Results and Discussion
The synthetic route followed is shown in
Scheme 1. One of the starting compounds, 3-phenyl-3-methyl-1-(2-chloro-1-oxoethyl)cyclobutane, was synthesized according to the general procedure described in [
8], starting from acrolein and dry chlorine, and subsequently isobutylene. Benzofuran derivatives were prepared from 2-hydroxy-5-bromobenzaldehyde or 2-hydroxy naphthalaldehyde and 3-phenyl-3-methyl-1-(2-chloro-1-oxoethyl) cyclobutane in different solvents.
The first reaction involves a nucleophilic substitution reaction in acetone between 3-phenyl-3-methyl-1-(2-chloro-1-oxoethyl) cyclobutane and the potassium salts of 2-hydroxy-5-bromo benzaldehyde or 2-hydroxynaphthalaldehyde. As a result of: (i) the strong nucleophilicity of the potassium salts of aldehydes, (ii) the absence of steric hindrance by 3-phenyl-3-methyl-1-(2-chloro-1-oxoethyl) cyclobutane and (iii) the increased positive charge of the positive center of the carbocation formed by the elimination of chloride from the latter, due to the inductive effect of chlorine atom, the acetone solvent provides a relatively mild temperature conditions for the formation of the expected substances with excellent yields. In
Scheme 1, the compounds
1,
3,
5,
7, and
9 have the R substituent, while
2,
4,
6,
8, and
10 include the R
1 one.
The first compound, benzofuran-2-yl-(3-methyl-3-phenylcyclobutyl) methanone (
1), is prepared in the presence of potassium carbonate in excellent yield (94%). It was expected that this compound should have two different isomers in the
cis and
trans configurations, due to the methyl and phenyl groups on the cyclobutane ring may. In fact, the starting material, 3-phenyl-3-methyl-1-(2-chloro-1-oxoethyl) cyclobutane itself is an 85:15 mixture of
cis and
trans isomeric structures [
8]. Unfortunately, only one isomer was separated, possible a result of the crystallization technique. In the second step, this compound was converted into its corresponding oxime
3 in 4:1 alcohol-water in the presence of pyridine by a well known method. The yield of the reaction was good (74%). As is known, oximes can be found in two different isomeric structures namely, the
syn and
anti configurations [
10,
11]. As expected, compound
3 (and similarly compound
4) shows two different isomeric structures. As can be seen from its
1H‑NMR spectra, both –CH
3 and oxime (N-OH) groups have double peaks. The oxime group has a descreening effect [
9], and thus the low-field signals are assigned to the
E-isomer, while the signals at the high-field are assigned to the
Z-isomer. Since the ratios of the
E- and
Z- isomers in the initial ketoximes and in the alkylation and acylation reaction products does not coincide in most cases, a partial
E/Z-isomerization probably takes place.
Reactions of 3 and 4 with epichlorohydrin, in the presence of KOH, gave oxime ethers 5 and 6. It is expected that these compounds must also be in different isomeric structures and it has been seen from the 1H-NMR spectra, that indeed these compounds are isomeric mixtures. Some functional groups of these compounds showed double peaks in the 1H-NMR spectra. Especially, substance 5 has two singlets at 1.57 and 1.62 ppm for –CH3 and four doublets at 4.12-4.50 ppm range for N-O-CH2- protons. When doublets are expanded, some additional doublets are shown. These results mean that these compounds have different isomeric structures resulting from cis and trans isomers on cyclobutane ring and syn and anti isomers on the oxime or the mixtures of these isomeric structures. Each one of the remaining four compounds 7, 8, 9 and 10 has only one isomeric structure.
The 13C-NMR resonances and IR data which are given in the Experimental section and the elemental analysis results confirm the identity of the compounds. All the compounds synthesized here are solid substances, stable at room temperature and not affected by atmospheric conditions over at least in two weeks. On the other hand, as these compounds contain cyclobutane, benzofuran, Schiff base, carbazone and thiazole functions in their molecules, they seem to be suitable candidates for further chemical modifications and may be pharmacologically active and useful as ligands in coordination chemistry.
For the biological evaluation of the compounds prepared some representative examples were screened against eight different microorganisms (six bacteria and two yeasts). The test results obtained are listed in
Table 1. Antifungal and antibacterial data for ampicillin and nystatin are also included in this Table for the purpose of comparison. The data show that these compounds generally exhibited moderate toxicity in the high concentrations towards many of the bacterias tested. Unexpectedly both of the substances tested showed no toxicity against all the yeasts. Biological activity studies of the substances on some other different bacterias and yeasts are in progress.
