Synthesis and Bioevaluation of 5-Fluorouracil Derivatives
by
Zhi-Yong Tian
1, 
Gang-Jun Du
2,
Song-Qiang Xie
2,
Jin Zhao
3,
Wen-Yuan Gao
1 and
Chao-Jie Wang
3,* 1
School of Pharmaceutical Science and Technology, Tianjin University, 92 Weijin Road, Nankai District, 300072, Tianjin, P. R. China
2
College of Pharmaceutical Science, Henan University, Jinming District, 475001, Kaifeng, Henan, P. R. China
3
Institute of Natural Products and Medicinal Chemistry, College of Chemistry and Chemical Engineering, Henan University, Jinming District, 475001, Kaifeng, Henan, P. R. China
*
Author to whom correspondence should be addressed.
Molecules 2007, 12(11), 2450-2457; https://doi.org/10.3390/12112450
Submission received: 9 August 2007
/
Revised: 9 October 2007
/
Accepted: 9 October 2007
/
Published: 6 November 2007
Abstract
:A series of six novel 5-fluorouracil derivatives 1-6 were synthesized and their structures confirmed by 1H- and 13C-NMR, MS and elemental analysis. The preliminary in vitro antitumor activities against B16, K562 and CHO cells and the in vivo inhibitions of liver cancer H22 demonstrated that some of these compounds effectively inhibit the growth of tumor cells, but the in vivo trials in mice revealed that the compounds also exhibited serious liver and lung tissue toxicity. The hydrolysis experiments indicated that this type of compound did not readily liberate 5-fluorouracil, as expected.
Introduction
5-Fluorouracil (5-Fu, 7), first synthesized in 1957 [1], is one of the antitumor agents frequently used for treating solid tumors such as breast, colorectal and gastric cancers [2]. 5-Fu is poorly tumor selective, and therefore its therapeutic use results in high incidences of bone marrow, gastrointestinal tract and central nervous system toxicity. To tackle these problems, numerous modifications of the 5-Fu structure have been performed. Thus, a series of 5-Fu prodrugs in which 5-Fu is attached to amino acids, peptides, phospholipids, and polymers have been reported [3,4,5,6,7]. These N-1 and/or N-3 substituted derivatives have exhibited improved pharmacological and pharmacokinetic properties, including increased bioactivity, selectivity, metabolic stability, absorption and lower toxicity.
Recently, the concept of mutual prodrugs in which two different antineoplastic agents are coupled directly or by means of a spacer has become well accepted [8,9,10,11,12,13,14,15,16,17]. This technique can be used to overcome many problems including poor solubility or absorption, patient acceptability, drug instability and toxicity, and especially drug resistance [11]. After the entry of a mutual prodrug into a cancer cell, the two active components can reach a target simultaneously and are liberated concomitantly, whereas they might be transported to the same site with different efficacy when administered individually. This kind of prodrugs can display a broader antitumor spectrum, less drug resistance and less toxicity [18].
Many DNA intercalators have been evaluated as antineoplastic agents in chemotherapy, where they constitute one of the most important drug classes. These agents are characterized by the presence of flat chromophores bearing π electron-deficient polyconjugated plane bound to polar groups. Their antitumor action results from DNA distortion and altered nuclear protein interaction as a consequence of reversible complex formation. Because the cellular target of 5-Fu is DNA, the combination of 5-Fu and some antineoplastic DNA binders through an ester bond [8,14] helps the resulting dual prodrug reach the target and optimize the efficiency of both 5-Fu and the DNA binders. According to this strategy, we prepared six novel 5-fluorouracil derivatives and evaluated their antitumor activity both in vitro and in vivo.
