Synthesis of 2-(4,6-Dimethoxy-1,3,5-triazin-2-yloxyimino) Derivatives: Application in Solution Peptide Synthesis

Abstract
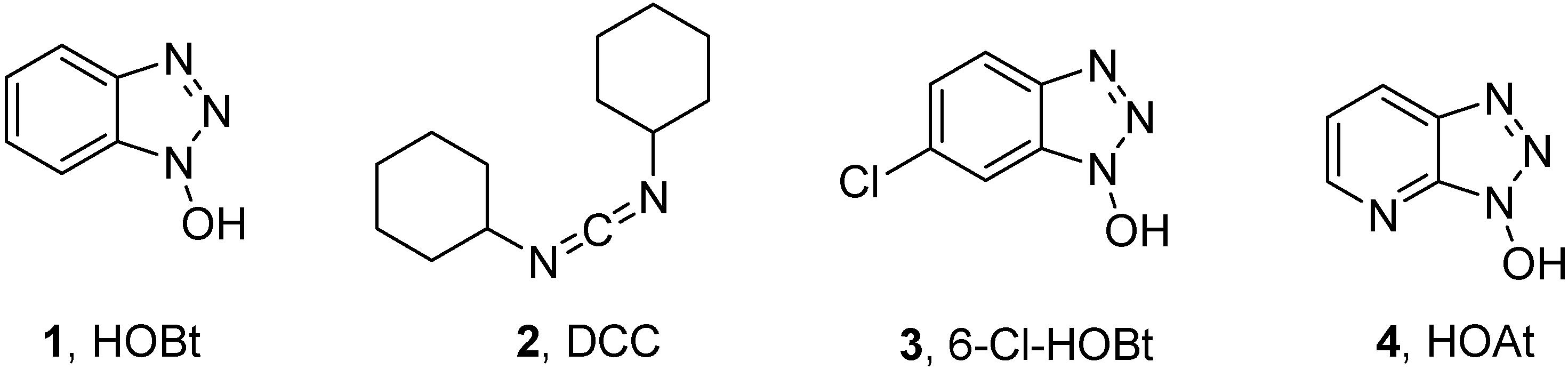
:1. Introduction



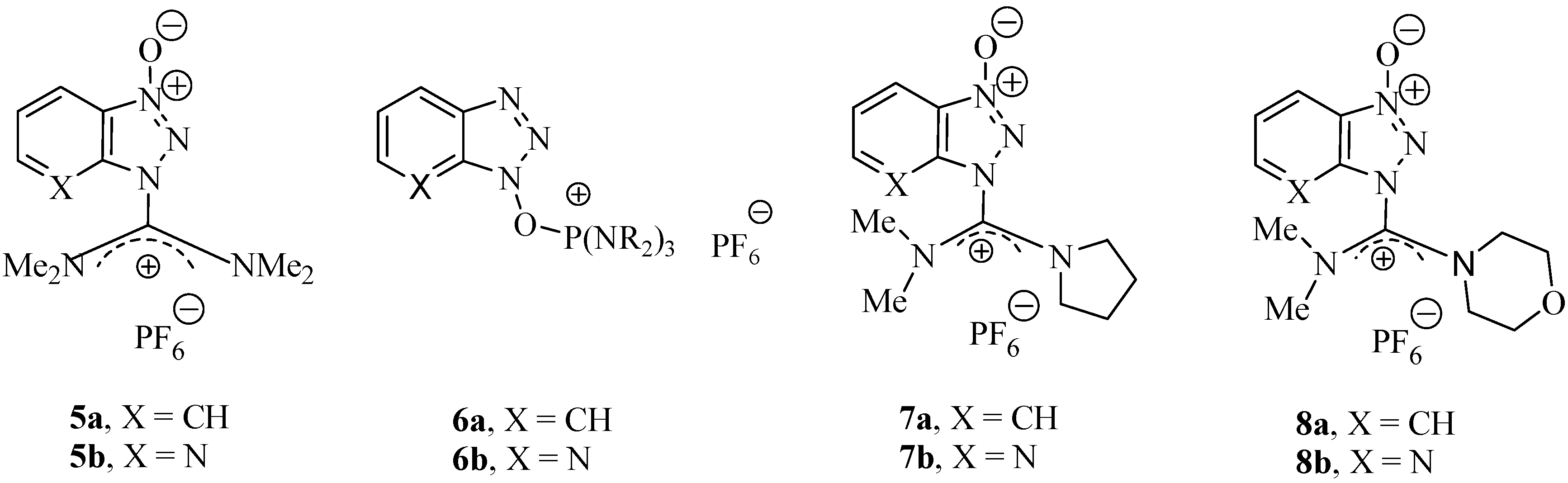
