Simple and Efficient Synthesis of 2,7-Difunctionalized-1,8-Naphthyridines

Department of Chemistry, Bengal Engineering and Science University, Shibpur, India
*
Author to whom correspondence should be addressed.
Molecules 2005, 10(8), 929-936; https://doi.org/10.3390/10080929
Submission received: 25 August 2004
/
Revised: 23 November 2004
/
Accepted: 6 January 2005
/
Published: 31 August 2005
{kind=link}
{kind=link}
{kind=link}
Abstract
:The syntheses in good yields of some new difunctionalized 1,8-naphthyridines 4, 6, 8 and 9 and a novel triethylene glycol ether-linked dinaphthyridine, 10a, along with the mononaphthyridine-linked ether alcohol 10b are described. An improved and milder method for the synthesis of 2,7-diamino-1,8-naphthyridine (14) is also reported.
Introduction
Naphthyridine or naphthyridone systems are of great importance due to their broad spectrum of biological activities. Substituted 1,8-naphthyridine compounds are used as antihypertensives, antiarrhythmics, herbicide safeners and also as immunostimulants [1,2,3]. We are interested in 2,7-difunctionalized-1,8-naphthyridines because of their aforesaid potential medicinal activity as well as for their use as important binding units in the molecular design of synthetic receptors [4]. This communication describes the first synthesis of four 2,7-difunctionalized-1,8-naphthyridines; viz. 2-amino-7-hydroxymethyl-1,8-naphthyridine (5), 2-amino-1,8-naphthyridine-7-carboxaldehyde (6), 2,7-dimethyl-4-methoxy-1,8-naphthyridine (8) and 4-methoxy-1,8-naphthyridine-2,7-dicarboxaldehyde (9) and a novel triethylene glycol ether-linked dinaphthyridine compound, 1,2-bis-[2-(2,7-dimethyl-1,8-naphthyridin-4-yloxy)ethoxy]ethane (10a) along with the mononaphthyridine-linked ether alcohol (10b). A new synthesis of 2,7-diamino-1,8-naphthyridine 14 by a more efficient reaction under milder conditions is also reported here.
Results and Discussion
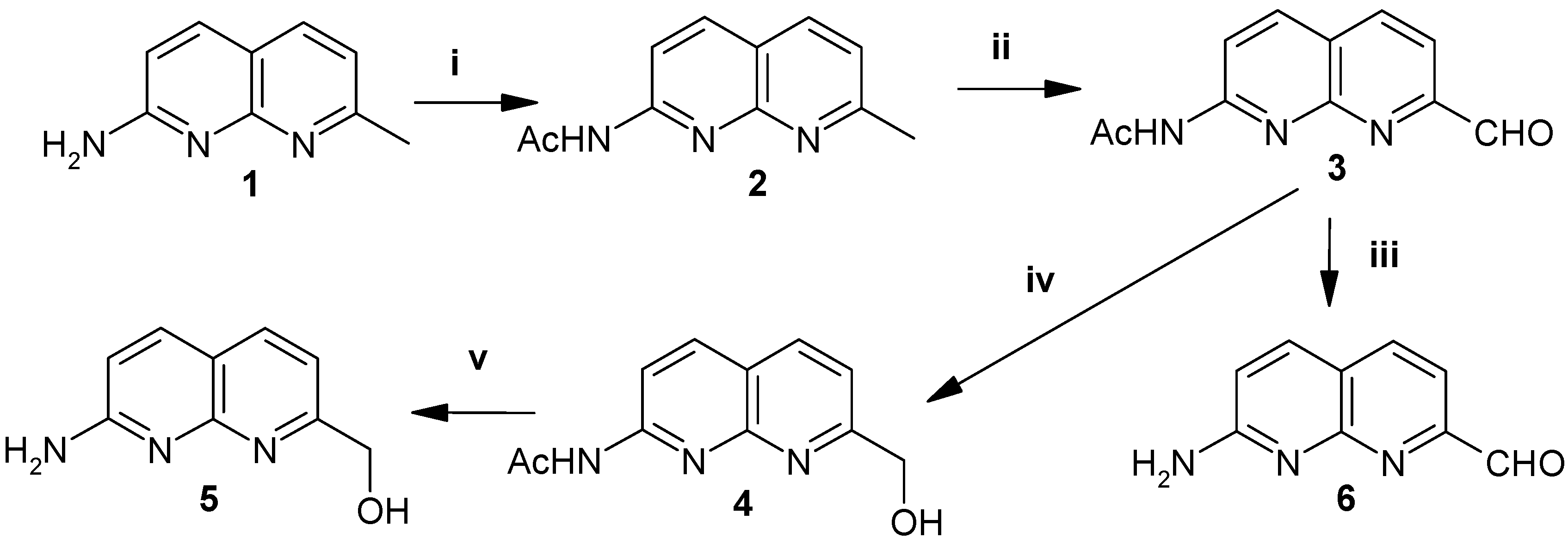
Naphthyridines 2-6 were synthesized starting from 2-amino-7-methylnaphthyridine (1), which is obtained by the condensation of commercially available 2,6-diaminopyridine and 3-oxo-butyraldehyde dimethyl acetal following a reported procedure [5]. The 7-methyl group of 2-acetylamino-7-methyl-1,8-naphthyridine (2) is oxidized with selenium dioxide in dioxane to the corresponding aldehyde 3, followed by deprotection of the N-acetyl group by hydrolysis with 1N hydrochloric acid, which afforded the desired 2-amino-1,8-naphthyridine-7-carboxaldehyde (6) in 85% yield (Scheme 1).
