An Efficient Synthesis of γ-Aminoacids and Attempts to Drive Its Enantioselectivity



Abstract
:Introduction

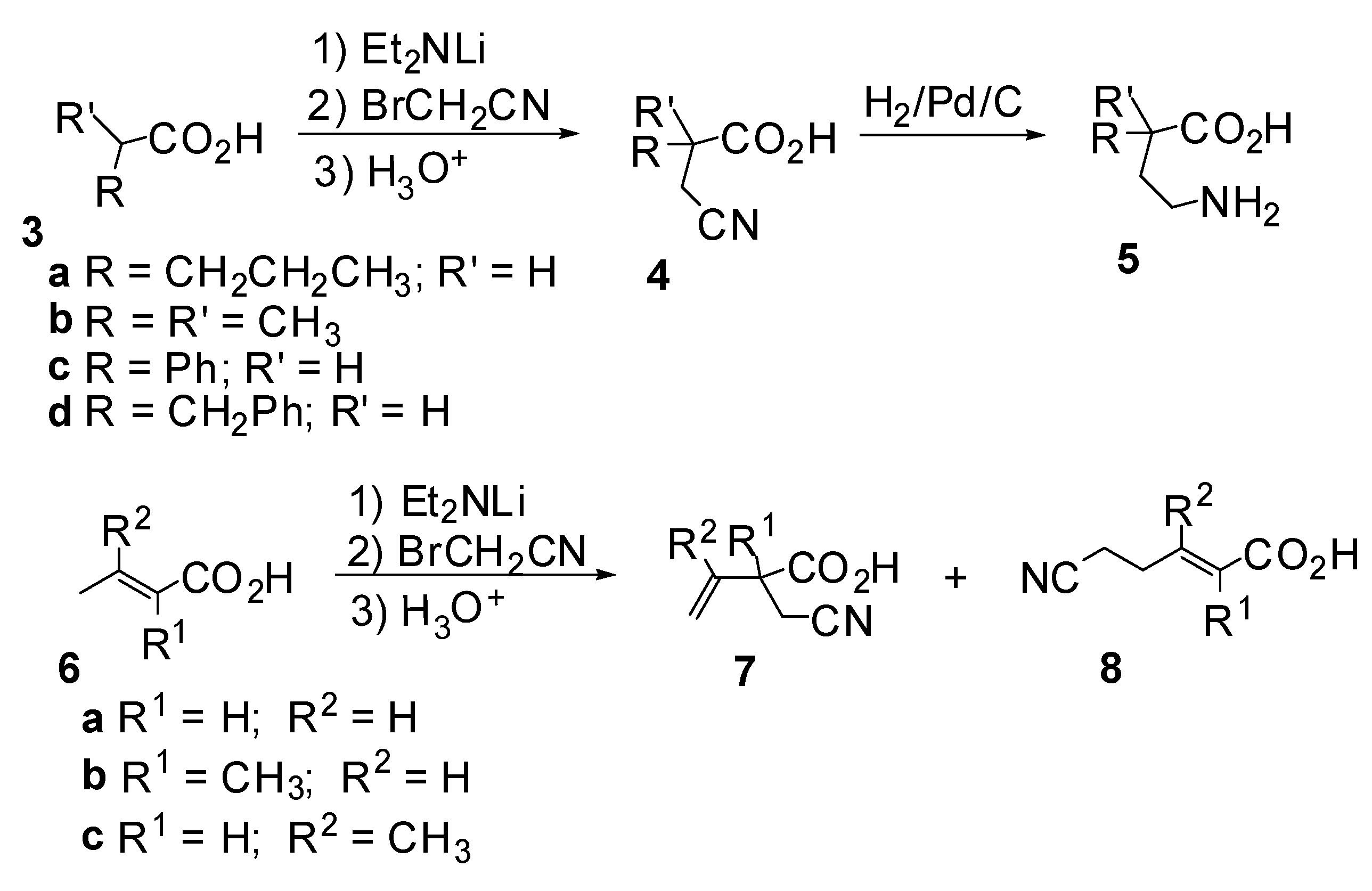
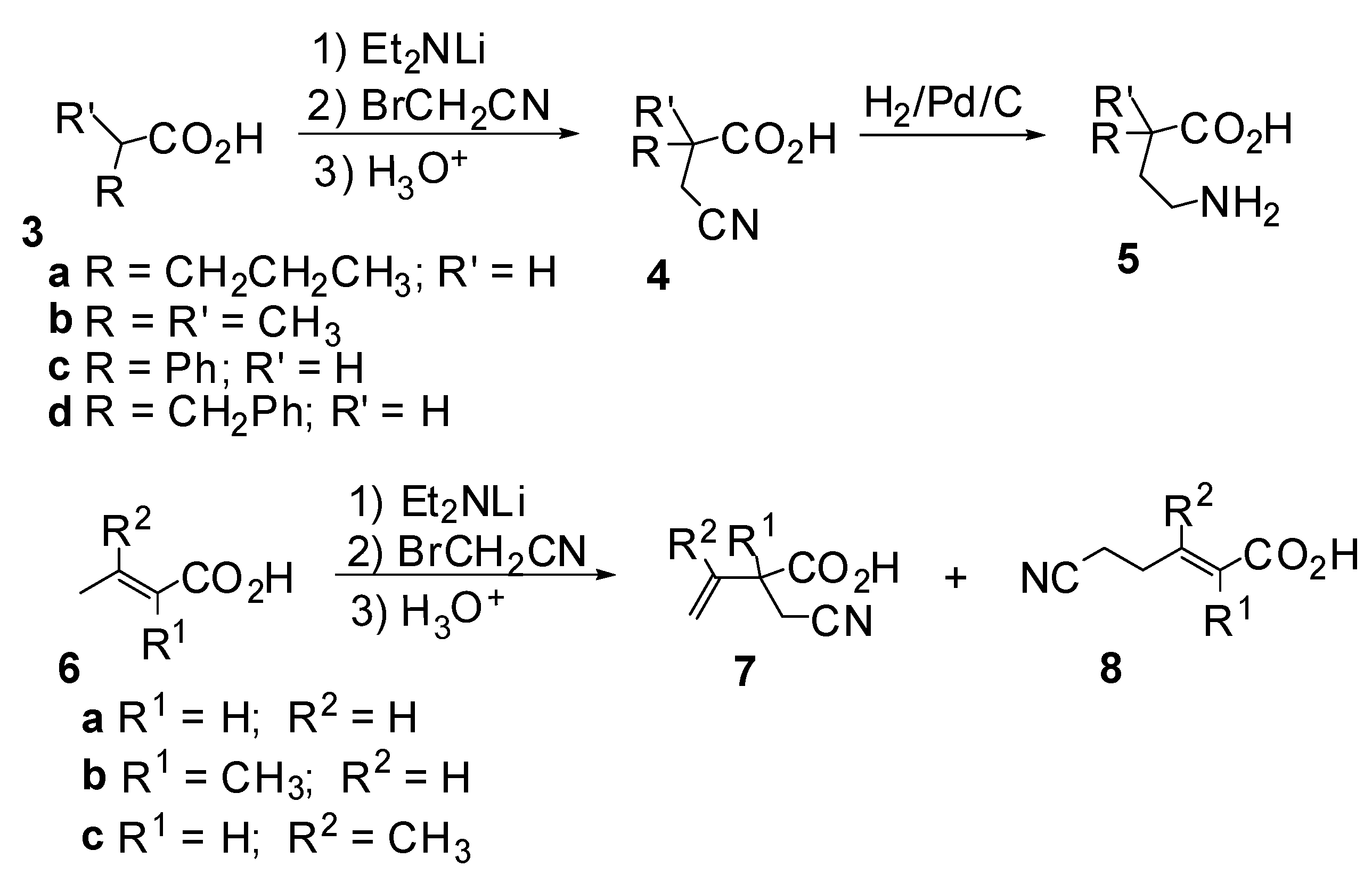
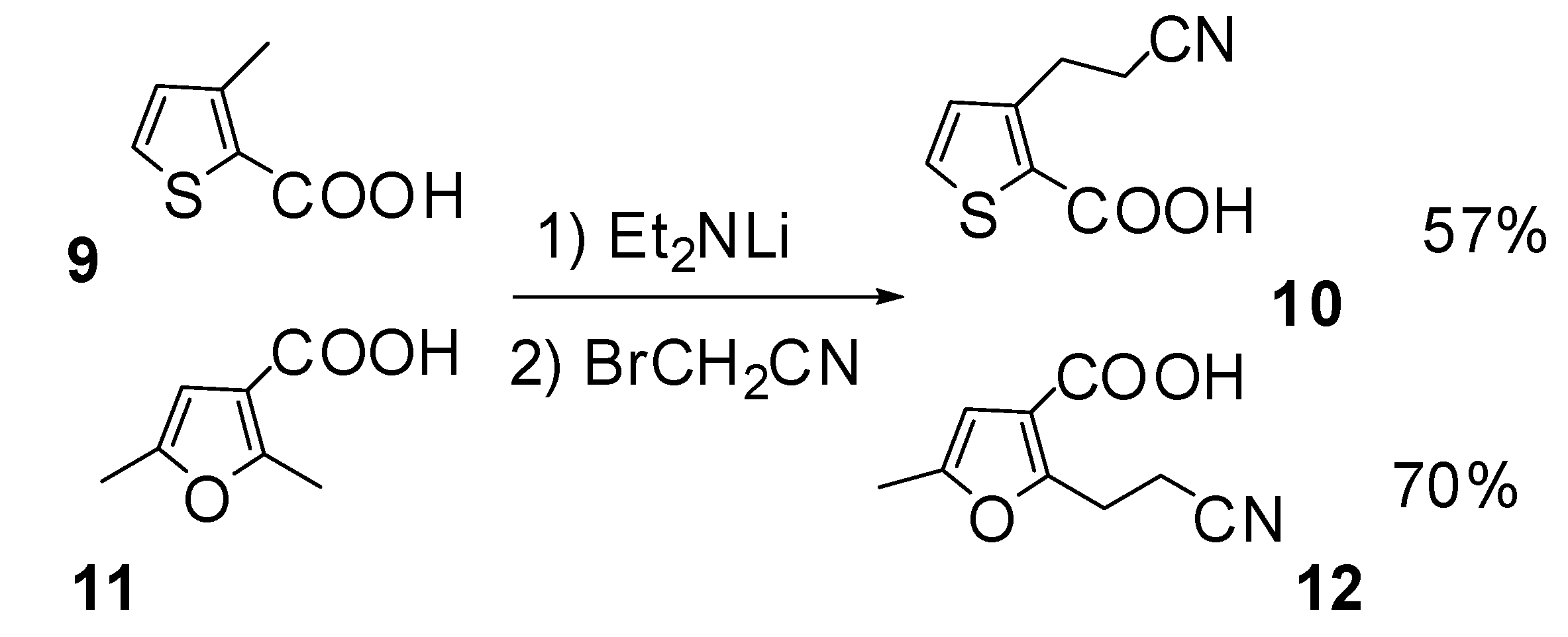
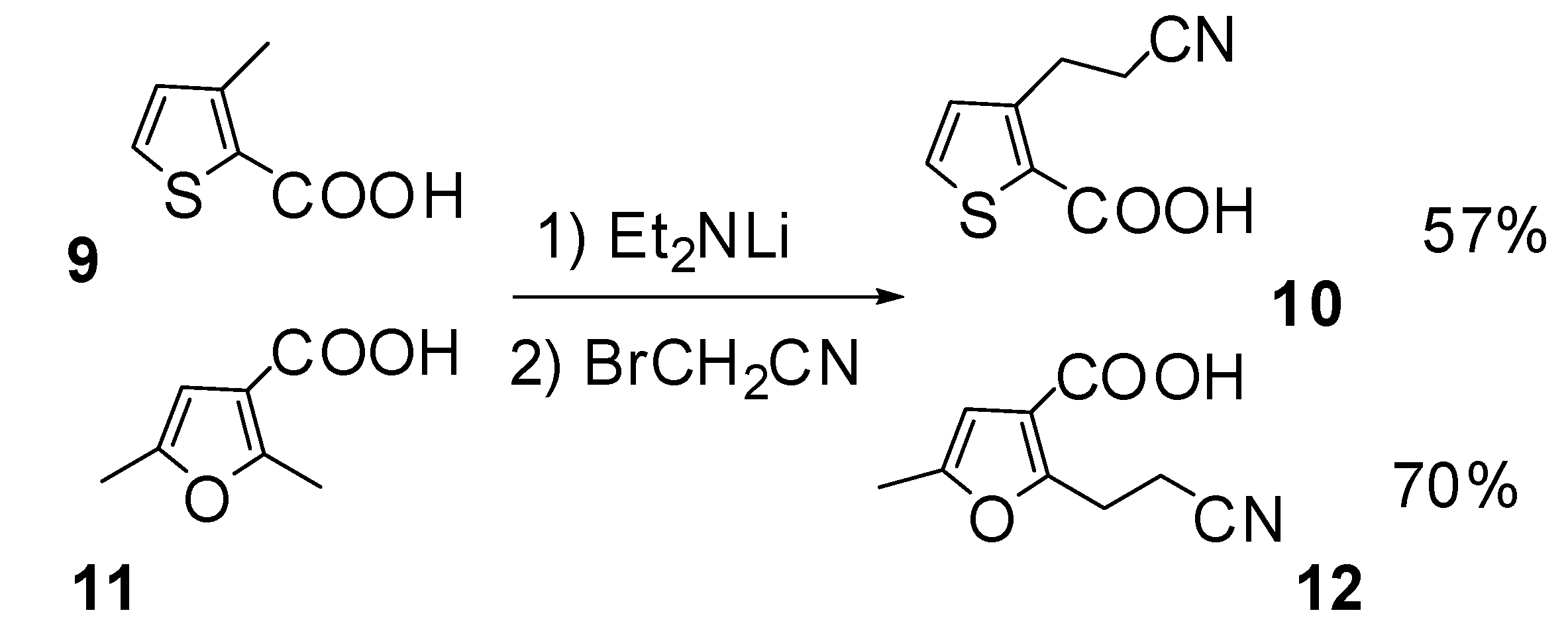
Results and Discussion

{kind=link}
{kind=link}
{kind=link}
| Entry | Acid | Amine | Eq. Amine | Time (h) | Yield (%) | Regioselectivity | |
|---|---|---|---|---|---|---|---|
| α (%) | γ (%) | ||||||
| 1 | 3a | Et2NH | 2 | 24 | 0 | ||
| 2 | 3a | i-Pr2NH | 2 | 24 | 0 | ||
| 3 | 3a | AZA* | 2 | 24 | 0 | ||
| 4 | 3a | Et2NH | 0.5 | 24 | 71 | ||
| 5 | 3a | i-PrCyNH* | 0.5 | 24 | 70 | ||
| 6 | 3b | Et2NH | 0.5 | 24 | 42 | ||
| 7 | 3c | Et2NH | 2 | 24 | 85 | ||
| 8 | 3c | Et2NH | 0.5 | 24 | 97 | ||
| 9 | 3c | i-PrCyNH* | 0.5 | 24 | 98 | ||
| 10 | 3d | Et2NH | 0.5 | 24 | 78 | ||
| 11 | 6a | Et2NH | 0.5 | 24 | 72 | 51** | 49 |
| 12 | 6b | Et2NH | 2 | 24 | 77 | 40 | 60 |
| 13 | 6b | Et2NH | 0.5 | 24 | 84 | 60 | 40 |
| 14 | 6c | Et2NH | 2 | 12 | 43 | 100 | 0 |
| 15 | 6c | Et2NH | 2 | 17 | 67 | 100 | 0 |
| 16 | 6c | Et2NH | 2 | 24 | 69 | 100 | 0 |
| 17 | 6c | Et2NH | 2 | 48 | 39 | 100 | 0 |
| 18 | 6c | Et2NH | 0.5 | 24 | 80 | 100 | 0 |
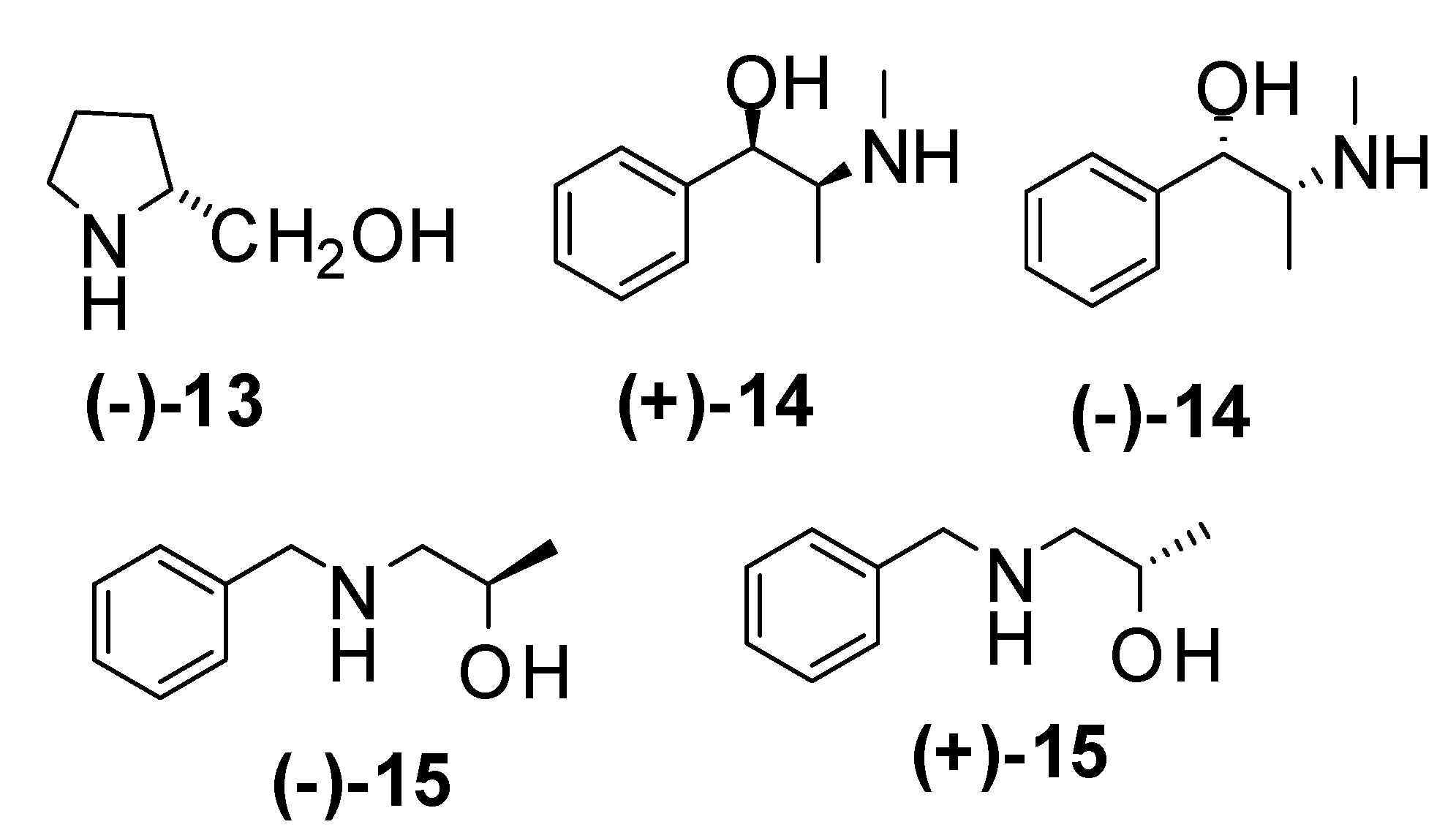
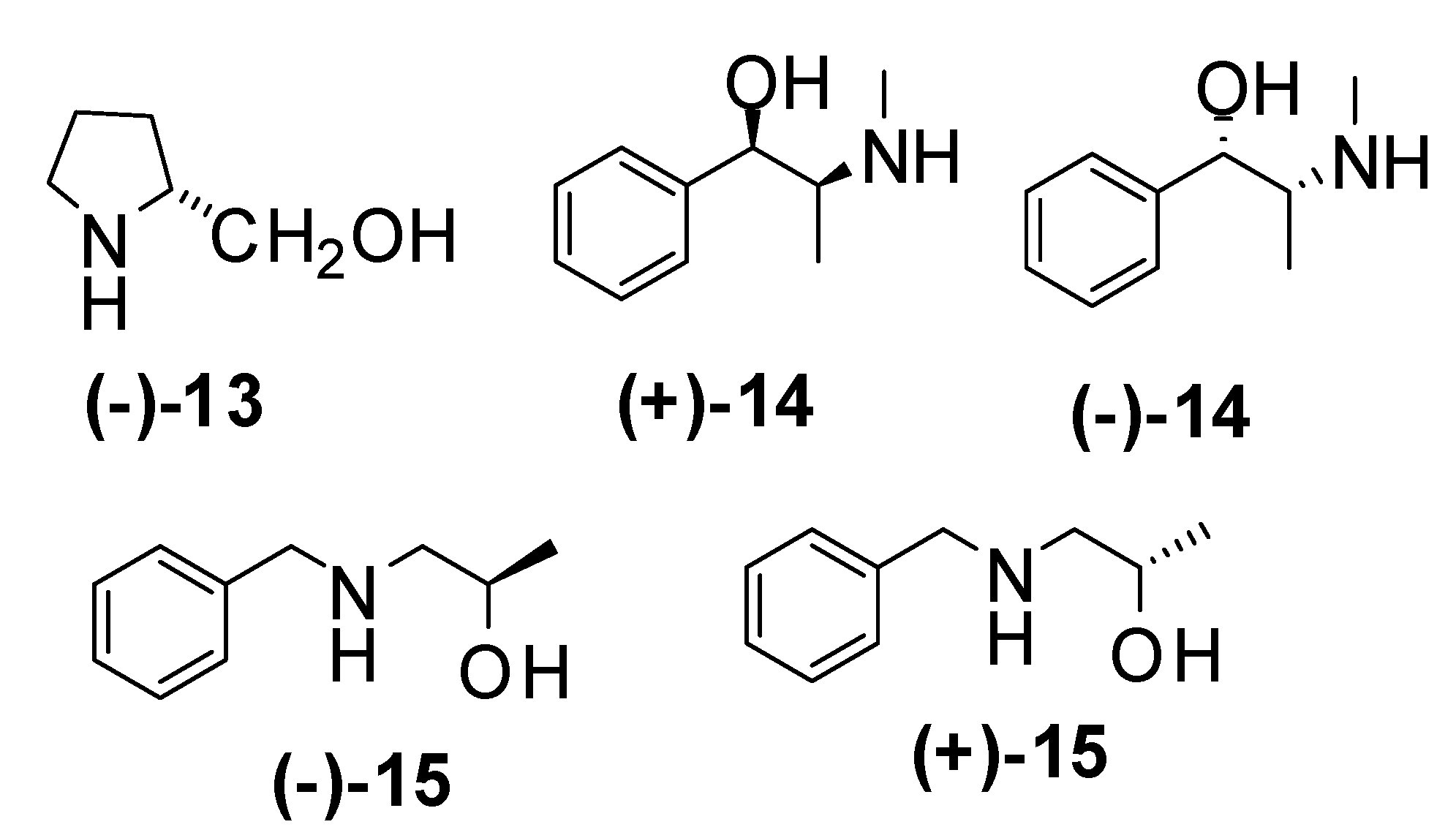
| Entry | Amine | Aditive | time/temp. h / ºC | Yield (%) | e.e. (%) | Major enantiomer |
|---|---|---|---|---|---|---|
| 1 | (-)-13 | 24 / 0 | 0 | |||
| 2 | Et2NH | (-)-13 | 24 / 0 | 0 | ||
| 3 | (+)-14 | 24 / -20 | 95 | 8 | (R)-5c | |
| 4 | (+)-14 | 24 / -78 | 88 | 8 | (R)-5c | |