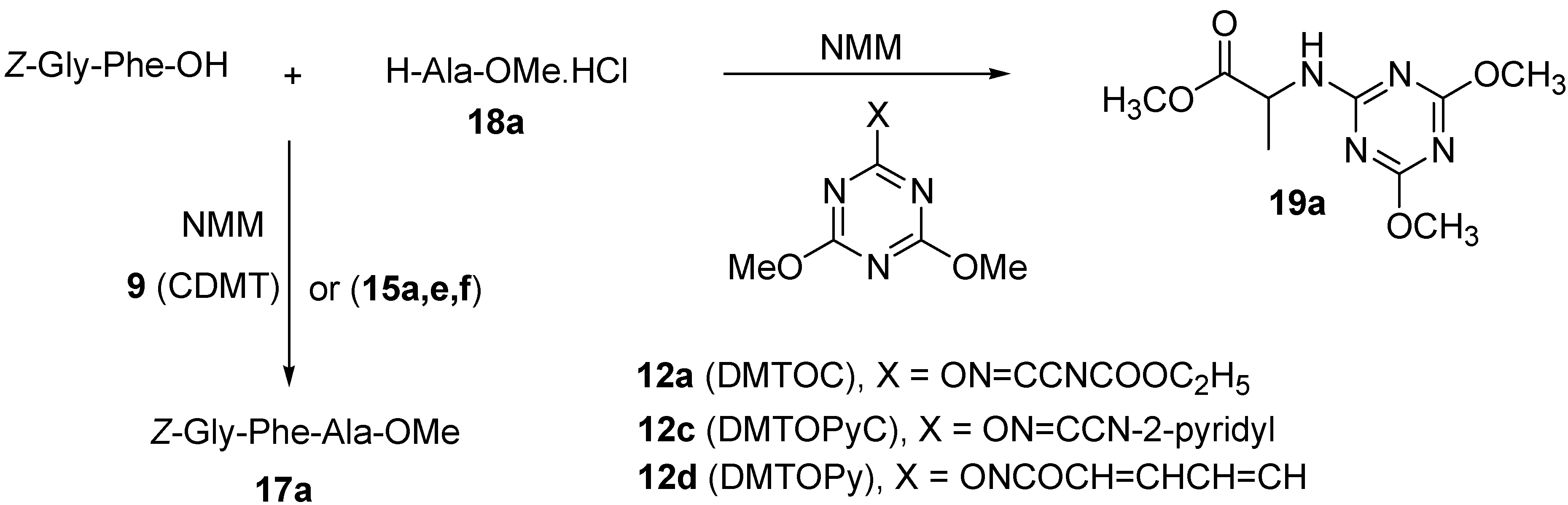
2. Results and Discussion




{kind=link}
{kind=link}
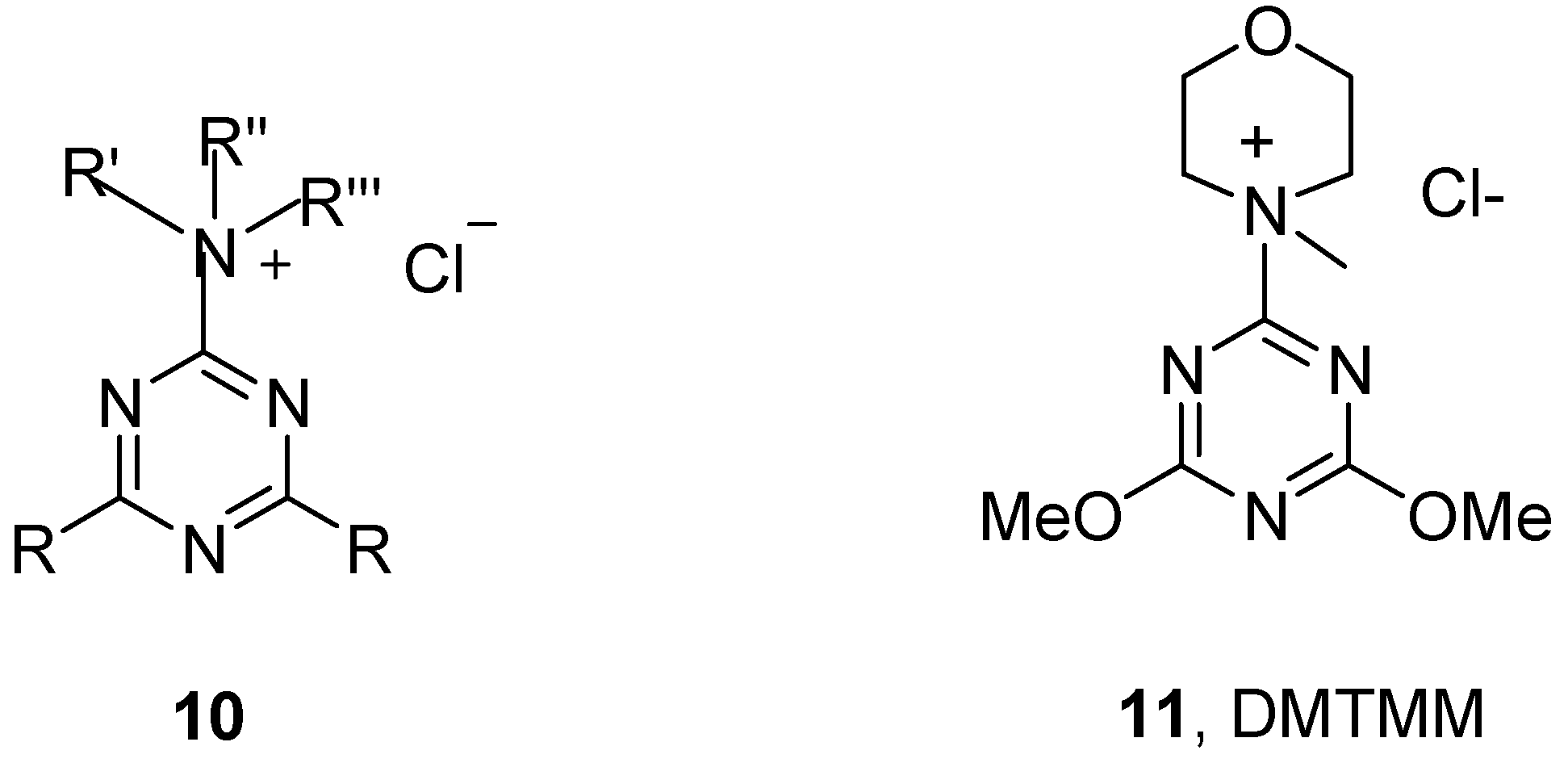{kind=link}