Scheme 1.

Reagents, conditions and yields: (i) acetic anhydride, 80°C, 12h., 87%. (ii) SeO2, dioxane, 50-55°C, 4h., 75%. (iii) 1N HCl, reflux, 0.5h., 85%. (iv) NaBH4, dry THF, 3-4h, 85%. (v) 1N NaOH, r.t.,12h., 80%.
The conversion of aldehyde 3 into 2-amino-7-hydroxymethyl-1,8-naphthyridine (5) was achieved by sodium borohydride reduction of the former to give 2-acetylamino-7-hydroxymethyl-1,8-naphthyridine (4), followed by mild alkaline hydrolysis (1N NaOH).
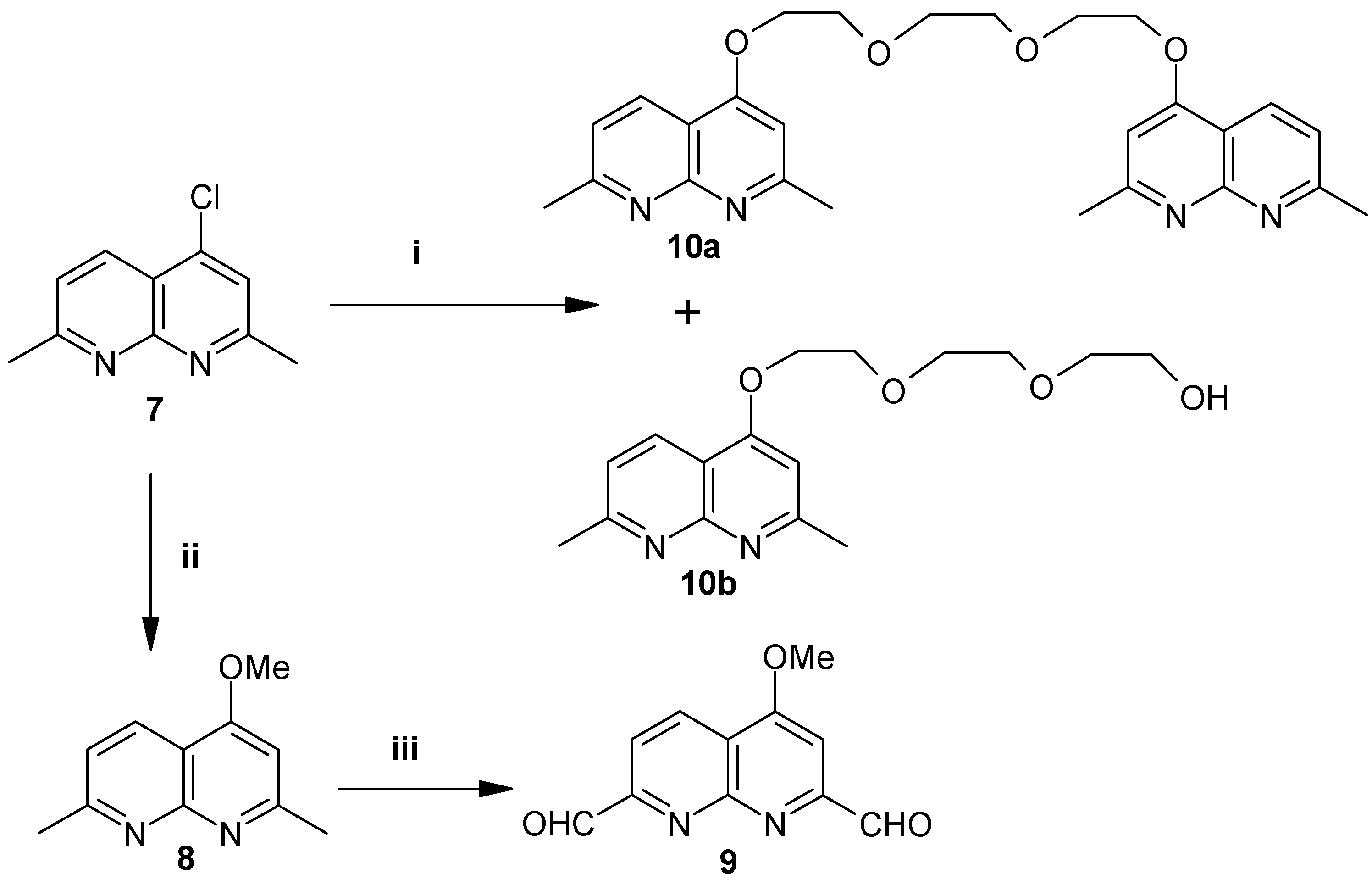
Compound 8 is made in excellent yield (95%) starting from 4-chloro-2,7-dimethyl-1,8-naphthyridine (7) [6] by stirring at room temperature with methanolic potassium hydroxide. Compound 8 is then oxidized with SeO2 in dioxane at room temperature to produce 4-methoxy-1,8-naphthyridine-2,7-dicarboxaldehyde (9) in 90% yield. The synthesis of the novel di- and mono-naphthyridines 10a-b with triglycol spacers is achieved by substitution of the chlorine atom by both two as well as only one of the hydroxyl functions of triethylene glycol, respectively (Scheme 2).
Scheme 2.

Reagents, conditions and yields: (i) Triethylene glycol, KOH, THF, 60°C, 80%. (ii) MeOH, KOH, 4h., 90%. (iii) SeO2, dioxane, r.t., 90%.
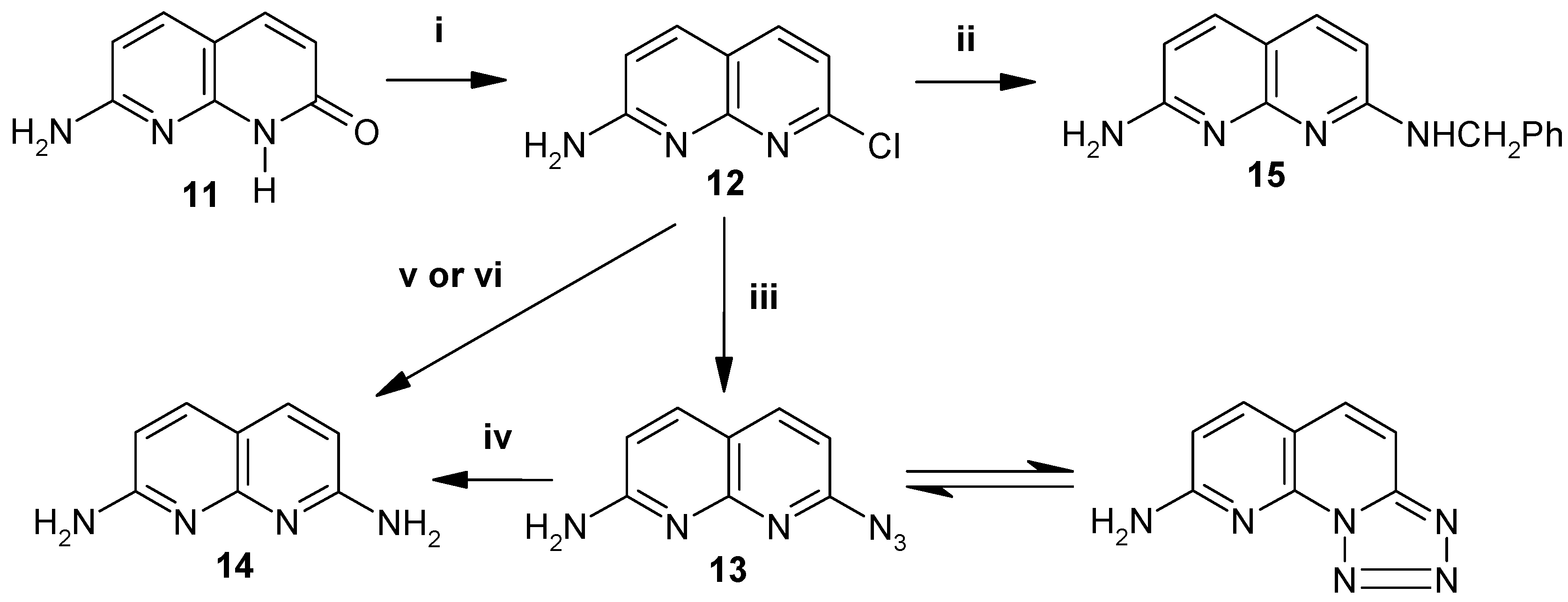
The synthesis of 2,7-diamino-1,8-naphthyridine 14 is described in Scheme 3. The advanced starting compound 11 was prepared according to the literature method [7, 8]. Compound 11 was then converted into the chloroamino intermediate 12 by direct reaction with POCl3 without conventional protection of the amino group. The substitution of chloro by azide was smoothly achieved by the treatment of 12 