| 5 | (+)-14 | 3 / -78 | 85* | 10 | (R)-5c | |
| 6 | (-)-14 | 3 / -78 | 73* | 0 | (S)-5c | |
| 7 | (-)-15 | 24 / -20 | 84 | 8 | (S)-5c | |
| 8 | (-)-15 | 24 / -78 | 71* | 6 | (S)-5c | |
| 9 | (+)-15 | 24 / -78 | 76 | 0 | (R)-5c | |
| 10 | (+)-14 | LiCl | 3 / -78 | 76* | 6 | (R)-5c |
| 11 | (+)-14 | LiBr | 3 / -78 | 78* | 7 | (R)-5c |
| 12 | (+)-14 | LiF | 3 / -78 | 75* | 6 | (R)-5c |
Conclusions
Experimental
General
General procedure for the synthesis of β-cyanoacids
Acknowledgements
References and Notes
- (a) McGeer, P. L.; McGeer, E. G. Basic Neurochemistry, 4th ed; Seigel, G., Agranoff, B., Albers R., W., Molinof, P., Eds.; Raven: New York, 1989. [Google Scholar] (b) Hansen, J. J.; Krogsgaardlarsen, P. Structural, Conformational and Sterochemical requirements of Central Excitatory Amino-Acid Receptors. Med. Res. Rev. 1990, 10, 55–94. [Google Scholar]
- (a) Lindquist, C. E. L.; Birnir, B. Graded response to GABA by native extrasynaptic GABA(A) receptors. J. Neurochem. 2006, 97, 1349–1356. [Google Scholar] [CrossRef] (b) Sinclair, J.; Granfeldt, D.; Pihl, J.; Millingen, M.; Lincoln, P.; Farre, C.; Peterson, L.; Orwar, O. A biohybrid dynamic random access memory. J. Am. Chem. Soc. 2006, 128, 5109–5113. [Google Scholar]
- Suman-Chauhman, N.; Webdale, L.; Hill, D. R.; Woodruff, G. N. Characterization of [3H]Gabapentin Binding to a Novel Site in Rat-Brain – Homogenate Binding-Studies. Eur. J. Pharmacol. Mol. Pharmacol. Sect. 1993, 244, 293–301. [Google Scholar] [CrossRef]