Results and Discussion
Chemistry
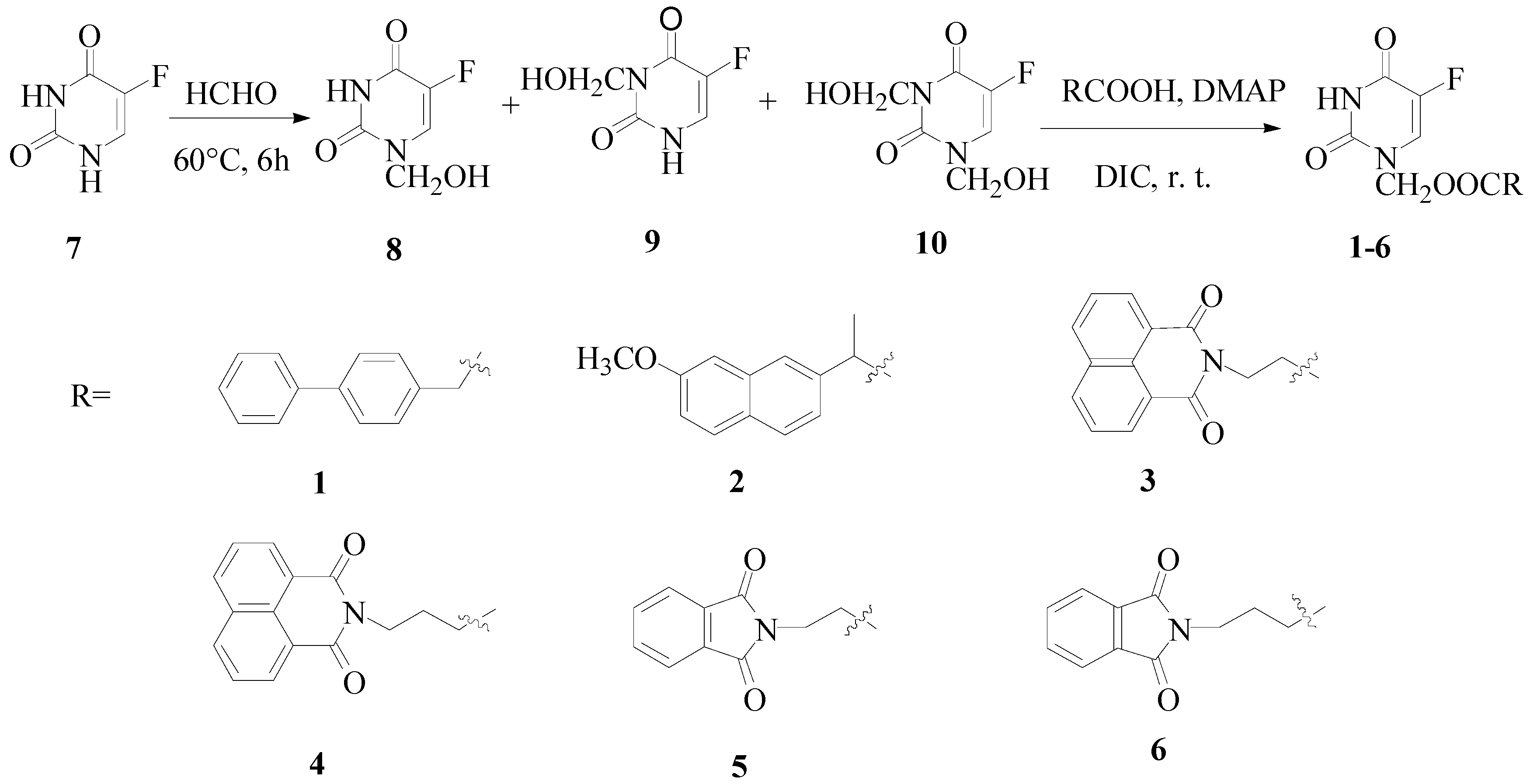
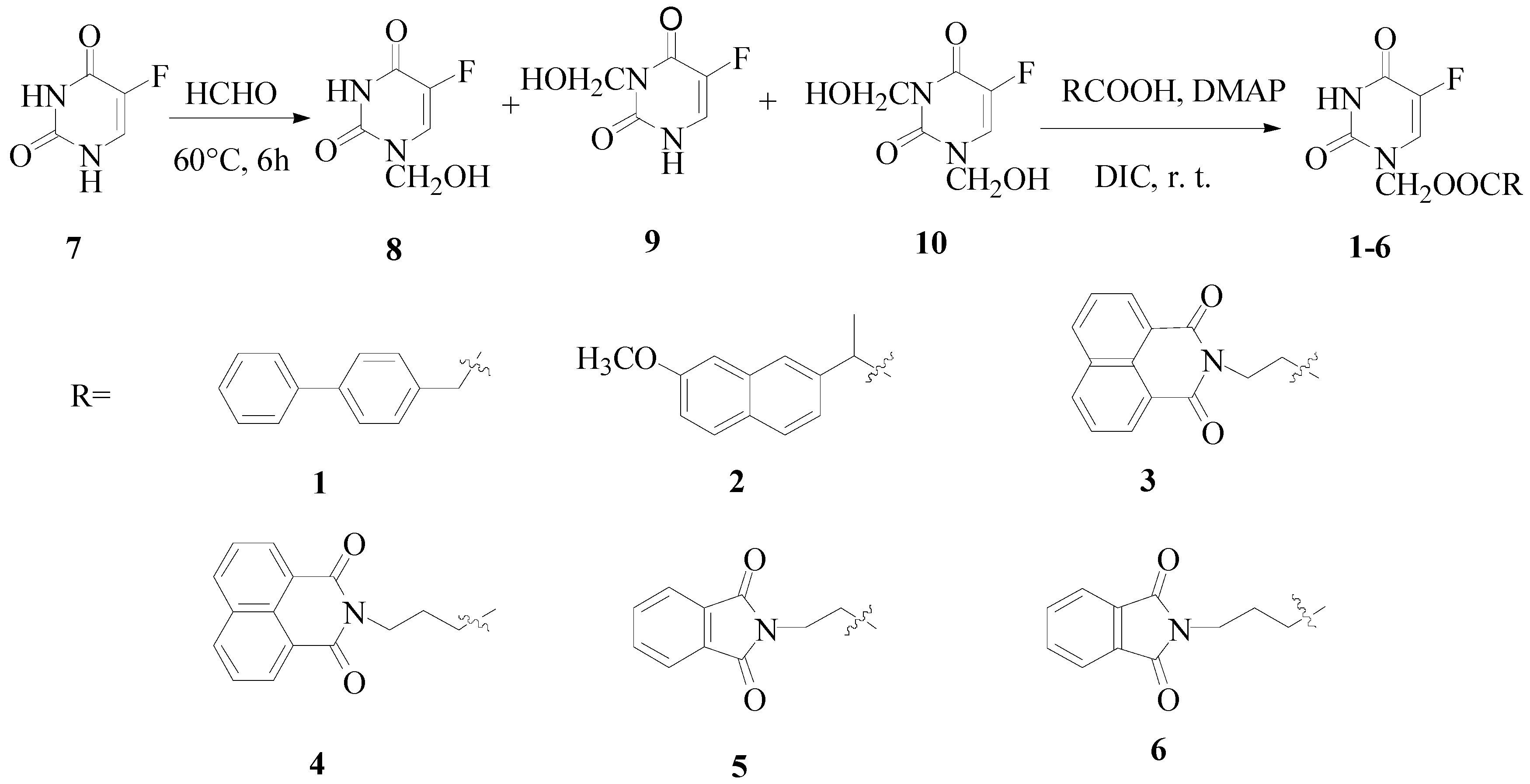
The synthetic route towards the target compounds 1-6 is shown in Scheme 1. 5-Fu was reacted with formaldehyde to form a mixture of N1-hydroxymethylene-5-fluorouracil, N3-hydroxymethylene-5- fluorouracil and N1,N3-dihydroxymethylene-5-fluorouracil. Without separation, the mixture was directly coupled with the appropriate carboxylic acids to give the target compounds. N,N'-dicyclo-hexylcarbodiimide (DCC) was the first reagent chosen to facilitate the esterification, but the resulting 1,3-dicyclohexylurea (DCU) byproduct was hard to remove by flash chromatograph and recrystallization. On the other hand, when N,N'-diisopropylcarbodiimide (DIC) was used, the target compounds could be purified through flash chromatograph and recrystallization from acetone.
Two kinds of drugs with flat structures were attached to 5-Fu. First, nonsteroidal antiinflammatory drugs such as naproxen and biphenylacetic acid, which have been reported to inhibit the growth of cancer cells. In addition, naproxen and biphenylacetic acid can bind with DNA [19]. Second, imides, which also exhibit efficient DNA-binding properties and antitumor activities on a variety of murine and human tumor cells [20]. The imides were first converted to the corresponding carboxylic acids through the reactions of 1,8-naphthalic or phthalic anhydride with relevant amino acids [21], then the imide moiety was linked to 5-Fu via an (acyloxy)methylene group, known to be easily removable [3,8]. All structures were verified by 1H-NMR, 13C-NMR, MS and elemental analysis.
Scheme 1.
Synthesis of compounds 1-6.