with sodium azide in DMF at 60 °C to produce 13 in 90% yield, while the reaction of 12 with benzylamine lead to 15 in 50% yield. The target compound 14 was then synthesized, albeit in poor yield (25%), by refluxing compound 12 with NaNH2 in xylene. However, excellent yields (98%) of 14 can be achieved via the reduction of the azide derivative 13 with Zn/AcOH.
Scheme 3.

Reagents, conditions and yields. (i) POCl3, reflux, 4h., 60%. (ii) PhCH2NH2, 120°C, 6h., 50%. (iii) NaN3, DMF, 60°C, 4h., 90%. (iv) Zn, AcOH, reflux, 3h., 98%. (v) NaNH2, xylene, reflux, 25%. (vi) ammonolysis.
Compound 14 is also obtained by direct ammonolysis of compound 12 at high temperature in a sealed tube. But care must be taken during this procedure as it was found to be dangerous and the yield is also poor compared to that of azide reduction with Zn/AcOH .
Conclusions
We have synthesized a series of functionalized naphthyridines and novel triethylene glycol-linked mono- and di-naphthyridines in good yields by simple and efficient procedures. All the naphthyridines and the intermediates were well characterized by spectroscopic means. A new, efficient and practical method under mild conditions for the synthesis of 2,7-diamino-1,8-naphthyridine (14) has also been developed, which also improves the yield for the synthesis of this compound.
Acknowledgements
Financial assistance from the Department of Science and Technology (DST), and the Council of Scientific and Industrial Research (CSIR), Govt. of India is gratefully acknowledged. S.J., and A.K.A. thank CSIR and A.C.M. thanks UGC for research fellowships.
Experimental
General