- See for example: (a) Burgos-Lepley, C. E.; Thompson, L. R.; Kneen, C. O.; Osborne, S. A.; Bryans, J. S.; Capiris, T.; Suman-Chauhan, N.; Dooley, D. J.; Donovan, C. M.; Field, M. J.; Vartanian, M. G.; Kinsora, J. J.; Lotarski, S. M.; El-Kattan, A.; Walters, K.; Cherukury, M.; Taylor, C. P.; Wustrow, D. J.; Schwarz, J. B. Carboylate bioisosteres of Gabapentin. Bioorg. Med. Chem. Lett. 2006, 16, 2333–2336. [Google Scholar] [CrossRef] (b) Denis, J-N.; Tchertchian, S.; Tomassini, A.; Vallée, Y. The reaction of propiolate acetylides with nitrones. Synthesis of γ-amino-α,β-ethyleninc acid derivatives. Tetrahedron Lett. 1997, 38, 5503–5506. [Google Scholar] (c) Azam, S.; D’Souza, A. A.; Wyatt, P. B. J. Enantioselective synthesis of 2-substituted-4-aminobutanoic acid (GABA) analogues via cyanomethyl methylation of chiral enolates. J. Chem. Soc. Perkin Trans. I 1996, 621–627. [Google Scholar] (d) Deng, J.; Duan, Z.C.; Huang, J.D.; Hu, X.P.; Wang, D.Y.; Yu, S.B.; Xu, X.F.; Zheng, Z. Rh-catalyzed asymmetric hydrogenation of gamma-phthalimido-substituted alpha,beta-unsaturated carboxylic acid esters: An efficient enantioselective synthesis of beta-aryl-gamma-amino acids. Org. Lett. 2007, 9, 4825–4828. [Google Scholar] (e) Winkler, M.; Knall, A.C.; Kulterer, M.R.; Klempier, N. Nitrilases catalyze key step to conformationally constrained GABA analogous gamma-amino acids in high optical purity. J. Org Chem. 2007, 72, 7423–7426. [Google Scholar] (f) Ordonez, M.; Cativiela, C.; Kulterer, M.R.; Klempier, N. Stereoselective synthesis of gamma-amino acids. Tetrahedron: Asymmetry 2007, 18, 3–99. [Google Scholar]
