The Synthesis of 1-(4-Triethoxysilyl)phenyl)-4,4,4-trifluoro-1,3-butanedione, a Novel Trialkoxysilane Monomer for the Preparation of Functionalized Sol-gel Matrix Materials

Abstract
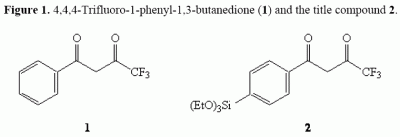
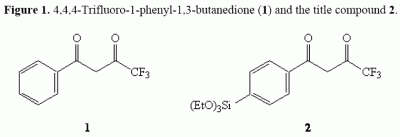
:
{kind=link}
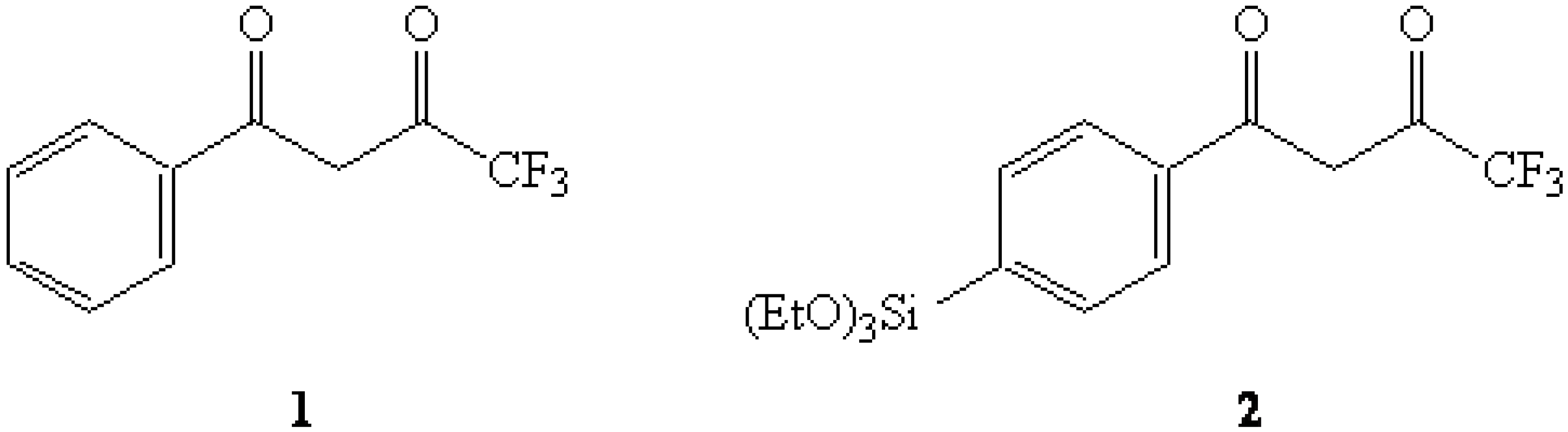{kind=link}
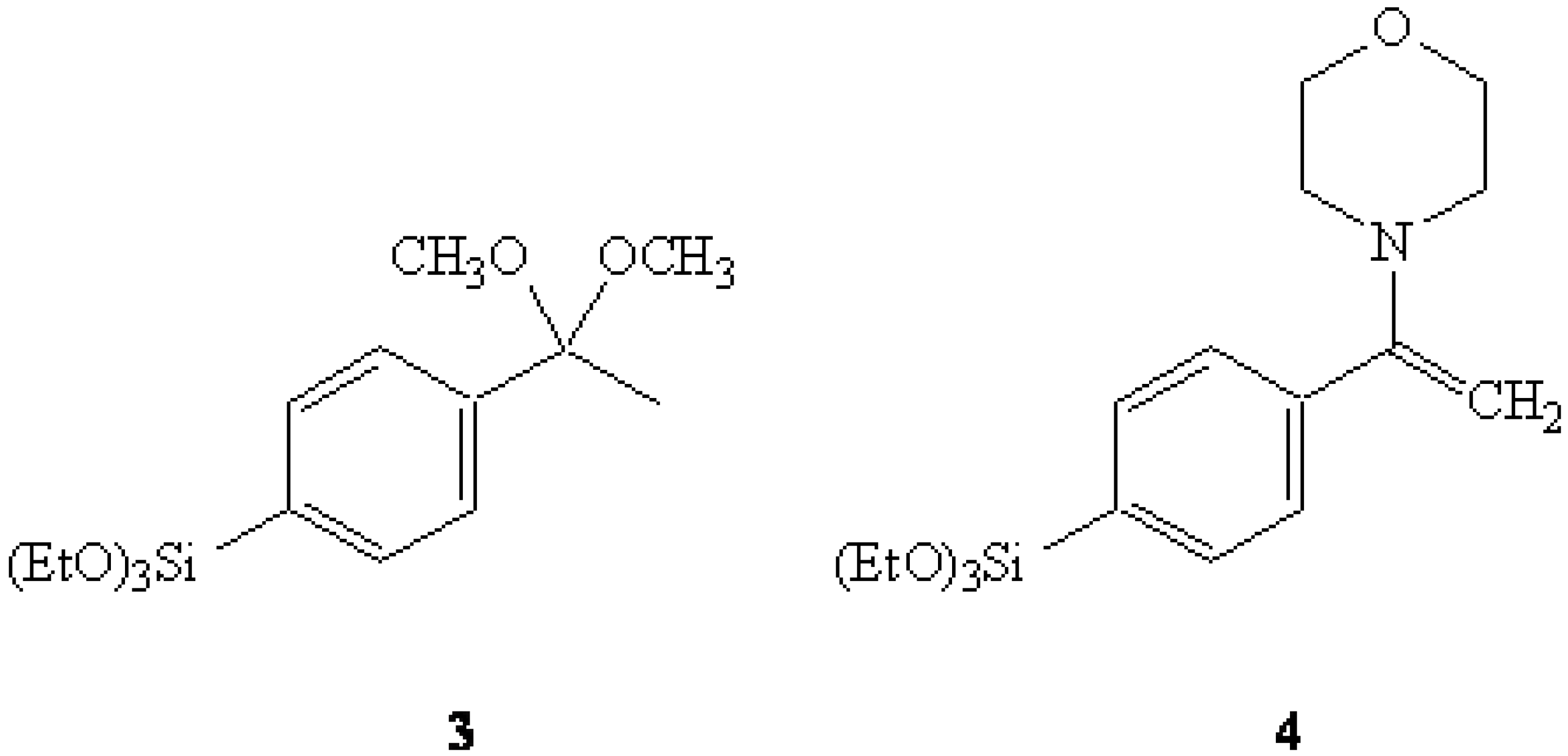{kind=link}
{kind=link}
Introduction

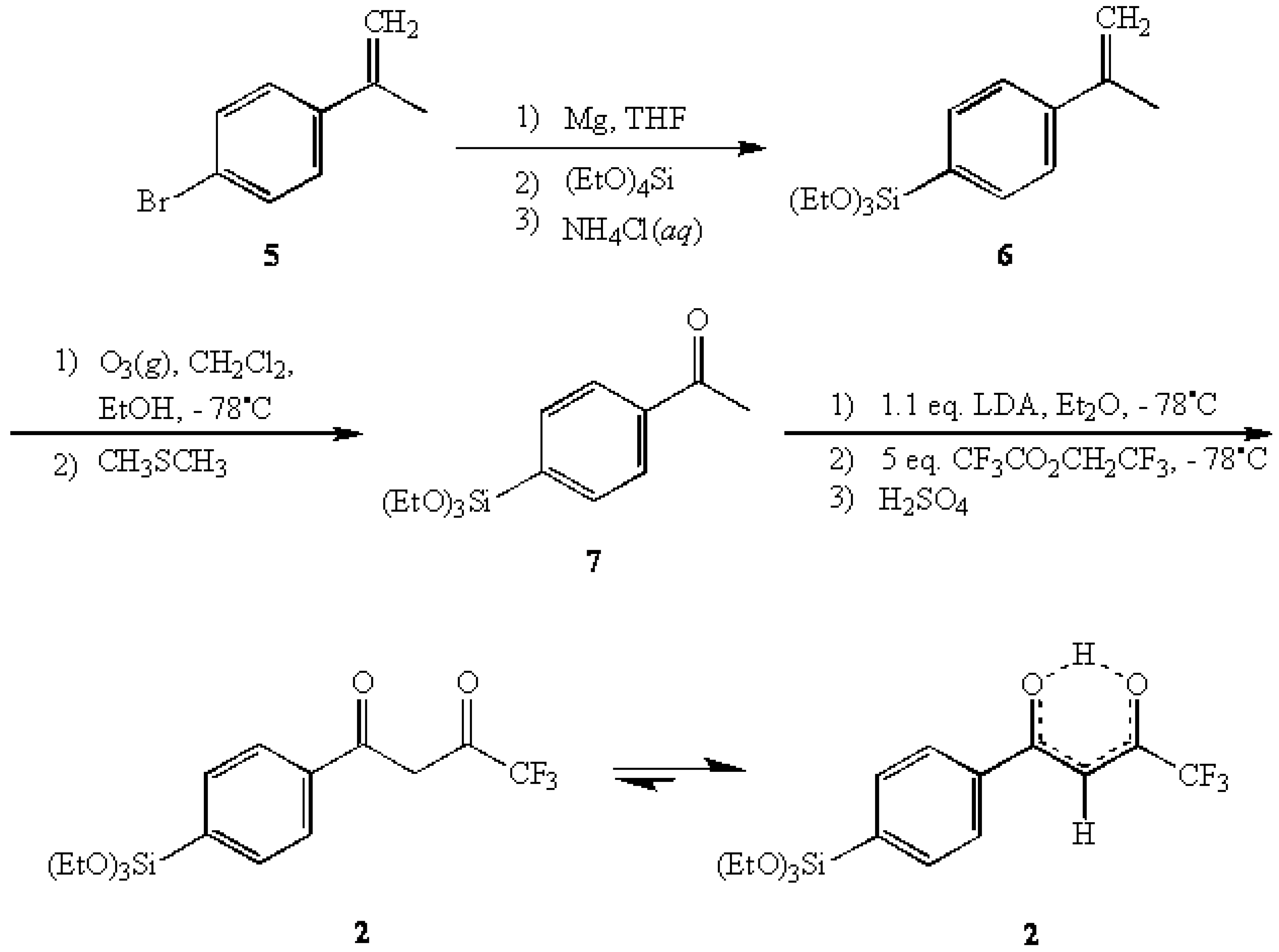
Results and Discussion


Conclusions
Experimental
General
Acknowledgements
References and Notes
- Collinson, M. Sol-gel strategies for the preparation of selective materials for chemical analysis. Crit. Rev. Anal. Chem. 1999, 29, 289–311. [Google Scholar] [CrossRef]
- Dickey, F. H. The Preparation of Specific Adsorbents. Proc. Natl. Acad. Sci. USA 1949, 35, 227–229. [Google Scholar] [CrossRef]
- Dickey, F. H. Specific Adsorption. J. Phys. Chem. 1955, 59, 695–707. [Google Scholar] [CrossRef]
- Sasaki, D. Y.; Rush, D. J.; Daitch, C. E.; Alalm, T. M.; Assink, R. A.; Ashely, C. S.; Brinker, J. C.; Shea, K. J. In Molecular and Ionic Recognition with Imprinted Polymers, ACS Symposium Series 703; Bartsch R., A., Maeda, M., Eds.; American Chemical Society: Washington DC, USA, 1998; pp. 314–324. [Google Scholar]
