Simple and Efficient Microwave Assisted N-Alkylation of Isatin

Abstract
:Introduction
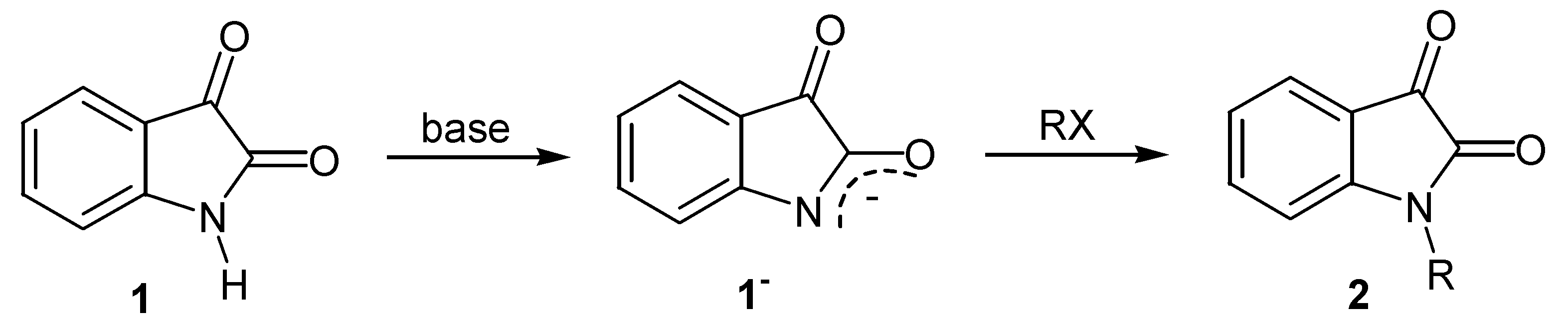

| Comp. | R | Comp. | R |
|---|---|---|---|
| 2a | Me | 2g | CH2CO2Et |
| 2b | Et | 2h | CH(CO2Et)2 |
| 2c | n-Bu | 2i | CH2CONHi-Pr |
| 2d | CH2Ph | 2j | CH2CON(Me)Ph |
| 2e | CH2CH=CHPh | 2k | CH2(CH2)2CO2Et |
| 2f | CH2CH2Br | 2l | CH2COPh |

{kind=link}
{kind=link}
Results and Discussion
| Entry | Cpd. [a] | Reagents | Base | Solvent | Microwave | Conventional heating | |||
| min/watts | Yield(%) | h/ºC | yield (%) | ||||||
| 1 | 2a | 1 | IMe [b] | K2CO3 | DMF | 3/300 | 95 | 1/70 | 80 |
| 2 | 2b | 1 | IEt [b] | K2CO3 | DMF | 3/300 | 90 | 1.30/70 | 78 |
| 3 | 2c | 1 | Br-nBu | K2CO3 | DMF | 3/400 | 90 | 2/70 | 85 |
| 4 | 2c | Na+1- | Br-nBu | DMF | 5/500 | 69 | |||
| 5 | 2d | 1 | ClCH2Ph | K2CO3 | DMF [c] | 5/200 | 96 | 1/120 | 82 |
| 6 | 2d | Na+1- | ClCH2Ph | - | DMF | 5/400 | 66 | ||
| 7 | 2d | Na+1-/Al2O3 | ClCH2Ph | - | - | 4/700 [d] | 62 [e] | ||
| 8 | 2e | 1 | BrCH2CH=CHPh | K2CO3 | DMF [c] | 2/200 | 86 | 4/70 | 62 |
| 9 | 2e | Na+1- | BrCH2CH=CHPh | - | DMF | 3/300 | 67 | ||
| 10 | 2f | 1 | BrCH2CH2Br | K2CO3 | DMF | 2/200 | 50 [f] | 2/70 | 40 [g] |
| 11 | 2f | 1 | BrCH2CH2Br [h] | K2CO3 | DMF | 3/300 | 15 [i] | ||
| 12 | 2g | 1 | ClCH2CO2Et | K2CO3 | DMF [c] | 3/200 | 76 | 2/85 | 68 |
| 13 | 2h | 1 | BrCH(CO2Et)2 | K2CO3 | DMF | 3/200 | 55 [j] | 4/70 | 25 |
| 14 | 2i | 1 | ClCH2CONHi-Pr | K2CO3 | DMF | 4/200 | 86 | 2/90 | 81 |
| 15 | 2i | Na+1- | ClCH2CONHi-Pr | - | DMF | 4/300 | 58 | ||
| 16 | 2i | Na+1-/Al2O3 | ClCH2CONHi-Pr | - | - | 10/400 [d] | 44 [e] | ||
| 17 | 2j | 1 | ClCH2CON(Me)Ph | K2CO3 | NMP [k] | 3/200 | 94 | 2/90 | 83 |
| 18 | 2j | Na+1- | ClCH2CON(Me)Ph | - | DMF | 5/300 | 43 | ||
| 19 | 2k | 1 | Cl(CH2)3CO2Et | K2CO3 | NMP | 4/400 | 56 [l] | 3/120 | 38 |
| 20 | 2k | Na+1- | Cl(CH2)3CO2Et | - | DMF | 6/500 | 28 | ||
