Zhang, J.; Wang, B.; Fang, L.; Zhang, H.; Wang, X.; Yang, S.; et al. Vitamin D Receptor Inhibits NLRP3 Activation by Impeding Its BRCC3-Mediated Deubiquitination. Front. Immunol. 2019, 10, 2783. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Liu, J.; Zhao, Y.; Xu, N.; Lv, E.; Ci, C.; Li, X. 1,25-dihydroxyvitamin D3 ameliorates lupus nephritis through inhibiting the NF-κB and MAPK signalling pathways in MRL/lpr mice. BMC Nephrol. 2022, 23, 243. [Google Scholar] [CrossRef]
- Piancone, F.; Saresella, M.; Marventano, I.; La Rosa, F.; Santangelo, M.A.; Caputo, D.; Mendozzi, L.; Rovaris, M.; Clerici, M. Monosodium Urate Crystals Activate the Inflammasome in Primary Progressive Multiple Sclerosis. Front. Immunol. 2018, 9, 983. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Ai, L.; Sun, Y.; Yang, B.; Chen, S.; Wang, Q.; Kuang, H. Role of NLRP3 Inflammasome in Lupus Nephritis and Therapeutic Targeting by Phytochemicals. Front. Pharmacol. 2021, 12, 621300. [Google Scholar] [CrossRef]
- Tsai, P.Y.; Ka, S.M.; Chang, J.M.; Chen, H.C.; Shui, H.A.; Li, C.Y.; Hua, K.F.; Chang, W.L.; Huang, J.J.; Yang, S.S.; et al. Epigallocatechin-3-gallate prevents lupus nephritis development in mice via enhancing the Nrf2 antioxidant pathway and inhibiting NLRP3 inflammasome activation. Free Radic. Biol. Med. 2011, 51, 744–754. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.J.; Klionsky, D.J.; Zhang, H. Podocytes and autophagy: A potential therapeutic target in lupus nephritis. Autophagy 2019, 15, 908–912. [Google Scholar] [CrossRef]
- Wu, Q.; Tang, Y.; Hu, X.; Wang, Q.; Lei, W.; Zhou, L.; Huang, J. Regulation of Th1/Th2 balance through OX40/OX40L signalling by glycyrrhizic acid in a murine model of asthma. Respirology 2016, 21, 102–111. [Google Scholar] [CrossRef]
- Wang, Y.Y.; Li, H.T.; Lu, Y.; Jia, X.Y.; Li, Y.L.; Chen, S.; Chai, J.X.; Zhang, J.J.; Liu, D.; Xie, C.H. [Protective effects of glycyrrhizic acid against lupus nephritis in MRL/lpr mice]. Nan Fang Yi Ke Da Xue Xue Bao 2017, 37, 957–961. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.; Zhang, R.Y.; Song, L. Beneficial effect of magnolol on lupus nephritis in MRL/lpr mice by attenuating the NLRP3 inflammasome and NF-κB signaling pathway: A mechanistic analysis. Mol. Med. Rep. 2017, 16, 4817–4822. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Wang, J.; Zhou, M.; Li, M.; Li, M.; Tan, H. Curcumin attenuates murine lupus via inhibiting NLRP3 inflammasome. Int. Immunopharmacol. 2019, 69, 213–216. [Google Scholar] [CrossRef] [PubMed]
- Holzinger, D.; Kessel, C.; Omenetti, A.; Gattorno, M. From bench to bedside and back again: Translational research in autoinflammation. Nat. Rev. Rheumatol. 2015, 11, 573–585. [Google Scholar] [CrossRef] [PubMed]
- Landolt-Marticorena, C. The need for preclinical biomarkers in systemic autoimmune rheumatic diseases. J. Rheumatol. 2015, 42, 152–154. [Google Scholar] [CrossRef] [PubMed]
- Kawakami, A.; Endo, Y.; Koga, T.; Yoshiura, K.I.; Migita, K. Autoinflammatory disease: Clinical perspectives and therapeutic strategies. Inflamm. Regen. 2022, 42, 37. [Google Scholar] [CrossRef] [PubMed]
- Alehashemi, S.; Goldbach-Mansky, R. Human Autoinflammatory Diseases Mediated by NLRP3-, Pyrin-, NLRP1-, and NLRC4-Inflammasome Dysregulation Updates on Diagnosis, Treatment, and the Respective Roles of IL-1 and IL-18. Front. Immunol. 2020, 11, 1840. [Google Scholar] [CrossRef]
- Maecker, H.T.; Lindstrom, T.M.; Robinson, W.H.; Utz, P.J.; Hale, M.; Boyd, S.D.; Shen-Orr, S.S.; Fathman, C.G. New tools for classification and monitoring of autoimmune diseases. Nat. Rev. Rheumatol. 2012, 8, 317–328. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Neamțu, M.; Bild, V.; Vasincu, A.; Arcan, O.D.; Bulea, D.; Ababei, D.-C.; Rusu, R.-N.; Macadan, I.; Sciucă, A.M.; Neamțu, A. Inflammasome Molecular Insights in Autoimmune Diseases. Curr. Issues Mol. Biol. 2024, 46, 3502-3532. https://doi.org/10.3390/cimb46040220
Neamțu M, Bild V, Vasincu A, Arcan OD, Bulea D, Ababei D-C, Rusu R-N, Macadan I, Sciucă AM, Neamțu A. Inflammasome Molecular Insights in Autoimmune Diseases. Current Issues in Molecular Biology. 2024; 46(4):3502-3532. https://doi.org/10.3390/cimb46040220
Chicago/Turabian StyleNeamțu, Monica, Veronica Bild, Alexandru Vasincu, Oana Dana Arcan, Delia Bulea, Daniela-Carmen Ababei, Răzvan-Nicolae Rusu, Ioana Macadan, Ana Maria Sciucă, and Andrei Neamțu. 2024. "Inflammasome Molecular Insights in Autoimmune Diseases" Current Issues in Molecular Biology 46, no. 4: 3502-3532. https://doi.org/10.3390/cimb46040220