Melting points (m.p) were recorded on a Toshniwal hot-coil stage melting point apparatus and are uncorrected. NMR spectra were recorded in CDCl3 (unless otherwise mentioned) with TMS as the internal standard on Bruker AM 200 MHz and 300 MHz NMR instruments. Chemical shifts are given in δ (ppm) scale and J values in Hz respectively. IR spectra were measured in KBr disks with a Perkin Elmer (Model 883) spectrometer. Mass spectra (JEOL JMS600), and elemental analyses were obtained from the IICB, and the IACS (Kolkata) respectively. All solvents were dried prior to use by common methods. Silica gel (60-120 mesh) has been used for all chromatographic purifications. Starting materials were either commercially available (purchased from Fluka and Aldrich) or synthesized according to the cited literature procedures.
2-Acetylamino-7-methyl-1,8-naphthyridine (2): Distilled acetic anhydride (0.5 mL) was added to 2-methyl-7-amino-1,8-naphthyridine (1, 0.158 g, 1.0 mmol) and the reaction mixture was stirred overnight at 80 °C. The excess acetic anhydride was removed under vacuum. To this, diethyl ether was added, and the solid separated was collected by filtration. The solid was washed thoroughly with NaHCO3 solution, dried and crystallized from chloroform-methanol to afford the title compound 2 (0.174g, 87%) as a yellow solid; m.p. 280 °C [lit. [5] 279-281 °C]; 1H-NMR (200 MHz) δ: 8.70 (bs, 1H), 8.44 (d, 1H, J = 8.8 Hz), 8.12 (d, 1H, J = 8.8 Hz), 8.00 (d, 1H, J = 8.2 Hz), 7.30 (d, 1H, J = 7.5 Hz), 2.74 (s, 3H), 2.27 (s, 3H); IR (cm-1): 1503, 1606, 1697, 3005, 3176; Anal. Calcd for C11H11N3O: required C, 65.65; H, 5.50; N, 20.88. Found: C, 65.58; H, 5.42; N, 20.95.