| 21 | 2k | Na+1-/Al2O3 | Cl(CH2)3CO2Et | - | - | 8/800 [d] | [m,e] | ||
| 22 | 2l | 1 | BrCH2COPh | K2CO3 | DMF | 7/160 | 53 [n] | 2/70 | 22 [o] |
| 23 | 2l | Na+1- | BrCH2COPh | - | DMF | 4/320 | 62 [p] | ||
Conclusions
Experimental
General
General procedure for synthesis of compounds 2 employing conventional heating
General procedures for synthesis of compounds 2 employing MW irradiation
Method A: Generating the isatin anion (1-) in situ
Method B: Employing preformed isatin sodium salt
Method C: Employing supported reagents
Physical properties of compounds 2
Acknowledgements
References and Notes
- (a) Sumpter, W.C. The chemistry of isatin. Chem. Rev. 1944, 34, 393–434. [Google Scholar] [CrossRef] (b) Popp, F.D. The chemistry of isatin. Adv. Heterocyclic Chem. 1975, 18, 1–58. [Google Scholar] (c) da Silva, J.F.M.; Garden, S.J.; da C. Pinto, A. The chemistry of isatin: a review from 1975 to 1999. J. Braz. Chem. Soc. 2001, 12, 273–324. [Google Scholar]
- Some recent examples: (a) Bursavich, M.G.; Gilbert, A. M.; Lombardi, S.; Georgiadis, K.E.; Reifenberg, E.; Flannery, C. R.; Morris, E.A. 5’-Phenyl-3’H-spiro[indoline-3,2’-[1,3,4]thiadiazol]-2-one inhibitors of ADAMTS-5 (Aggrecanase-2). Bioorg. Med. Chem. Lett. 2007, 17, 5630–5633. [Google Scholar] [CrossRef] (b) Jarrahpour, A.; Khalili, D. Synthesis of some mono- and bis-spiro-β-lactams of benzylisatin. Tetrahedron Lett. 2007, 48, 7140–7143. [Google Scholar] (c) Basavaiah, D.; Reddy, K.R. Simple and One-Pot Protocol for Synthesis of Indene-spiro-oxindoles Involving Tandem Prins and Friedel-Crafts Reactions. Org. Lett. 2007, 9, 57–60. [Google Scholar]
- Vine, K.L.; Locke, J.M.; Ranson, M.; Pyne, S.G.; Bremner, J.B. An Investigation into the Cytotoxicity and Mode of Action of Some Novel N-Alkyl-Substituted Isatins. J. Med. Chem. 2007, 50, 5109–5117. [Google Scholar] [CrossRef] and references cited therein.