- Makote, R.; Collinson, M. Template Recognition in Inorganic-Organic Hybrid Films Prepared by the Sol-Gel Process. Chem. Mater. 1998, 10, 2440. [Google Scholar] [CrossRef]
- Makote, R.; Collinson, M. Dopamine recognition in templated silicate films. Chem. Comm. 1998, 3, 425. [Google Scholar] [CrossRef]
- Kepley, L. J.; Crooks, R. M.; Ricco, A. J. A. selective SAW-based organophosphonate chemical sensor employing a self-assembled, composite monolayer: a new paradigm for sensor design. Anal. Chem. 1992, 64, 3191–3193. [Google Scholar] [CrossRef]
- Hierlemann, A.; Ricco, A. J.; Bodenhofer, K.; Gopel, W. Effective Use of Molecular Recognition in Gas Sensing: Results from Acoustic Wave and in Situ FT-IR Measurements. Anal. Chem. 1999, 71, 3022–3035. [Google Scholar] [CrossRef]
- Thomas, R. C.; Hierlemann, A.; Staton, A. W.; Hill, M.; Ricco, A. J. Reflectance Infrared Spectroscopy on Operating Surface Acoustic Wave Chemical Sensors during Exposure to Gas-Phase Analytes. Anal. Chem. 1999, 71, 3615–3621. [Google Scholar] [CrossRef]
- Stork, G.; Brizzolara, A.; Landesman, H.; Szmuszkovicz, J.; Terrell, R. The Enamine Alkylation and Acylation of Carbonyl Compounds. J. Am. Chem. Soc. 1963, 85, 207–222. [Google Scholar] [CrossRef]