- See for example: (a) Martin, C. J.; Rawson, D. J.; Williams, J. M. J. The preparation of enenatiomerically enriched γ-aminoacids (GABAs) using palladium catalyzed allylic substitution. Tetrahedron: Asymmetry 1998, 9, 3723–3730. [Google Scholar] (b) Dryanska, V.; Pashkuleva, I. A simple and efficient synthesis of gamma-aminobutyric acid (GABA). Org. Prep. Proced. Int. 1999, 31, 232–236. [Google Scholar] (c) Dryanska, V.; Pashkuleva, I.; Angelov, V. A convenient synthesis of threo-4-amino-3, 4-diphenylbutanoic acid and its derivatives. J. Chem. Res.-S 2003, 89–90. [Google Scholar] (d) Kohler, F.; Gais, H.J.; Raabe, G. Asymmetric synthesis of highly substituted gamma-amino acids from allyltitanium sulfoximines. Org. Lett. 2007, 9, 1231–1234. [Google Scholar]
- (a) Calmès, M.; Escale, F.; Martinez, J. Synthesis of N-Boc-(R)-α-phenyl-γ-aminobutyric acid using and in situ diastereoselective protonation strategy. Tetrahedron: Asymmetry 2002, 13, 293–296. [Google Scholar] [CrossRef] (b) Camps, P.; Muñoz-Torrero, D.; Sánchez, L. Stereoselective synthesis of both enantiomers of N-Boc- α-phenyl-γ-aminobutyric acids. Tetrahedron: Asymmetry 2004, 15, 311–321. [Google Scholar] (c) Bergner, I.; Opatz, T. alpha-Aminonitriles and alpha-(alkylideneamino)nitriles can serve as readily available alpha-aminocarbanion equivalents. Their conjugate addition to alpha,beta-unsaturated esters followed by reduction furnishes polysubstituted gamma-amino acid esters in moderate to high yield. Synthesis 2007, 918–928. [Google Scholar] (d) Amruta Reddy, P.; Hsiang, B.C.H.; Latifi, T.N.; Hill, M.W.; Woodward, K.E.; Rothman, S.M.; Ferrendelli, J.A.; Covey, D.F. 3,3-Dialkyl- and 3-alkyl-3-benzyl-substituted 2-pyrrolidinones: a new class of anticonvulsant agents. J. Med. Chem. 1996, 39, 1898–1906. [Google Scholar]