- Zhou, L.; Liu, Y.; Zhang, W.; Wei, P.; Huang, C.; Pei, J.; Yuan, Y.; Lai, L. Isatin Compounds as Noncovalent SARS Coronavirus 3C-like Proteasa Inhibitors. J. Med. Chem. 2006, 49, 3440–3443. [Google Scholar] [CrossRef] and references cited therein.
- (a) Chu, W.; Rothfuss, J.; d’Avignon, A.; Zeng, C.; Zhou, D.; Hotchkiss, R.S.; Mach, R.H. Isatin Sulfonamide Analogs Containing a Michael Addition Acceptor: A new Class of Caspase 3/7 Inhibitors. J. Med. Chem. 2007, 50, 3751–3755. [Google Scholar] [CrossRef] and references cited therein; (b) Kopka, K.; Faust, A.; Keul, P.; Wagner, S.; Breiholz, H.-J.; Höltke, C.; Schober, O.; Schäfers, M.; Levkau, B. 5-Pyrolidinylsulfonyl Isatins as a Potential Tool for the Molecular Imagin of Caspases in Apoptosis. J. Med. Chem. 2006, 49, 6704–6715. [Google Scholar] and references cited therein.
- For some examples: (a) Baiocchi, L.; Giannangeli, M.; Rossi, V.; Ambrogi, V.; Grandolini, G.; Perioli, L. Synthesis and antimicrobial activity of some new indolo[2,1-b]quinazolin-6(12H)ones. Il Farmaco 1993, 48, 487–501. [Google Scholar] (b) Meth-Cohn, O.; Goon, S. Synthetic Apllications of Umpoled Vilsmeier Reagents. A new Simple One-Pot Route to Isatins from Formanilides. Tetrahedron Lett. 1996, 37, 9381–9384. [Google Scholar]
- Casey, L. A.; Galt, R.; Page, M. I. The Mechanisms of Hydrolysis of the β-Lactam Isatin and its Derivatives. J. Chem. Soc. Perkin Trans. 2 1993, 23–28. [Google Scholar] [CrossRef]
- Garden, S.J.; Torres, J.C.; da Silva, L.E.; Pinto, A.C. A Convenient Methodology for the N-Alkylation of Isatin Compounds. Synth. Commun. 1998, 28, 1679–1689. [Google Scholar] [CrossRef]
- Shuttleworth, S.J.; Nasturica, D.; Gervais, C.; Siddiqui, M.A.; Rando, R.F.; Lee, N. Parallel Synthesis of Isatin-Based Serine Protease Inhibitors. Bioorg. Med. Chem. Lett. 2000, 10, 2501–2504. [Google Scholar] [CrossRef]
- Torisawa et al. developed a mild base combination of CuCO3/Cs2CO3 for N-alkylation of the labile 5-nitroisatin: Torisawa, Y.; Nishi, T.; Minamikawa, J.-I. An Efficient Conversion of 5-Nitroisatin Into 5-Nitroindole Derivative. Bioorg. Med. Chem. Lett. 2001, 11, 829–832. [Google Scholar] [CrossRef]
- Gedye, R.; Smith, F.; Westaway, K.; Ali, H.; Baldisera, L.; Laberge, L.; Rousell, J. The Use of Microwave Ovens for Rapid Organic Synthesis. Tetrahedron Lett. 1986, 27, 279–282. [Google Scholar] [CrossRef]
- Giguere, R. J.; Bray, T. L.; Duncan, S. M.; Majetich, G. Application of Commercial Microwave ovens to Organic Synthesis. Tetrahedron Lett. 1986, 27, 4945–4948. [Google Scholar] [CrossRef]
- Relevant reviews of microwave assisted reactions: (a) Lidström, P.; Tierney, J.; Wathey, B.; Westman, J. Microwave assisted organic synthesis –a review. Tetrahedron 2001, 57, 9225–9283. [Google Scholar] (b) Kappe, C.O. Controlled Microwave Heating in Modern Organic Synthesis. Angew. Chem. Int. Ed. 2004, 43, 6250–6284. [Google Scholar] (c) Bose, A.K.; Manhas, M.S.; Ganguly, S.N.; Sharma, A.H.; Banik, B.K. MORE Chemistry for Less Pollution: Applications for Process Development. Synthesis 2002, 1578–1591. [Google Scholar] (d) Xu, Y.; Guo, Q.-X. Synthesis of Heterocyclic Compounds under Microwave Irradiation. Heterocycles 2004, 63, 903–974. [Google Scholar]
- N-Benzylation of isatin under MW irradiation employing the salt of isatin generated from isatin and K2CO3 in water and irradiating at high power to eliminate the solvent was reported in 2004 [15]. However, our attempts to reproduce this reaction failed, and a change of the red isatin solution to yellow as the result of the ring cleavage in aqueous basic media was observed [7].