In vitro antitumor activity
All target compounds 1-6 were evaluated for their antitumor activity in vitro against K562, B16, and CHO cell lines by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-2H-tetrazoliumbromide (MTT) assay method [6,22]. The reference drug used was 5-fluorouracil. The activity of the samples and the reference drug was assayed under identical conditions at concentrations of 0.1, 1, 10, 30 and 50 µM. The lower its IC50 value, the more efficiently a compound kills cancer cells. As shown in Table 1, all compounds’ in vitro activity was significantly decreased because the N-1 position, the active area of 5-Fu, was occupied. Compounds 2 and 6 still possessed some moderate to potent inhibition rates on the three tested cell lines and compounds 3, 4 and 5 exhibited some selectivity against B16 and/or CHO cells.

{kind=link}
| Sample | IC50 (µM) | ||
|---|---|---|---|
| K562 | B16 | CHO | |
| 5-Fu | 0.27 | 0.23 | 0.35 |
| 1 | 16.83 | 12.15 | 12.86 |
| 2 | 43.25 | 36.93 | >50 |
| 3 | >50 | 3.72 | 32.30 |
| 4 | 30.46 | 4.44 | 16.83 |
| 5 | >50 | 3.97 | 7.41 |
| 6 | 14.48 | 7.35 | 6.86 |
In vivo antitumor activity
The in vivo antitumor activities of three compounds on mice bearing liver cancer H22 are listed in Table 2. The T/C ratio is used as an index of the antitumor activity: T/C (%) = (C − T) / C x 100 % (T: Average tumor weight of treated mice; C: Average tumor weight of control mice). As shown in Table 2, compounds 2, 5 effectively inhibited tumor growth (ratio of tumor inhibition >40%) although they were less active than 5-Fu. Compound 2 was special because it was ineffective against all three cell types in vitro but was effective in vivo. We also found that surprisingly all three tested compounds caused more liver and lung tissue toxicity in mice than 5-Fu. This could happen if the hydrolysis of compound 2, which contained only two clinically used drugs (5-Fu and naproxen), occurred readily under physiological conditions, liberating toxic formaldehyde. To verify this the hydrolysis of compound 2 was monitored by UV spectroscopy at physiological pH, but no 5-Fu was detected, thus indicating that the (acyloxy)methylene group in target compounds is stable [8], and target compounds might exert their biological functions as a new kind of “drugs”.
| Sample (T/C, %) | Dose (µmol/kg) | Weight of tumor (g) | Ratio of tumor inhibition |
|---|---|---|---|
| Control | 0 | 1.63±0.31 | 0 |
| 5-Fu | 150 | 0.59±0.19 | 64.07 |
| 2 | 150 | 0.95±0.30 | 41.64 |
| 5 | 150 | 0.89±0.53 | 45.17 |
| 6 | 150 | 1.26±0.54 | 22.85 |
Conclusions
In conclusion, a novel series of 5-fluorouracil derivatives was synthesized and some compounds were found to be active against tumor cell lines in vitro and in vivo. Three tested compounds caused more in vivo toxicities on the liver and lung tissues of mice than 5-Fu and were less effective than 5-Fu. This might be explained by the difficult hydrolysis of target compounds to give free 5-fluorouracil under physiological pH conditions.
Experimental
General
Melting points were determined on an X-6 hot stage microscope and were uncorrected. Mass spectra were obtained on an ESQUIRE-LC instrument. 1H-NMR spectra and 13C-NMR were run in DMSO-d6, with TMS as the internal standard, on a Bruker AV-400 instrument operating at 400 MHz. Elemental analysis was performed with a Gmbe VarioEL Elemental analysis instrument.
Synthesis of the target compounds 1-6
Synthesis of compounds 1-6 was accomplished as shown in Scheme 1. 5-Fu (7, 1.3 g, 10 mmol), formaldehyde (37 wt.%, aq., 1.26 g, 15.5 mmol) and water (10 g) were added to a round-bottom flask, which was then immersed in water bath at 60°C and agitated for 6 h. The product solution was concentrated under reduced pressure to give an oily mixture containing 8-10. The oil was dissolved in dry acetonitrile (50 mL), and the corresponding carboxylic acids (10 mmol), DMAP (N,N-dimethylpyridin-4-amine, 80 mg, 14 mmol) and DIC (2.2 mL, 14 mmol) were added and the mixture was stirred at room temperature for 48-72 h. The white precipitate was filtered off and the solvent was removed by evaporation. The residue was dissolved in ethyl acetate (30 mL) and washed by diluted hydrochloric acid (pH = 3-4), saturated aqueous NaHCO3 (pH = 7-8) and water. The organic layer was collected, dried over sodium sulphate, filtered and finally concentrated under reduced pressure. The target compounds were obtained through flash column chromatography: petroleum ether-acetone = 7:3 and recrystalization from acetone [8,14].