2-Acetylamino-1,8-naphthyridine-7-carboxaldehyde (3): To a stirred solution of selenium dioxide (0.11 g, 1.0 mmol) in dioxane (10 mL) containing 2-3 drops of water, compound 2 (0.201 g, 1.0 mmol) was added and heated for 4 h at 50-55 °C. The hot solution was filtered through a pad (2-3 cm) of Celite and the solvent was removed through the short path distillation. The residue was extracted with chloroform and washed well with water. The organic layer after evaporation and column chromatography purification gave the desired compound 3 (0.161g, 75%); m.p 215 °C; 1H-NMR (200 MHz) δ: 10.23 (s, 1H), 8.70 (d, 1H, J = 8.9 Hz), 8.52 (bs, 1H), 8.37 (d, 1H, J = 3.1 Hz), 8.34 (d, 1H, J = 4.0 Hz), 8.13 (d, 1H, J = 8.1 Hz), 2.32 (s, 3H); 13C-NMR (50 MHz) δ: 193.41, 169.59, 154.51, 139.36, 138.22, 123.11, 117.52, 117.07, 25.00; IR (cm-1): 1607, 1703, 3060, 3186; Anal. Calcd for C11H9N3O2: required C, 61.39; H, 4.21; N, 19.52. Found: C, 61.31; H, 4.25; N, 19.45.
2-Acetylamino-7-hydroxymethyl-1,8-naphthyridine (4): To a solution of 3 (0.215 g, 1.0 mmol) in dry THF (10 mL), sodium borohydride (0.016 g, 0.433 mmol) was added and stirred under nitrogen for half an hour. THF was then removed and the excess borohydride was decomposed by dropwise addition of 2.5 N HCl. The mixture was extracted with choloroform. The organic layer was washed with water, dried (Na2SO4) and evaporated under reduced pressure to give the title compound 4 (0.184 g, 85%) which was sufficiently pure for the next step. M.p. 240 °C (decomp.); 1H-NMR (200 MHz) δ: 8.69 (bs, 1H), 8.52 (d, 1H, J = 8.8 Hz), 8.19 (d, 1H, J = 8.8 Hz), 8.11 (d, 1H, J = 8.2 Hz), 7.30 (d, 1H, J = 8.2 Hz), 4.96 (s, 2H), 3.41 (bs, 1H, OH), 2.46 (s, 3H); IR (cm-1): 1296, 1389, 1696, 3203, 3391, 3733; Anal. Calcd for C11H11N3O2: required C, 60.82; H, 5.10; N, 19.34. Found: C, 60.72; H, 5.14; N, 19.40.
2-Amino-7-hydroxymethyl-1,8-naphthyridine (5): Compound 4 (0.217 g, 1.0 mmol) was taken up in 1N NaOH (3 mL) and stirred overnight. The reaction mixture was then neutralized with acetic acid, and extracted with 5% methanol in chloroform solution. The organic layer was dried (Na2SO4) and then evaporated under reduced pressure to give the desired product 5 (0.140g, 80%); m.p. 240 °C; 1H-NMR (10% DMSO-d6 in CDCl3, 200 MHz) δ: 7.73 (d, 1H, J = 8.0 Hz), 7.63 (d, 1H, J = 8.0 Hz), 6.99 (d, 1H, J = 8.0 Hz), 6.64 (d, 1H, J = 8.0 Hz), 5.80 (bs, 2H), 4.63 (s, 2H), 2.67 (bs, 1H, OH); MS (FAB): m/z (%): 176 (MH+, 100), 159 (20), 146 (40), 75 (95), 57 (75); Anal. Calcd for C9H9N3O: required C, 61.70; H, 5.18; N, 23.99. Found: C, 61.71; H, 5.20; N, 24.02.
2-Amino-1,8-naphthyridine-7-carboxaldehyde (6): A mixture of compound 3 (0.215 g, 1.0 mmol) and 1N HCl (1.0 mL) in dioxane (10 mL) was refluxed for 30 min. Then the solution was cooled to room temperature, and neutralized carefully with NaOH solution. The precipitate formed was filtered, which afforded the desired compound 6 (0.147 g, 85%); 1H-NMR (200 MHz) δ: 10.19 (s, 1H), 8.1 (d, 1H, J = 8.0 Hz), 7.94 (d, 1H, J = 8.0 Hz), 7.84 (d, 1H, J = 8.0 Hz), 6.88 (d, 1H, J = 8.0 Hz), 5.18 (bs, 2H); IR (cm-1): 1305, 1387, 1701, 3332; MS (FAB): m/z (%): 173 (M+, 65), 159 (30), 145 (45), 117 (25), 97 (35), 81 (45), 69 (95), 57 (100); Anal. Calcd for C9H7N3O: required C, 62.42; H, 4.07; N, 24.26. Found: C, 62.40; H, 4.10; N, 24.28.