- Fraenkel, G.; Geckle, J. M. Influence of substituents on NMR and barriers to rotation in tert-benzyllithium compounds. J. Am. Chem. Soc. 1980, 102, 2869–2880. [Google Scholar] [CrossRef]
- Seymour, D.; Wolfstirn, K. B. Preparation and Properties of Trimethylsilylmethanol. J. Am. Chem. Soc. 1948, 70, 1177–1179. [Google Scholar] [CrossRef]
- For an alternate preparation of 7 via a Rh(I)-catalyzed silylation of 4-iodoacetophenone, see: Murata, M.; Ishikura, M.; Nagata, M.; Watanabe, S.; Masuda, Y. Rhodium(I) Catalyzed Silylation of Aryl Halides with Triethoxysilane: Practical Synthetic Route to Aryltriethoxysilanes. Org. Lett. 2002, 4, 1843–1845. [Google Scholar] [CrossRef]
- For an alternate preparation of 7 via a Pd(0)-catalyzed silylation of 4-iodoacetophenone, see: Manoso, A. S.; DeShong, P. Improved Synthesis of Aryltriethoxysilanes via Palladium(0)-Catalyzed Silylation of Aryl Iodides and Bromides with Triethoxysilane. J. Org. Chem. 2001, 66, 7449–7455. [Google Scholar] [CrossRef]
- Zayia, G. H. First General Method for Direct Formylation of Kinetically-Generated Ketone Enolates. Org. Lett. 1999, 1, 989–991. [Google Scholar] [CrossRef]