Table 1.
Antimicrobial effect of the compounds*
Table 1.
Antimicrobial effect of the compounds*
| Compound | B. s. LM622 | E. a. CCM 2531 | E. c. ATCC 25922 | L. m. SCOTTA | P. a. DSM 50071 | S. a. COWAN I | C. a. FMC 17 | S. c. FMC 16 |
|---|
| 7 | 9 | 6 | 9 | 9 | — | 9 | — | — |
| 8 | 8 | 10 | 8 | 10 | 8 | — | — | — |
| A.50 | 34 | — | 33 | 46 | — | 43 | — | — |
| B.50 | — | — | — | — | — | — | 18 | 22 |
Experimental
General
Benzene, acrolein, benzaldehyde and amines were purchased from Merck and used as received. Melting points were determined on a Thomas Hoover melting point apparatus and are uncorrected. The IR spectra were measured with Mattson 1000 FT-IR spectrophotometer. The 1H-NMR spectra were recorded on a Varian-Gemini 200 MHz spectrometer and are reported in ppm (δ) relative to tetramethylsilane (TMS) as the internal standard and 13C-NMR (50.34 MHz) is referenced to deuterochloroform (CDCl3). Elemental analyses were determined on a LECO CHNSO-932 auto elemental analysis apparatus. Analyses (TLC) were performed on precoated 0.2 mm Merck kieselgel 60 F254 plates, visualizing with 254 nm UV lamp. Column chromatography was performed using Merck silica gel, 70-230 mesh. Solvents were dried and purified by known conventional methods prior to use. As the preparation for the ten compounds are identical in pairs, we describe below the synthesis of 1, 3, 5, 7 and 9 as representative examples for all the compounds.
General procedure for the synthesis of ketones 1 and 2.
In a two necked 250 mL flask equipped with a reflux condenser, a mixture of 5-bromo-salicylaldehyde (4.02 g, 20 mmol), potassium carbonate (2.76 g, 20 mmol) and 3-phenyl-3-methyl-1-(2-chloro-1-oxoethyl) cyclobutane (4.45 g, 20 mmol) in acetone (100 mL) was refluxed for 8 h. The solvent was removed under reduced pressure and the residue was extracted with diethyl ether, the etheral phase dried over anhydrous MgSO4, filtered and the solvent removed under reduced pressure. Solid substance 1 thus obtained was crystallized from acetone.
(5-Bromo-1-benzofuran-2-yl)(3-methyl-3-phenylcyclobutyl)methanone (1). Yield: 6.93 g (94%); m.p.: 134-135 °C (from acetone); IR (KBr): 1674 (C=O), 1255 (C-O, on furan ring), 542 (C-Br) cm-1; 1H‑NMR (200 MHz; CDCl3) δ: 1.64 (s, 3H, CH3), 2.49 (m, 2H, CH2), 2.81 (m, 2H, CH2), 4.08 (q, 1H, >CH-), 7.15-7.83 (m, 9H, aromatics); 13C-NMR (50.34 MHz; CDCl3) δ: 193.74(C=O), 156.19, 154.90, 152.99, 133.04, 130.97, 130.34, 127.68, 126.64, 126.11, 118.93, 115.98, 113.77, 40.88, 38.97, 38.84, 32.54. Anal. Calcd. For C20H17BrO2: C 65.05, H 4.64. Found: C 65.04, H 4.60.
(3-Methyl-3-phenylcyclobutyl)(naphtho[2,1-b]furan-2-yl)methanone (2). Yield: 1.18 g (74%); m.p.: 148 °C (from ethanol); IR (KBr): 1674 (C=O), 1255 (C-O, on furan ring) cm-1; 1H-NMR (200 MHz; CDCl3) δ: 1.69 (s, 3H, CH3), 2.53 (m, 2H, CH2), 2.88 (m, 2H, CH2), 4.17 (q, 1H, >CH-), 7.17-8.18 (m, 12H, aromatics); 13C-NMR (50.34 MHz; CDCl3) δ: 193.36 (C=O), 155.97, 153.75, 153.17, 132.53, 131.72, 131.08, 130.32, 130.21, 129.38, 127.62, 127.51, 126.69, 125.37, 124.95, 114.78, 113.73, 40.91, 39.13, 38.66, 32.54. Anal. Calcd. For C20H17BrO2: C 84.68, H 5.92. Found: C 84.57, H 5.61.