- (a) Brun, E.M.; Gil, S.; Mestres, R.; Parra, M. Lithium enediolates and dienediolates of carboxylic acids in synthesis: Alkylation with secondary halides. Tetrahedron 1998, 54, 15305–15320. [Google Scholar] [CrossRef] (b) Aurell, M. J.; Gil, S.; Mestres, R.; Parra, M.; Parra, L. Alkylation of lithium dienediolates of butenoic acids. Regioselectivity effects of structure and leaving group of the alkylating agent. Tetrahedron 1998, 54, 4357–4366. [Google Scholar]
- Gil, S.; Parra, M.; Rodriguez, P. A simple synthesis of γ-aminoacid. Tetrahedron Lett. 2007, 48, 3451–3453. [Google Scholar] [CrossRef]
- Brun, E.M.; Gil, S.; Parra, M. Enantioselective alpha-alkylation of unsaturated carboxylic acids using a chiral lithium amide. Tetrahedron: Asymmetry 2001, 12, 915–921. [Google Scholar] [CrossRef]
- (a) Thomson, C. M. Dianion Chemistry in Organic Synthesis; CRC Press: Boca Raton, FL, 1994; pp. 88–129. [Google Scholar] (b) Gil, S.; Parra, M. Dienediolates of carboxylic acids in synthesis. Recent advances. Curr. Org. Chem. 2002, 6, 283–302. [Google Scholar] (c) Gil, S.; Parra, M. Reactivity control of dianions of carboxylic acids. Synthetic applications. Recent Res. Devel. Org. Chem. 2002, 6, 449–481. [Google Scholar]
- Clayden, I. Organolithiums: selectivity for synthesis; Pergamon Press: Oxford, 2002; p. 73. [Google Scholar]