(5-Fluoro-2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)methyl-2-(biphenyl-4-yl)acetate (1). Yield: 31.1%; m. p.: 180.5-182°C; 1H-NMR δ: 12.00 (s, 1H, NH of 5-Fu), 7.3-8.1 (m, 10H, 9×ArH, CH=CF of 5-Fu), 5.58 (s, 2H, CH2O), 3.7 (s, 2H, CH2C=O); 13C-NMR δ: 170.89, 157.37 (d, JCCF = 26 Hz), 149.22, 139.78, 139.41 (d, JCF = 229 Hz), 138.84, 132.92, 130.00, 129.42 (d, JCCF = 34 Hz), 128.89, 127.38, 126.61, 126.57, 70.86, 40.22; ESI-MS: m/z 377 (M++Na); Anal. calcd. for C19H15FN2O4: C, 64.40; H, 4.27; N, 7.91; found: C, 64.33; H, 4.29; N, 7.81.
(5-Fluoro-2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)methyl-2-(7-methoxynaphthalen-2-yl)propanoate (2). Yield: 60.0%; m. p.: 171.5-172.5°C; 1H-NMR δ:11.96 (s, 1H, NH of 5-Fu), 7.10-8.1 (m, 7H, 6xArH, CH=CF of 5-Fu), 5.60 (s, 2H, CH2O), 3.92 (s, 3H, CH3O), 3.84 (q, 1H, J = 6.6 Hz, CH3CH), 1.40 (d, J = 7.6 Hz, 3H, CH3CH); 13C-NMR δ: 173.53, 157.28 (d, JCCF = 26 Hz), 157.23, 149.09, 139.34 (d, JCF = 229 Hz), 135.02, 133.33, 129.34 (d, JCCF = 34 Hz), 129.08, 128.34, 127.00, 126.10, 125.64, 118.79, 105.70, 70.84, 55.15, 44.13, 18.13; ESI-MS: m/z 395 (M++Na); Anal. calcd. for C19H17FN2O5: C, 61.29; H, 4.60; N, 7.52; found: C, 61.36; H, 4.33; N, 7.29.
(5-Fluoro-2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)methyl-3-(1,8-naphthalimido)propanoate (3). Yield: 20.0%; m. p.: 215.5-217°C; 1H-NMR δ: 11.93 (s, 1H, NH of 5-Fu), 8.39-8.51 (4H, m, ArH), 8.02 (d, 1H, J = 6.0 Hz, CH=CF), 7.83-7.87 (2H, m, ArH), 5.56 (s, 2H, OCH2), 4.29 (t, 2H, NCH2), 2.74 (t, 2H, CH2CO); 13C-NMR δ: 170.72, 163.3, 157.43 (d, JCCF = 26 Hz), 149.20, 139.34 (d, JCF = 229 Hz), 134.40, 131.24, 130.68, 129.43 (d, JCCF =34 Hz), 127.31, 127.14, 121.84, 70.42, 35.56, 32.19; ESI-MS : m/z 434 (M++Na); Anal. calcd. for C20H14FN3O6∙0.30H2O: C, 57.64; H, 3.53; N, 10.08; found: C, 57.71; H, 3.34; N, 9.75.
(5-Fluoro-2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)methyl-4-(1,8-naphthalimido)butyrate (4). Yield: 20.0%; m. p.: 233.5-235°C; 1H-NMR δ: 11.97 (s, 1H, NH of 5-Fu), 8.12-8.43 (m, 4H, ArH), 8.07 (d, 1H, J = 6.0 Hz, CH=CF), 7.83-7.85 (m, 2H, ArH), 5.54 (s, 2H, OCH2), 4.06 (t, 2H, NCH2), 2.37 (t, 2H, CH2CO), 1.84-1.91 (m, 2H, CH2CH2CO); 13C-NMR δ: 172.23, 163.59, 157.46 (d, JCCF = 25 Hz), 149.27, 139.31 (d, JCF = 229 Hz), 134.23, 133.96, 131.23, 130.61, 129.58 (d, JCCF = 34 Hz), 127.26, 122.07, 70.23, 39.11, 30.58, 22.50; ESI-MS: m/z 448 (M++ Na); Anal. calcd. for C21H16FN3O6∙0.39H2O: C, 58.33; H, 3.91; N, 9.72; found: C, 58.73; H ,3.93; N, 9.26.