General procedure for the synthesis of ketoximes 3 and 4.
Compound 1 (3.689 g, 10 mmol), hydroxylamine hydrochloride (0.781 g, 12 mmol) and pyridine (5 mL) were dissolved in ethyl alcohol (50 mL). The mixture was refluxed to complete the reaction while monitoring its course by IR. After cooling to room temperature, the mixture was poured into water. The solid 3 thus separated was filtered off, washed with water several times and crystallized from ethanol.
(Z)-(5-Bromo-1-benzofuran-2-yl)(3-methyl-3-phenylcyclobutyl)methanone oxime (
3). Yield: 3.57 g (93%); m.p.: 193 °C (from ethanol); IR (KBr): 3201 (N-OH), 1602 (C=N), 1251 (C-O, on furan ring), 990 (N‑O, oxime) [
8,
9], 544 (C-Br) cm
-1.
1H-NMR (200 MHz; DMSO-d
6) δ: 1.49 and 1.54 (two s, 3H, CH
3), 2.42-2.63 (m, 4H, CH
2), 3.92 (q, 1H, >CH-), 7.10-7.96 (m, 9H, aromatics), 11.75 and 12.03 (two s, 1H, N-OH);
13C-NMR (50.34 MHz; DMSO-d
6) δ: 155.17, 154.58, 153.74, 153.23, 149.62, 148.65, 132.08, 131.82, 130.37, 129.94, 127.01, 126.40, 126.24, 126.15, 125.52, 117.27, 117.18, 115.19, 113.70, 106.62, 42.17, 41.60, 40.50, 40.37, 40.29, 40.23, 40.08, 32.10, 31.91. Anal. Calcd. For C
20H
18BrNO
2: C 62.51, H 4.72, N 3.65. Found: C 62.35, H 4.81, N 3.67.
(Z)-(3-Methyl-3-phenylcyclobutyl)(naphtho[2,1-b]furan-2-yl)methanone oxime (4). Yield: 1.81 g (94%); m.p.: 205-208 °C (from ethanol); IR (KBr): 1674 (C=O), 1255 (C-O, on furan ring) cm-1, 1H-NMR (200 MHz; CDCl3) δ: 1.49 and 1.69 (s, 3H, CH3), 2.59-2.81(m, 4H, CH2), 4.06 (q, 1H, >CH-), 7.14-7.99 (m, 12H, aromatics), 11.64 and 11.88 (two s, 1H, N-OH; 13C-NMR (50.34 MHz; CDCl3) δ: 154.09, 153.42, 152.05, 147.86, 132.44, 130.79, 130.27, 130.18, 129.39, 128.72, 127.29, 126.88, 126.73, 126.62, 125.84, 125.47, 124.87, 115.14, 114.24, 41.22, 40.73, 33.30, 32.23; Anal. Calcd. For C24H21NO2: C 81.10, H 5.96, N 3.94. Found: C 81.00, H 5.96, N 3.77.
General procedure for the synthesis of oxyranyl compounds 5 and 6.
In a two necked 250 mL reaction flask equipped with a reflux condenser and a dropping funnel, 3 (3.83 g, 10 mmol) was dissolved in dry acetone (50 mL). To this solution powdered KOH (1.68 g, 30 mmol) was added portions over a 1 h period. Subsequently, epichlorohydrin (2 mL, 30 mmol) was added in 15 minutes and the solution was refluxed for 8 h. The solvent was removed under reduced pressure and the residue was treated with diethyl ether, the etheral phase dried over anhydrous MgSO4, filtered and the ether was removed under reduced pressure. Thus obtained solid substance 5 was crystallized from ethanol.
(Z)-(5-Bromo-1-benzofuran-2-yl)(3-methyl-3-phenylcyclobutyl)methanone O-(oxiran-2-yl-methyl)oxi-me (
5). Yield: 3.04 g (69%); m.p.: 139-142 °C (from ethanol); IR (KBr): 1594 (C=N), 1253 (C-O, on furan ring), 1053 and 1031 (C-O, epoxide), 979 (N-O, oxime) [
8,
9], 541 (C-Br) cm
-1.