- Juaristi, E.; Beck, A. K.; Hansen, J.; Matt, T.; Mukhopadhyay, T.; Simson, M.; Seebach, D. Enantioselective Aldol and Michael Additions of Achiral Enolates in the presence of chiral lithium amides and amines. Synthesis 1993, 1271–1290. [Google Scholar]
- (a) Streitwieser, A.; Husemann, M.; Kim, Y-J. Aggregation and reactivity of the dilithium and dicesium N-diolates of 1-naphthyl acetic acid. J. Org. Chem. 2003, 68, 7937–7942. [Google Scholar] [CrossRef] (b) Eames, J.; Suggate, M.J. Recent developments in the transfer of chirality within enolate alkylation reactions. Angew. Chem. Int. Ed. 2005, 44, 186–189. [Google Scholar] (c) Sott, R.; Granader, J.; Hilmersson, G. Solvent-dependent mixed complex formation –NMR studies and asymmetric addition reactions of lithiumacetonitrile to benzaldehyde mediated by chiral lithium amides. Chem. Eur. J. 2002, 8, 2081–2087. [Google Scholar] (d) Sotoca, E.; Bouillon, J. P.; Gil, S.; Parra, M.; Portella, C. Reaction of lithium enediolates with perfluoroketene dithioacetals. Synthesis of α-trifluoromethyl γ-dicarboxylic acid derivatives. Tetrahedron 2005, 61, 4395–4402. [Google Scholar]
- (a) Brun, E. M.; Gil, S.; Mestres, R.; Parra, M. Dienediolates of α,β-unsaturated carboxylic acids in synthesis: A new synthetic method to 2-pyridones. Synlett. 1999, 1088–1090. [Google Scholar] (b) Brun, E. M.; Gil, S.; Mestres, R.; Parra, M. A new synthetic method to 2-pyridones. Synthesis 2000, 273–280. [Google Scholar] (c) Brun, E. M.; Gil, S.; Parra, M. New approach to condensed pyrid-2-ones. Arkivoc 2002, X, 80–89. [Google Scholar]