(5-Fluoro-2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)methyl-3-(phthalimido)propanoate (5). Yield: 46.7%; m. p.: 233-235°C; 1H-NMR δ: 11.92 (s, 1H, NH of 5-Fu), 8.10 (d, 1H, J = 6.0 Hz, CH=CF), 7.82 (m, 4H, 4×ArH), 5.53 (s, 2H, OCH2), 3.81 (t, 2H, NCH2), 2.72 (t, 2H, CH2CO); 13C-NMR δ: 170.84, 168.04, 157.92 (d, JCCF = 26Hz), 149.69, 139.83 (d, JCF = 229 Hz), 134.90, 131.99, 129.91 (d, JCCF = 34 Hz), 123.48, 70.93, 33.82, 32.74; ESI-MS: m/z 384 (M++Na); Anal. calcd. for C16H12FN3O6: C, 53.19; H, 3.35; N, 11.63; found: C, 53.16; H, 3.22; N, 11.37.
(5-Fluoro-2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)methyl-4-(phthalimido)butyrate (6). Yield: 71.5%; m. p.: 181.5-184°C; 1H-NMR δ: 11.99 (s, 1H, NH of 5-Fu), 8.09 (d, 1H, J = 6.0 Hz, CH=CF), 7.76-7.84 (m, 4H, 4×ArH), 5.55 (s, 2H, CH2O), 3.60 (t, 2H, NCH2), 2.43 (t, 2H, CH2CO),1.82-1.86 (m, 2H, CH2); 13C-NMR δ: 172.58, 168.29, 157.90 (d, JCCF = 26 Hz), 149.71, 139.83 (d, JCF = 229 Hz), 134.67, 132.04, 129.96 (d, JCCF = 34 Hz), 123.31, 70.82, 36.89, 30.81, 23.33; ESI- MS: m/z 398 (M++ Na); Anal. calcd. for C17H14FN3O6: C, 54.40; H, 3.76; N, 11.20; found: C, 54.54; H, 3.56; N, 11.91.
In vitro antitumor activity experiments
Cell lines (K562, B16, CHO) were obtained from the American Type Culture Collection (ATTC) and cultured in RPMI1640, supplemented with 10% heat-inactivated fetal calf serum and antibiotics (penicillin, 100 units/mL; streptomycin sulfate, 100 μg/mL) at 37°C, in an atmosphere of 95% air and 5% CO2 under humidified condition. All chemicals were purchased from Sigma, unless otherwise indicated. RPMI1640 and fetal calf serum (FCS) were purchased from Gibco. Stock solution (10 µM) of samples was prepared in dimethylsulfoxide (DMSO) and diluted with various concentrations with serum-free culture medium. The in vitro antitumor activity of compounds 1-6 and 5-Fu were determined by MTT assay method [10]. Briefly, exponentially growing B16 or CHO cells were seeded in 96-well plates (5000 to each well) and allowed to attach overnight. After 24 h, the cells were treated with indicated concentrations of samples for 48 h, and then MTT (100 μL, 1 mg/mL) was added. After incubation for 4 h at 37°C, the MTT solution was removed and the crystals of viable cells were dissolved with DMSO (150 μL) in each well; 4000/well exponentially growing K562 were seeded in 96-well plates and treated with indicated concentrations of samples for 48 h, and then MTT (10 mg/mL, 10 μL) was added. After incubation for 4 h at 37°C the crystals of viable cells were dissolved overnight with SDS (sodium dodecylsulfonate, 10%, 100 μL) in each well. The absorbance spectra were measured on an ELISA Processor II Microplate Reader at a wavelength of 570 nm. The percentage of cytotoxicity was defined with treated and untreated cell lines. The 50% antitumor activity dose (IC50) was defined as the concentration of samples that reduced the absorbance of the treated cells by 50% [22].
In vivo antitumor activity experiments
To evaluate the in vivo antitumor activity of 5-fluorouracil derivatives 2, 5 and 6 mice bearing H22 tumor cells were used. Kunming mice (n = 10) were first injected subcutaneously (s.c.) with liver cancer H22 cell line (2×105 cells/mL). The mice bearing the liver cancer H22 tumor cell line were then injected intraperitoneally (i.p.) with water solutions of compounds 2, 5 or 6 for 8 days. For comparison, antitumor activities of 5-fluorouracil (5-Fu) also were tested by the same method. A control group was treated with H22 tumor cells along with the same volume of NaCl (0.9 wt.%, aq.). The ratio (T/C) obtained by dividing the number of mice treated with 5-fluorouracil derivatives (T) showing tumor inhibition to that of mice in the control groups (C) was used as the antitumor activity index [22].