1H-NMR (200 MHz; CDCl
3) δ: 1.57 and 1.62 (two s, 3H, CH
3), 2.47-2.74 (m, 4H, CH
2), 2.87 (t,
J=6.9 Hz, 2H, -CH
2-O), 3.33 (m, 1H, >CH-O), 3.90 (q, 1H, >CH-), 4.12-4.50 (four d, 2H, N-O-CH
2-), 7.13-7.77 (m, 9H, aromatics);
13C-NMR (50.34 MHz; CDCl
3) δ: 154.21, 154.12, 151.00, 149.54, 132.27, 130.99, 130.16, 127.28, 126.71, 126.14, 118.15, 114.97, 114.86, 77.73, 51.98, 46.79, 41.14, 40.58, 33.26, 32.33. Anal. Calcd. For C
23H
22BrNO
2: C 65.10, H 5.23, N 3.30. Found: C 65.20, H 4.97, N 3.21.
(Z)-(3-Methyl-3-phenylcyclobutyl)(naphtho[2,1-b]furan-2-yl)methanone O-(oxiran-2-yl-methyl)oxime (6). Yield: 0.28 g (67%); m.p.: 205-207 °C (from ethanol); IR (KBr): 1674 (C=O), 1255 (C-O, on furan ring) cm-1; 1H-NMR (200 MHz; CDCl3) δ: 1.66 (s, 3H, CH3), 2.53-2.75 (m, 4H, CH2), 2.86 (t, -CH2-O), 3.28 (m, 1H, >CH-O), 4.00 (q, 1H, >CH-), 4.21-4.53 (four d, 2H, N-O-CH2-), 7.17-8.23 (m, 12H, aromatics); 13C-NMR (50.34 MHz; CDCl3) δ: 154.26, 153.41, 151.29, 148.07, 132.43, 130.76, 130.15, 129.33, 128.69, 127.24, 126.85, 126.77, 125.80, 125.53, 114.82, 114.64, 114.21, 77.56, 52.14, 46.92, 41.18, 40.72, 33.34, 32.35.Anal. Calcd. For C27H25NO2: C 82.00 5, H 6.37, N 3.54. Found: C 82.21, H 6.44, N 3.29.
General procedure for the synthesis of thiosemicarbazones 7 and 8.
A mixture of 1 (0.369 g, 1.0 mmol), thiosemicarbazide (0.091 g, 1.0 mmol) and p-toluene sulphonic acid (0.010 g, as catalyst) in absolute ethanol (50 mL) was refluxed. The course of the reaction was monitored by TLC. After the solvent was removed under reduced pressure, the residue was treated with water and solid product 7 thus obtained was recrystallized from methanol.
(Z)-(5-Bromo-1-benzofuran-2-yl)(3-methyl-3-phenylcyclobutyl)methanone thiosemicarbazone (7). Yield: 0.42 g (94%); m.p.: 219 °C (from ethanol); IR (KBr,): 3432-3155 (four sharp peaks, thiosemicarbazide), 1598 (C=N), 1244 (C-O, on furan ring), 1045 (C=S), 547 (C-Br) cm-1. 1H-NMR (200 MHz; CDCl3) δ: 1.61 (s, 3H, CH3), 2.47-2.72 (m, 4H, -CH2-), 3.70 (q, 1H, >CH-), 6.32 (broad s, 2H, -NH2), 7.02-7.80 (m, 9H, aromatics), 10.82 (s, 1H, -NH-); 13C-NMR (50.34 MHz; CDCl3) δ: 181.48 (C=S), 155.30, 153.37, 151.87, 141.16, 132.15, 130.39, 130.33, 127.66, 126.71, 126.56, 119.41, 115.52, 111.74, 41.00, 40.82, 34.96, 32.55. Anal. Calcd. For C21H20BrN3OS: C 57.02, H 4.56, N 9.50, S 7.25. Found: C 57.35, H 4.67, N 9.80, S 7.44.
(Z)-(3-Methyl-3-phenylcyclobutyl)(naphtho[2,1-b]furan-2-yl)methanone thiosemicarbazone (8). Yield: 0.38 g (91%); m.p.: 217 °C (from ethanol); IR (KBr): 1674 (C=O), 1255 (C-O, on furan ring) cm-1; 1H-NMR (200 MHz; CDCl3) δ: 1.65 (s, 3H, CH3), 2.53-2.77 (m, 4H, CH2), 3.84 (q, 1H, >CH-), 6.31 (broad s, 2H, -NH2), 7.15-8.21 (m, 12H, aromatics), 10.99 (s, 1H, -NH-); 13C-NMR (50.34 MHz; CDCl3) δ: 181.38 (C=S), 154.84, 153.52, 150.36, 141.69, 132.71, 131.12, 130.70, 130.39, 129.48, 129.26, 127.62, 126.61, 126.53, 125.33, 124.22, 114.32, 111.67, 41.13, 40.86, 34.98, 32.63. Anal. Calcd. For C25H23N3OS: C 72.61, H 5.61, N 10.16, S 7.75. Found: C 72.98, H 5.68, N 10.23, S 7.82.