- (a) Brun, E. M.; Casades, I.; Gil, S.; Mestres, R.; Parra, M. New conditions for the generation of dianions of carboxylic acids. Tetrahedron Lett. 1998, 39, 5443–5446. [Google Scholar] [CrossRef] (b) Gil, S.; Torres, M.; Ortúzar, N.; Wincewicz, R.; Parra, M. Eficient addition of acid enediolates to epoxides. Eur. J. Org. Chem. 2004, 2160–2165. [Google Scholar]
- Enders, D; Gröbner, R.; Raaba, G. Enantioselective synthesis of 2-substituted 5-, 6- and 7-membered lactams via α-alkylation of their chiral N-dialkylamino derivatives. Synthesis 1996, 941–948. [Google Scholar] [CrossRef]
- Murakata, M.; Yasukata, T.; Aoki, T.; Nakajima, M.; Koga, K. Stereoselective reactions. 30. Enantioselective alkylation of the lithium enolates of 6-membered cyclic ketones using tetradentated chiral amines in the presence of lithium bromide. Tetrahedron 1998, 54, 2449–2458. [Google Scholar] [CrossRef]
- Domingo, L.R.; Gil, S.; Parra, M.; Saez, J.A.; Torres, M. Experimental and theoretical investigations for the tandem alkvlation-isomerization reactions between unsaturated carboxylic acids and allyl halides. Tetrahedron 2003, 59, 6223–6239. [Google Scholar]
- (a) O’Brien, P. Recent advances in asymmetric synthesis using chiral lithium amide bases. J. Chem. Soc. Perkin Trans I 1998, 1439–1457. [Google Scholar] [CrossRef] (b) Duguet, N.; Harrison-Marchand, A.; Maddamno, J’y; Tomioka, K. Enantioselective conjugate addition of a lithium ester enolate catalyzed by chiral lithium amides. Org. Lett. 2006, 8, 5745–5748. [Google Scholar] (c) Matsubara, H.; Maeda, L.; Sugiyama, H.; Ryn, I. Chiral and achiral lithium amides having a fluorous ponytail: Preparation and evaluation as a recycling reagent for lithium enolate generation. Synthesis 2007, 2901–2912. [Google Scholar]