Hydrolysis experiments
Ultraviolet and visible spectral measurements were performed with a Varian DMS 300 spectrophotometer. Buffers (trishydroxymethylaminomethane-HCl, Tris-HCl, pH = 7.4) were made in our laboratory. The UV method is defined as recording the absorbance changes over time for reactions in which the absorbance of substrate and product differ maximally at a particular wavelength. This method was used to study the activation of compound 2 [8]. Reactions were performed on aliquots of buffer solution (3 mL) in a round-bottom flask by addition of compound 2 in chloroform at 25°C for 2 hours. Release of 5-Fu of compound 2 was determined from the absorbance at 266 nm. Initial concentration of all substrates studied by the UV method was 5× 10-5 M. No absorbance of 5-Fu was observed at 266 nm.
Acknowledgments
This work was supported by Henan Natural Science Foundation (0423031800)
References
- Heidelberger, C.; Chaudhuri, N. K.; Danneberg, P.; Mooren, D.; Griesbach, L. Fluorinated pyrimidines, a new class of tumor-inhibitory compounds. Nature 1957, 179, 663–666. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, D.; James, R. D. Integrating the oral fluoropyrimidines into the management of advanced colorectal cancer. Eur. J. Cancer 2001, 37, 826–834. [Google Scholar] [CrossRef] [PubMed]
- Ozaki, S.; Watanabe, Y.; Hoshiko, T.; Mizuno, H.; Ishikawa, K.; Mori, H. 5-Fluorouracil derivatives (IV): Synthesis of antitumor active acyloxyalkyl-5-fluouracils. Chem. Pharm. Bull. 1984, 32, 733–738. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F. M.; Yao, X. J.; Tian, X.; Tu, Y. Q. Synthesis and Biological Evaluation of New 4β-5-Fu-substituted 4'-Demethylepipodophyllotoxin Derivatives. Molecules 2006, 11, 849–875. [Google Scholar]
- Zhang, C. X.; Zhang, Z. B.; Chen, H. M.; Tang, C. C.; Chen, R. Y. synthesis of 1,2- and 1,3-Cyclic Phospholipid Conjugates of N1-(2-Furanidyl)-N3-(2-hydroxyethyl)-5-fluorouracil. Heteroatom. Chem. 1998, 9, 295–298. [Google Scholar] [CrossRef]
- Jung, E. Y.; Chung, I. D.; Lee, N. J.; Park, J. S.; Ha, C. S.; Cho, W. J. Syntheses, Antitumor Activities, and Antiangiogenesis of a Monomer and Its Medium Molecular Weight Polymers: Maleimidoethanoyl-5-fluorouracil and Its Polymers. J. Polym. Sci., Part A: Polym. Chem. 2000, 38, 1247–1256. [Google Scholar]
- Lee, J. S.; Jung, Y. J.; Kim, Y. M. Synthesis and Evaluation of N-Acyl-2-(5-Fluorouracil-1-yl)-D,L- Glycine as a Colon-Specific Prodrug of 5-Fluorouracil. J. Pharm. Sci. 2001, 90, 1787–1794. [Google Scholar] [CrossRef] [PubMed]
- Menger, F. M.; Rourk, M. J. Synthesis and Reactivity of 5-Fluorouracil/Cytarabine Mutual Prodrugs. J. Org. Chem. 1997, 62, 9083–9088. [Google Scholar]
- McElhinney, R. S.; McCormick, J. E.; Bibby, M. C.; Double, J. A.; Radacic, M.; Dumont, P. Nucleoside Analogs. 14. The synthesis and antitumor activity in mice of molecular combinations of 5-fluorouracil and N-(2-chloroethyl)-N-nitrosourea moieties separated by a three-carbon chain. J. Med. Chem. 1996, 39, 1403–1412. [Google Scholar]
- Mori, M.; Hatta, H.; Nishimoto, S. Stereoelectronic effect on one-electron reductive release of 5-fluorouracil from 5-fluoro-1-(2´-oxocycloalky)uracils as a new class of radiation-activated antitumor prodrugs. J. Org. Chem. 2000, 65, 4641–4647. [Google Scholar] [CrossRef] [PubMed]