General procedure for the synthesis of thiazolyl hydrazones 9 and 10.
A mixture of of 7 (0.442, 1.0 mmol) and dichloroacetone (0.127 g, 1.0 mmol) in dry acetone (40 mL) was stirred at room temperature. The course of the reaction was monitored by TLC. After completing the reaction, the solution was then made alkaline with an aqueous solution of NH3 (5%). The precipitate was filtered off, washed with same NH3 solution several times, dried in air and crystallized from EtOH and dried and stored in a desiccator over CaCl2.
(Z)-(5-Bromo-1-benzofuran-2-yl)(3-methyl-3-phenylcyclobutyl)methanone[4-(chloromethyl)-1,3-thiazol-2-yl]hydrazone (9). Yield: 0.29 g (57%); m.p.: 199 °C (from acetone); IR (KBr): 3351 (-NH-), 1598 (C=N), 1258 (C-O, on furan ring), 762 (C-Cl), 542 (C-Br) cm-1; 1H-NMR (200 MHz; CDCl3) δ: 1.63 (s, 3H, CH3), 2.46-2.79 (m, 4H, CH2), 3.68 (q, 1H, >CH-), 4.54 (s, 2H, -CH2-Cl), 6.67 (s, 1H, aromatic in thiazole), 6.94-7.77 (m, 9H, aromatics), 10.99 (broad s, 1H, -NH-); 13C-NMR (50.34 MHz; CDCl3) δ: 171.66, 153.51, 151.78, 141.26, 132.39, 130.88, 130.64, 130.14, 128.28, 127.12, 126.17, 126.04, 125.90, 124.78, 114.27, 111.75, 43.41, 41.23, 40.49, 34.88, 32.38; Anal. Calcd. For C24H21ClBrN3OS: C 55.99, H 4.11, N 8.16, S 6.23. Found: C 56.02, H 4.21, N 8.04, S 6.33.
(Z)-(3-Methyl-3-phenylcyclobutyl)(naphtho[2,1-b]furan-2-yl)methanone[4-(chloromethyl)-1,3-thiazol- 2-yl]hydrazone (10). Yield: 0.30 g (61%); m.p.: 207 °C (from acetone); IR (KBr): 1674 (C=O), 1255 (C-O, on furan ring) cm-1; 1H-NMR (200 MHz; CDCl3) δ: 1.68 (s, 3H, CH3), 2.52-2.87 (m, 4H, -CH2-), 3.84 (s, 1H, >CH-), 4.56 (s, 2H, -CH2-Cl), 6.67 (s, 1H, aromatic in thiazole), 7.14-8.21 (m, 12H, aromatics), 10.85 (broad s, -NH-). 13C-NMR (50.34 MHz; CDCl3) δ: 172.24, 153.72, 151.09, 140.26, 132.66, 131.03, 130.63, 130.26, 129.93, 129.53, 129.05, 127.42, 127.33, 126.80, 126.41, 125.37, 124.51, 114.27, 110.30, 109.90, 43.61, 41.10, 40.59, 34.91, 32.34. Anal. Calcd. For C28H24ClN3OS: C 69.19, H 4.98, N 8.65, S 6.60. Found: C 69.09, H 4.98, N 8.82, S 6.63.
Preparation of microbial cultures
Microorganisms were provided from the culture collection of the Microbiology Laboratory of the Biological Sciences of Faculty of Science and Arts, Firat University, Turkey. In this work,
Bacillus subtilis LM622 (B.s.),
Enterobacter aeruginosa CCM 2531 (E.a.),
Eschericia coli ATCC 25922 (E.c.),
Listeria monocytogenes SCOTTA (L.m.),
Pseudomonas aeruginosa DSM 50071 (P.a.),
Staphilococcus aureus COWAN I (S.a.)