2,7-Dimethyl-4-methoxy-1,8-naphthyridine (8): To a methanolic solution of 2,7-dimethyl-4-chloro-1,8- naphthyridine (7, 0.193 g, 1.0 mmol), solid KOH (0.056 g, 1.0 mmol) was added and stirred for 4 h at 80 °C. The solvent was stripped off. The residue was added to water and extracted with dichloromethane. The solvent was evaporated to dryness to get the desired compound 8 (0.169 g, 90%); mp. 130 °C; 1H-NMR (200 MHz) δ: 8.31 (d, 1H, J = 8.3 Hz), 7.21 (d, 1H, J = 8.3 Hz), 6.60 (s, 1H), 4.01 (s, 3H), 2.73 (s, 3H), 2.70 (s, 3H); IR (cm-1): 1092, 1133, 1181, 1606; Anal. Calcd for C11H12N2O3: required C, 70.19; H, 6.43; N, 12.96. Found: C, 70.21; H, 6.45; N, 12.99.
4-Methoxy-1,8-naphthrydine-2,7-dicarboxaldehyde (9): To a stirred solution of 8 (0.188 g, 1.0 mmol) in dioxane (10 mL), SeO2 (0.33 g, 3.0 mmol) was added and stirring was continued for 12 h. The reaction mixture was filtered through a 4-5 cm pad of Celite-silica gel. Water (5 mL) was added to the dioxane solution, followed by extraction with dichloromethane. The organic solvent was evaporated under reduced pressure to give the desired compound 9 (0.194 g, 90%); m.p. 210 °C; 1H-NMR (200 MHz) δ: 10.32 (s, 1H), 10.26 (s, 1H), 8.81 (d, 1H, J = 8.2 Hz), 8.18 (d, 1H, J = 8.3 Hz), 7.54 (s, 1H), 4.20 (s, 3H); IR (cm-1): 1091, 1170, 1606, 1705; MS (EI): m/z (%): 216 (M+, 100), 188 (95), 173 (20), 158 (40), 129 (25), 90 (95), 71 (50), 57 (75); Anal. Calcd. for C11H8N2O3: required C, 61.11; H, 3.73; N, 12.96. Found: C, 61.10; H, 3.75; N, 13.00.
1,2-bis-[2-(2,7-dimethyl-1,8-naphthyridin-4-yloxy)ethoxy]ethane (10a) and 1,2-[2-(2,7-dimethyl-1,8- naphthyridin-4-yloxy)ethoxy]ethane (10b): A solution of triethylene glycol (0.150 g, 1.0 mmol), and KOH (0.112 g, 2.0 mmol) in dry THF (10 mL) was added the solution of compound 7 (0.386 g, 2.0 mmol) and the mixture was heated at 60 °C for 24 h. Then the solvent was removed to dryness and dichloromethane was added to the residue. The organic layer was washed with water, dried (Na2SO4) and solvent was removed under reduced pressure to give a brown gum. Column chromatography of the crude product eluting with 1% methanol in chloroform afforded first the di-naphthyridine polyether 10a (0.369 g, 80%) as an off-white solid. 1H-NMR (200 MHz) δ: 8.36 (d, 2H, J = 8.3 Hz), 7.22 (d, 2H, J = 8.3 Hz), 6.61 (s, 2H), 4.32 (t, 4H, J = 4.6 Hz), 3.96 (t, 4H, J = 4.6 Hz), 3.58 (t, 4H, J = 4.3 Hz), 2.73 (s, 6H), 2.70 (s, 6H); MS (FAB): m/z (%): 462 (M+, 5), 427 (10), 405 (10), 325 (5), 225 (10), 203 (100), 187 (20), 175 (40), 159 (10); Anal. Calcd. for C26H30N4O4: required C, 67.48; H, 6.53; N, 12.01. Found: C, 67.62; H, 6.45; N, 11.95. Further elution gave the mono-naphthyridine 10b (0.08 g, 15- 18%) as a light yellow semi-solid. 1H-NMR (300 MHz) δ: 8.41 (d, 1H, J = 8.4 Hz), 7.21 (d, 1H, J = 8.3 Hz), 6.69 (s, 1H), 4.38 (t, 2H, J = 4.4 Hz), 4.00 (t, 2H, J = 4.2 Hz), 3.80-3.76 (m, 4H), 3.74-3.70 (m, 2H), 3.62 (t, 2H, J = 4.4 Hz), 2.76 (s, 3H), 2.75 (s, 3H), 2.36 (s, 1H); MS (EI): m/z (%): 306 (M+, 5), 289 (10), 276 (10), 262 (10), 232 (20), 188 (25), 174 (100), 157 (20), 145 (65), 116 (25), 93 (20), 74 915); Anal. Calcd for C16H22N2O4: required C, 62.73; H, 7.23; N, 9.14. Found: C, 62.70; H, 7.26; N, 9.20.