- Matsuo, J.-i; Koga, K. Enantioselective alpha-alkylation of phenylacetic acid using a chiral bidentate lithium amide as a chiral auxiliary. Chem. Pharm. Bull. 1997, 45, 2122–2124. [Google Scholar] [CrossRef]
- (a) Parra, M.; Sotoca, E.; Gil, S. A convenient generation of acetic acid dianion. Eur. J. Org. Chem. 2003, 1386–1388. [Google Scholar] (b) Newton, R.; Marsden, S. P. Efficient synthesis of quaternary alpha-hydroxy acids by alkylation of alpha-ketoamide-derived dienediolates. Synthesis 2005, 3263–3270. [Google Scholar]
- Sample Availability: Samples of the compounds 4c, 4d, 7c, 7b and 12 are available from the authors.
© 2008 by MDPI (http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
Gil, S.; Parra, M.; Rodríguez, P. An Efficient Synthesis of γ-Aminoacids and Attempts to Drive Its Enantioselectivity. Molecules 2008, 13, 716-728. https://doi.org/10.3390/molecules13040716
Gil S, Parra M, Rodríguez P. An Efficient Synthesis of γ-Aminoacids and Attempts to Drive Its Enantioselectivity. Molecules. 2008; 13(4):716-728. https://doi.org/10.3390/molecules13040716
Chicago/Turabian StyleGil, Salvador, Margarita Parra, and Pablo Rodríguez. 2008. "An Efficient Synthesis of γ-Aminoacids and Attempts to Drive Its Enantioselectivity" Molecules 13, no. 4: 716-728. https://doi.org/10.3390/molecules13040716
APA StyleGil, S., Parra, M., & Rodríguez, P. (2008). An Efficient Synthesis of γ-Aminoacids and Attempts to Drive Its Enantioselectivity. Molecules, 13(4), 716-728. https://doi.org/10.3390/molecules13040716