- El Ashry, E.S.H.; Ramadan, E.S.; Abdel Hamid, H.M.; Hagar, M. Microwave Irradiation for Acceleration each Step for the Synthesis of 1,2,4-Triazino[5,6b]indole-3-thiols and their derivatives from Isatin and 5-Chloroisatin. Synlett 2004, 723–725. [Google Scholar]
- Solvent-free reactions represent eco-friendly approaches, recognized by their simplicity, manipulative ease of the operation, increased safety and economic advantages due to the absence of solvent. Some reviews: (a) Varma, R.S. Solvent-free Accelerated Organic Syntheses using Microwaves. Pure Appl. Chem. 2001, 73, 193–198. [Google Scholar] (b) Bougrin, K.; Loupy, A.; Soufiaoui, M. Microwave Assisted Solvent-free Heterocyclic Synthesis. J. Photochem. Photobiol. C: Photochem. Rev. 2005, 6, 139–167. [Google Scholar] [CrossRef]
- Vidal, T.; Petit, A.; Loupy, A.; Gedye, R.N. Re-examination of Microwave-Induced Synthesis of Phthalimides. Tetrahedron 2000, 56, 5473–5478. [Google Scholar] [CrossRef]
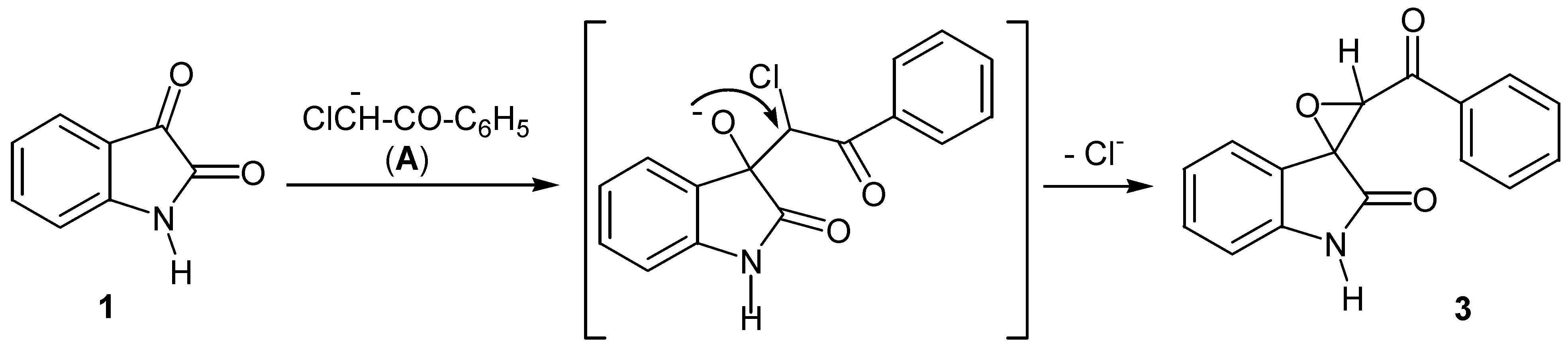
- Epoxide formation in the reaction of isatin with phenacyl halides in basic medium is the result of the presence of an acidic α-proton in the alkylating agent. This is a well-known reaction, producing the epoxide as the main product under certain conditions: (a) Ainley, A.D.; Robinson, R. The Epindoline Group. Part I. Trial of Various Methods for the Synthesis of Epindolidiones. J. Chem. Soc. 1934, 1508–1520. [Google Scholar] [CrossRef] (b) Black, D.S.C.; Wong, L.C.H. A Simple Synthesis of-2-Acyl Indoles from Isatins. J. Chem. Soc., Chem. Comun. 1980, 200. [Google Scholar]
- Varma, R. S.; Dahiya, R. An Expeditious and Solvent Free Synthesis of 2-Amino-Substituted Isoflav-3-enes Using Microwave Irradiation. J. Org. Chem. 1998, 63, 8038–8041. [Google Scholar] [CrossRef]
- Kingston, H.M.; Haswell, S. J. Microwave –Enhanced Chemistry; American Chemical Society: Washington DC, 1997; p. 25. [Google Scholar]
- Esmaili, A.E.; Bodaghi, A. New and efficient one-pot synthesis of functionalized-spirolactones mediated by vinyltriphenylphosphonium salts. Tetrahedron 2003, 59, 1169–1171. [Google Scholar] [CrossRef]