- Bill-Cai, T.; Tang, X.; Nagorski, J.; Brauschweiger, P. G.; Wang, P. G. Synthesis and cytotoxicity of 5-fluorouracil/diazeniumdiolate conjugates. Bioorg. Med. Chem. 2003, 11, 4971–4975. [Google Scholar]
- Domínguez, J. F.; Marchal, J. A.; Correa, A.; Carrillo, E.; Boulaiz, H.; Aránega, A.; Gallo, M. A.; Espinosa, A. Synthesis and evaluation of new 5-fluorouracil antitumor cell differentiating derivatives. Bioorg. Med. Chem. 2003, 11, 315–323. [Google Scholar]
- Díaz-Gavilán, M.; Gómez-Vidal, J. A.; Entrena, A.; Gallo, M. A.; Espinosa, A.; Campos, J. M. Study of the factors that control the ratio of the products between 5-fluorouracil, uracil, and tetrahydrobenzoxazepine O,O-acetals bearing electron-withdrawing groups on the nitrogen atom. J. Org. Chem. 2006, 71, 1043–1054. [Google Scholar]
- Liu, Z. F.; Stephen, R. Synthesis and release of 5-fluorouracil from poly (N-vinylpyrrolidinone) bearing 5-fluorouracil derivatives. J. Control. Rel. 2002, 81, 91–99. [Google Scholar]
- Shoichiro, O. Synthesis and antitumor activity of 5-fluorouracil derivatives. Medicinal Research Reviews 1996, 16, 51–86. [Google Scholar] [CrossRef] [PubMed]
- Malet-Martino, M.; Jolimaître, P.; Martino, R. The prodrugs of 5-fluorouracil. Curr. Med. Chem.- Anti-Cancer Agents. 2002, 2, 267–310. [Google Scholar]
- Campos, J.; Dominguez, J. F.; Gallo, M. A.; Espinosa, A.; Campos, J.; Dominguez, J. F.; Gallo, M. A.; Espinosa, A. From a Classic Approach in Cancer Chemotherapy Towards Differentiation Therapy: Acyclic and Cyclic Seven-Membered 5-Fluorouracil O, N-Acetals. Curr. Pharmaceut. Design. 2000, 6, 1797–1810. [Google Scholar]
- Marx, J. L. Drug resistance of cancer cells probed. Science 1986, 234, 818–820. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y. X.; Zhao, J.; Sun, X. Q.; Wang, C. J. Synthesis, Interaction with DNA and Bioactivity of N-Piperazinoalkylamide. Chin. J. Org. Chem. 2006, 26, 1066–1072. [Google Scholar]
- Braña, M. F.; Castellano, J. M.; Perron, D.; Maher, C.; Conlon, D.; Bousquet, P. F.; George, J.; Qian, X.-D.; Robinson, S. P. Chromophore-Modified Bis-Naphthalimides: Synthesis and Anti-tumor Activity of Bis-Dibenz[de,h]isoquinoline-1,3-diones. J. Med. Chem. 1997, 40, 449–454. [Google Scholar]
- Lee, N. J.; Kim, K. H.; Rhew, H.Y.; Choi, W. M.; Chung, I. D.; Cho, W. J. Synthesis and biological activity of medium molecular weight polymers containing 3,6-endo-methylene-1,2,3,6-tetrahydrophthalimido-butanoyl-5-fluorouracil. Polym. Int. 2000, 49, 1702–1708. [Google Scholar]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival. Application to proliferation and cytotoxicity assays. J. Immunol. Meth. 1983, 65, 55–63. [Google Scholar]
- Sample Availability: Contact the authors.
© 2007 by MDPI (http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
MDPI and ACS Style
Tian, Z.-Y.; Du, G.-J.; Xie, S.-Q.; Zhao, J.; Gao, W.-Y.; Wang, C.-J. Synthesis and Bioevaluation of 5-Fluorouracil Derivatives. Molecules 2007, 12, 2450-2457. https://doi.org/10.3390/12112450
AMA Style
Tian Z-Y, Du G-J, Xie S-Q, Zhao J, Gao W-Y, Wang C-J. Synthesis and Bioevaluation of 5-Fluorouracil Derivatives. Molecules. 2007; 12(11):2450-2457. https://doi.org/10.3390/12112450
Chicago/Turabian StyleTian, Zhi-Yong, Gang-Jun Du, Song-Qiang Xie, Jin Zhao, Wen-Yuan Gao, and Chao-Jie Wang. 2007. "Synthesis and Bioevaluation of 5-Fluorouracil Derivatives" Molecules 12, no. 11: 2450-2457. https://doi.org/10.3390/12112450