2-Amino-7-chloro-1,8-naphthyridine (12): A mixture of the compound 11 (4.0 g, 0.024 mol) in freshly distilled POCl3 (25 mL) was refluxed for 4h. Excess POCl3 was distilled off and the reaction mixture was poured onto ice-cold water. After neutralization with Na2CO3, a yellow solid appeared, which was collected by filtration and recrystallized from methanol-ether to give 12 (2.67 g, 60%). M.p. 170 °C; 1H-NMR (200 MHz) δ: 7.86 (d, 1H, J = 4.0 Hz), 7.82 (d, 1H, J = 4.0 Hz), 7.17 (d, 1H, J = 8.0 Hz), 6.75 (d, 1H, J = 8.0 Hz), 5.33 (bs, 2H); IR (cm-1): 1437, 1489, 1607, 1695, 3311; MS (FAB) (m/z): 179.5 (M+, 100%); Anal. Calcd. for C8H6N3Cl: required C, 53.49; H, 3.36; N, 23.39. Found: C, 53.41; H, 3.40; N, 23.50.
2-Amino-7-azido-1,8-naphthyridine (13): To a stirred solution of 12 (0.4 g, 2.22 mmol) in dry DMF (5 mL), sodium azide (0.289 g, 4.44 mmol) was added and the mixture stirred for 4h at 60 °C. The precipitated NaCl was filtered off. The filtrate was distilled under reduced pressure to remove DMF. The solid was washed well with water and air-dried to give the title compound 13 (0.248 g, 95%); mp. 275 °C; 1H-NMR (200 MHz) δ: 8.1 (d, 1H, J = 8.0 Hz), 7.76 (d, 1H, J = 8.0 Hz), 7.69 (d, 1H, J = 8.0 Hz), 6.9 (d, 1H, J = 8.0 Hz), 5.5 (bs, 2H); IR (cm-1): 1651, 3168, 3321.
2,7-Diamino-1,8-naphthyridine (14) from 13: To a stirred solution of the azo compound 13 (0.8 g, 4.3 mmol) in glacial acetic acid (12.72 mL), zinc dust (2.12 g) was added and refluxed for 3 h. A precipitate was obtained which was filtered. Acetic acid was removed under reduced pressure to give the desired pure product as a yellowish brown solid 14 (0.674 g, 98%); m.p. 222-223 °C [lit.[6] 223 °C]; 1H-NMR (DMSO-d6, 200 MHz) δ: 7.67 (d, 2H, J = 10.0 Hz), 6.53 (d, 2H, J = 8.0 Hz), 7.25 (bs, 4H); IR (cm-1): 1267, 1385, 1641, 2925, 3427; MS (FAB) (m/z): 160 (M+, 52%); Anal. Calcd for C8H8N4: required C, 59.98; H, 5.03; N, 34.97. Found: C, 59.95; H, 5.10; N, 35.00.
2,7-Diamino-1,8-naphthyridine (14) from 12: To a stirred solution of sodium amide (1.086 g, 0.025 mol) in dry xylene (100 mL) compound 12 (1.0 g, 0.005 mol) was added and heated at 1300C under nitrogen for 12 h. The precipitate obtained was filtered and poured onto ice to quench excess sodium amide. Water was removed by distillation under reduced pressure and the solid was extracted with methanol-chloroform (15:85) to afford the desired product 14 (0.21 g, 25%) identical in all respects with the above.