- Bauer, D.J.; Sadler, P.W. 1-Substituted Isatin-thiosemicarbazones, their Preparation and Pharmaceutical Preparations Containing them. Brit. Pat. 1964, 975357, [Chem. Abstr. 1965, 62, 6462c]. [Google Scholar]
- Majumdar, K.C.; Kundu, A. K.; Chatterjee, P. 1-Alkylisatins via Aldol-Retro-aldol Condensation. J. Chem. Res. (S) 1996, 460–461. [Google Scholar]
- Brittain, D. R.; Wood, R. Pharmaceutical Spiro-hydantoin Derivatives. Eur. Pat. Appl.EP 66378. [Chem. Abstr. 1983, 98, 179379d].
- Muthusamy, S.; Arulananda Babu, S.; Nethaji, M. A Facile Regioselective Construction of Spiro epoxi-bridged tetrahydropyranona Frameworks. Tetrahedron 2003, 59, 8117–8127. [Google Scholar] [CrossRef]
- Blanco, M.M.; Dal Maso, M.; Shmidt, M.S.; Perillo, I.A. Reaction of Isatin-1-acetamides with Alkoxides: Synthesis of Novel 1,4-Dihydro-3-hydroxy-4-oxo-2-quinolinecarboxamides. Synthesis 2007, 829–834. [Google Scholar]
- Rekhter, M.A. Direct N-Alkylation of Isatin by Halomethyl Ketones. Chem. Heterocycl. Comp. 2005, 41, 1119–1120. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds are available from the authors.
© 2008 by MDPI(http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
Shmidt, M.S.; Reverdito, A.M.; Kremenchuzky, L.; Perillo, I.A.; Blanco, M.M. Simple and Efficient Microwave Assisted N-Alkylation of Isatin. Molecules 2008, 13, 831-840. https://doi.org/10.3390/molecules13040831
Shmidt MS, Reverdito AM, Kremenchuzky L, Perillo IA, Blanco MM. Simple and Efficient Microwave Assisted N-Alkylation of Isatin. Molecules. 2008; 13(4):831-840. https://doi.org/10.3390/molecules13040831
Chicago/Turabian StyleShmidt, María Sol, Ana María Reverdito, Lautaro Kremenchuzky, Isabel Amalia Perillo, and María Mercedes Blanco. 2008. "Simple and Efficient Microwave Assisted N-Alkylation of Isatin" Molecules 13, no. 4: 831-840. https://doi.org/10.3390/molecules13040831
APA StyleShmidt, M. S., Reverdito, A. M., Kremenchuzky, L., Perillo, I. A., & Blanco, M. M. (2008). Simple and Efficient Microwave Assisted N-Alkylation of Isatin. Molecules, 13(4), 831-840. https://doi.org/10.3390/molecules13040831