Candida albicans FMC 17 (C.a.), and
Saccharomyces cerevisiae FMC 16 (S.c.) were used to investigate the bacteriological and antifungal activities of selected substances. The bacteria and yeast strains were nourished in nutrient broth (Difco) and in malt extract broth (Difco), and incubated for 24 and 48 h, respectively. In the Disc Diffusion method, sterile Mueller Hinton Agar (Oxoid) for bacteria and Saburoud Dextrose Agar for yeast were separately inoculated with the test microorganisms. The compounds, dissolved in DMSO as 500 µg/disc solutions, were placed in wells (6 mm diameter) in the agar media and the plates were incubated at 32 °C for bacteria (18-24 h) and at 25° C for yeast (72 h) [
12]. The resulting inhibition zones on the plates were measured in mm after 48 h (
Table 1). The control samples only contained DMSO. The data reported in
Table 1 represent the average of three experiments.

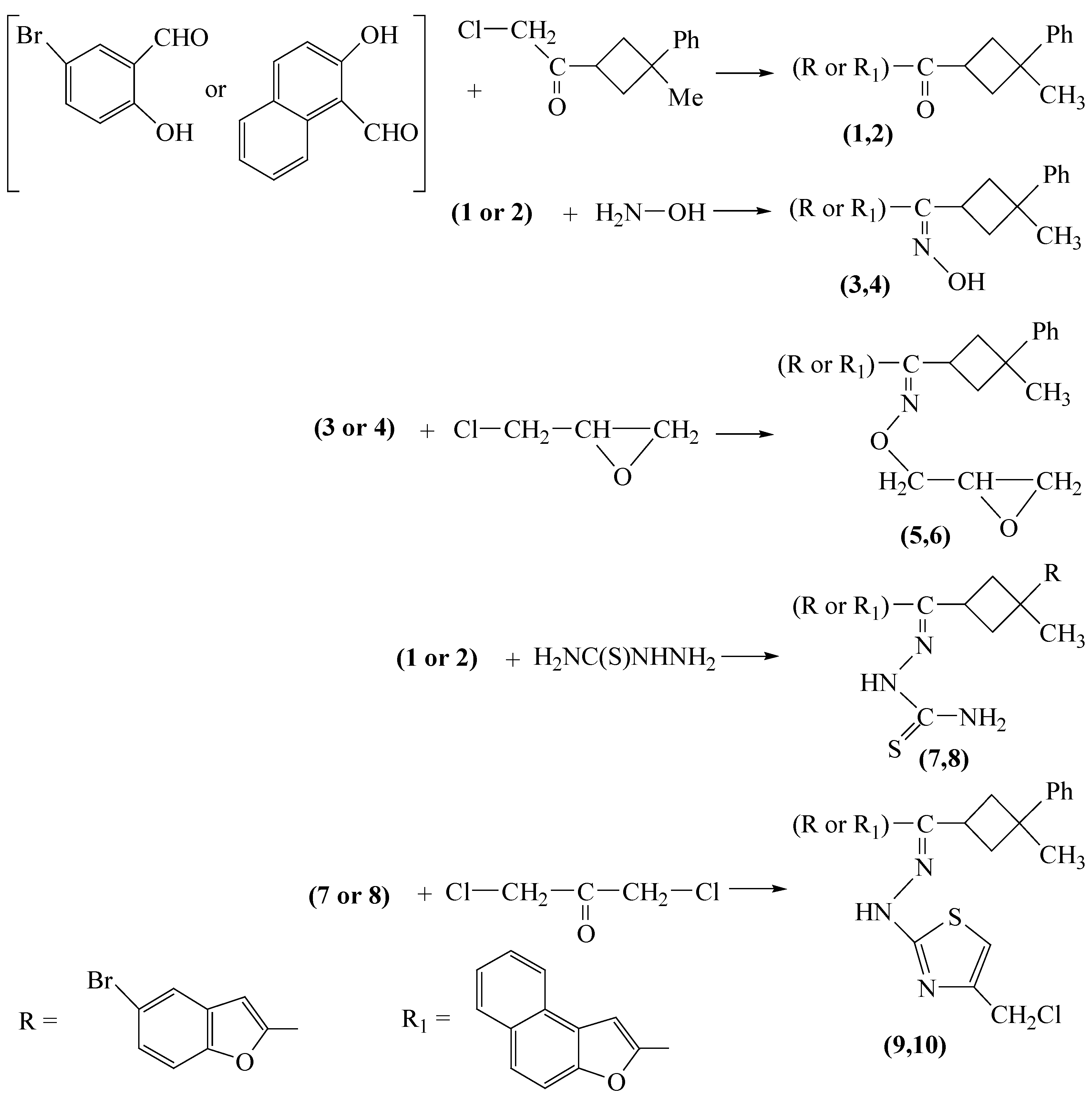

{kind=link}