2-Amino-7-benzylamino-1,8-naphthyridine (15): A suspension of 2-Amino-7-chloro-1,8-naphthyridine (12, 0.175 g, 1.0 mmol), and distilled benzylamine (0.5 mL) was heated for 6h at 120 °C. The sticky precipitate was washed with water followed by acetone and it was then air-dried. The crude solid was recrystallized from methanol which afforded pure compound 15 (0.118 g, 50%); m.p > 300 °C; 1H-NMR (200 MHz) δ: 7.65-7.57 (m, 2H), 7.40-7.29 (m, 6H), 6.48 (d, 1H, J = 8.5 Hz), 6.38 (d, 1H, J = 8.6 Hz), 5.60 (bs, 2H), 4.73 (d, 2H, J = 4.0 Hz); IR (cm-1): 1526, 1627, 1660, 3173, 3351; Anal. Calcd for C15H14N4: required C, 71.97; H, 5.63; N, 22.38. Found: C, 71.90; H, 5.60; N, 22.42.
References
- Tomcufcik, A. S.; Meyer, W. E.; Marsico, J. W. Eur. Pat. Appl. EP 446604. 1991. US Appl. 494387, 1990; [Chem. Abstr. 1992, 116, 235628p].
- Saupe, T.; Schaefer, P.; Meyer, N.; Wuerzer, B.; Westphalen, K. O. Ger. Offen. DE 3907937. 1990. [Chem. Abstr. 1991, 114, 81808s].
- Cotrel, C.; Guyon, C.; Roussel, G.; Taurand, G. Eur. Pat. Appl. EP 208621. 1987. FR Appl. 85/10619, 1985; [Chem. Abstr. 1987, 107, 39780g].
- Goswami, S.; Mukherjee, R. Molecular recognition: A simple dinaphthyridine receptor for urea. Tetrahedron Lett. 1997, 38, 1619–1622. [Google Scholar] Goswami, S.; Ghosh, K. Mukherjee, Recognition of insoluble tartaric acid in chloroform. Tetrahedron 2001, 57, 4987–4993. [Google Scholar] Hamilton, A. D.; Pant, N. Nucleotide base recognition: ditopic binding of guanine to a macrocyclic receptor containing naphthyridine and naphthalene units. J. Chem. Soc. Chem. Commun. 1988, 765, 765–766. [Google Scholar] [PubMed]
- Brown, E. V. 1,8-Naphthyridines. I. Derivatives of 2- and 4-methyl-1,8-naphthyridines. J. Org. Chem. 1965, 30, 1607–1610. [Google Scholar] [CrossRef]
- Collin, J-P.; Youinou, M-T. Synthesis, X-ray molecular structure and electrochemical properties of a binuclear copper(I) complex with 2,7-diphenyl-azo-1,8-naphthyridine. Inorg. Chim. Acta. 1992, 201, 29–34. [Google Scholar]
- Chandler, C. J.; Deady, L. W.; Reiss, J. A.; Tzimos, V. J. The synthesis of macrocyclic polyether- diesters incorporating 1,10-phenanthrolino and 1,8-naphthyridino subunits. J. Heterocycl. Chem. 1982, 19, 1017–1019. [Google Scholar] [CrossRef]
- Carboni, S.; Settimo, A. D.; Pirisino, G. Synthesis in the 1,8-naphthyridine series. Ann. Chim. (Rome) 1964, 54, 883–890, [Chem. Abstr. 1965, 63, 5620]. [Google Scholar]
- Carboni, S.; Settimo, A. D.; Ferrarini, P. L.; Ciantelli, P. L. Investigation of some tetrazole derivatives of 1,8-naphthyridines. J. Heterocycl. Chem. 1970, 7, 1037–1043. [Google Scholar] [CrossRef]
- Samples Availability: Available from the authors.
© 2005 by MDPI (http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
MDPI and ACS Style
Goswami, S.; Mukherjee, R.; Mukherjee, R.; Jana, S.; Maity, A.; Adak, A. Simple and Efficient Synthesis of 2,7-Difunctionalized-1,8-Naphthyridines. Molecules 2005, 10, 929-936. https://doi.org/10.3390/10080929
AMA Style
Goswami S, Mukherjee R, Mukherjee R, Jana S, Maity A, Adak A. Simple and Efficient Synthesis of 2,7-Difunctionalized-1,8-Naphthyridines. Molecules. 2005; 10(8):929-936. https://doi.org/10.3390/10080929
Chicago/Turabian StyleGoswami, S., R. Mukherjee, R. Mukherjee, S. Jana, A. Maity, and A. Adak. 2005. "Simple and Efficient Synthesis of 2,7-Difunctionalized-1,8-Naphthyridines" Molecules 10, no. 8: 929-936. https://doi.org/10.3390/10080929