- Claus, R.; Schreiber, S. Ozonolytic Cleavage of Cyclohexene to Terminally Differentiated Products: Methyl 6-Oxohexonate, 6,6-Dimethoxyhexanal, Methyl 6,6-Dimethoxyhexanoate. Org. Synth. 1990, Collective Volume VII, 168. [Google Scholar]
- Sample Availability: Contact the authors.
© 2008 by the authors. Licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Peeples, C.J.; Earni, R.R.; DiCesare, J.C. The Synthesis of 1-(4-Triethoxysilyl)phenyl)-4,4,4-trifluoro-1,3-butanedione, a Novel Trialkoxysilane Monomer for the Preparation of Functionalized Sol-gel Matrix Materials. Molecules 2008, 13, 2601-2607. https://doi.org/10.3390/molecules13102601
Peeples CJ, Earni RR, DiCesare JC. The Synthesis of 1-(4-Triethoxysilyl)phenyl)-4,4,4-trifluoro-1,3-butanedione, a Novel Trialkoxysilane Monomer for the Preparation of Functionalized Sol-gel Matrix Materials. Molecules. 2008; 13(10):2601-2607. https://doi.org/10.3390/molecules13102601
Chicago/Turabian StylePeeples, Christopher J., Raghu Ram Earni, and John C. DiCesare. 2008. "The Synthesis of 1-(4-Triethoxysilyl)phenyl)-4,4,4-trifluoro-1,3-butanedione, a Novel Trialkoxysilane Monomer for the Preparation of Functionalized Sol-gel Matrix Materials" Molecules 13, no. 10: 2601-2607. https://doi.org/10.3390/molecules13102601