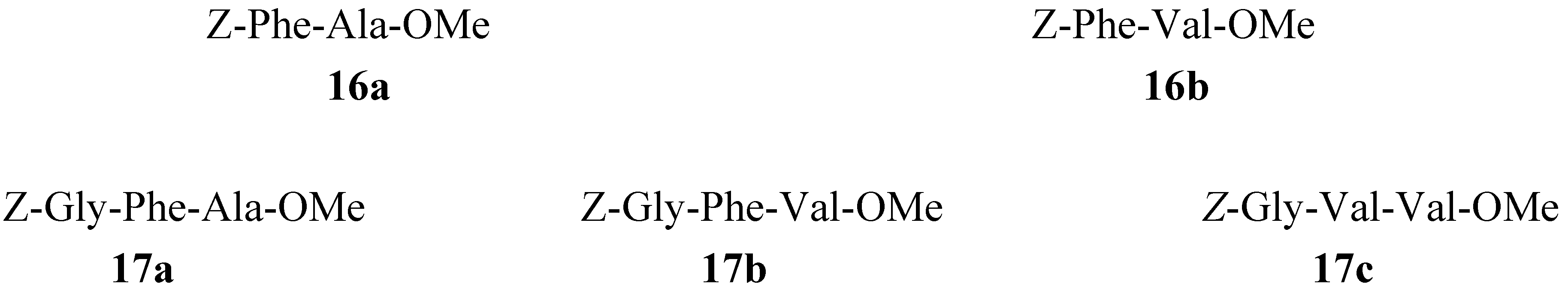{kind=link}
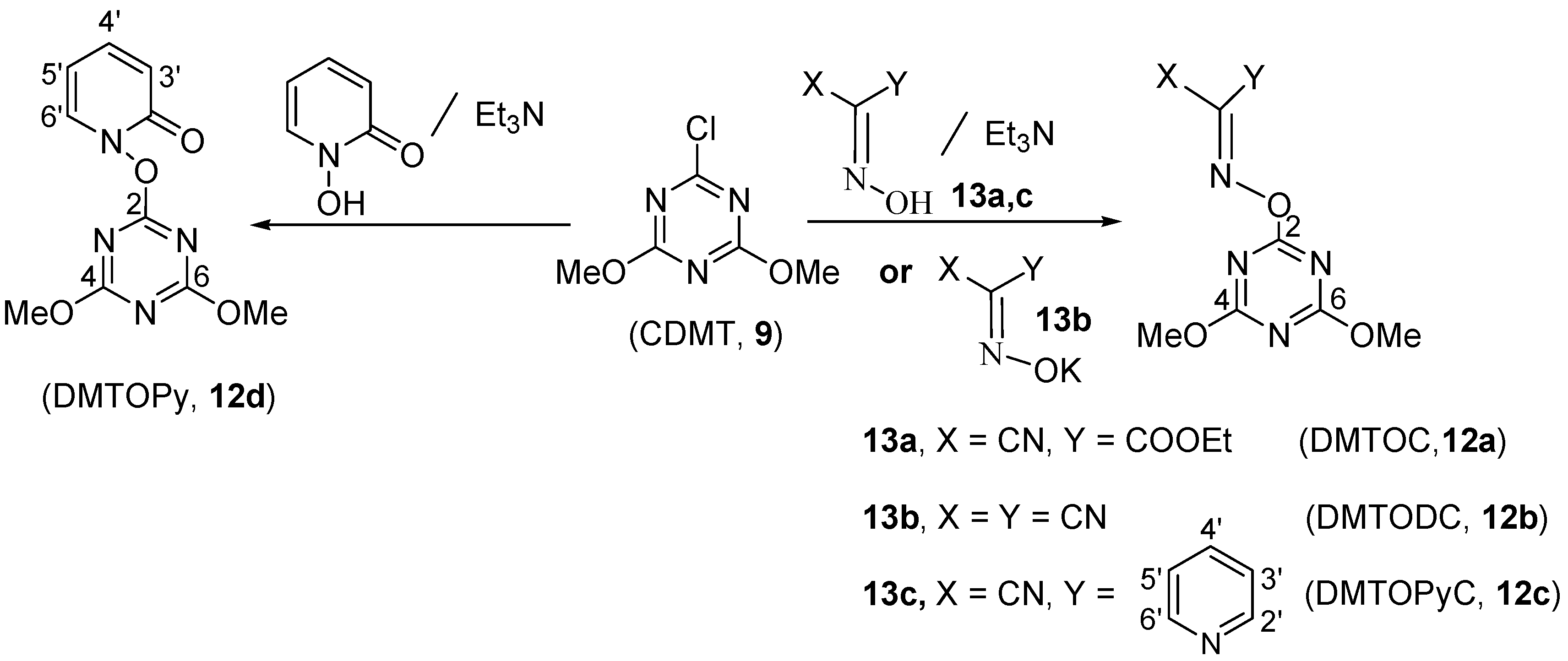{kind=link}
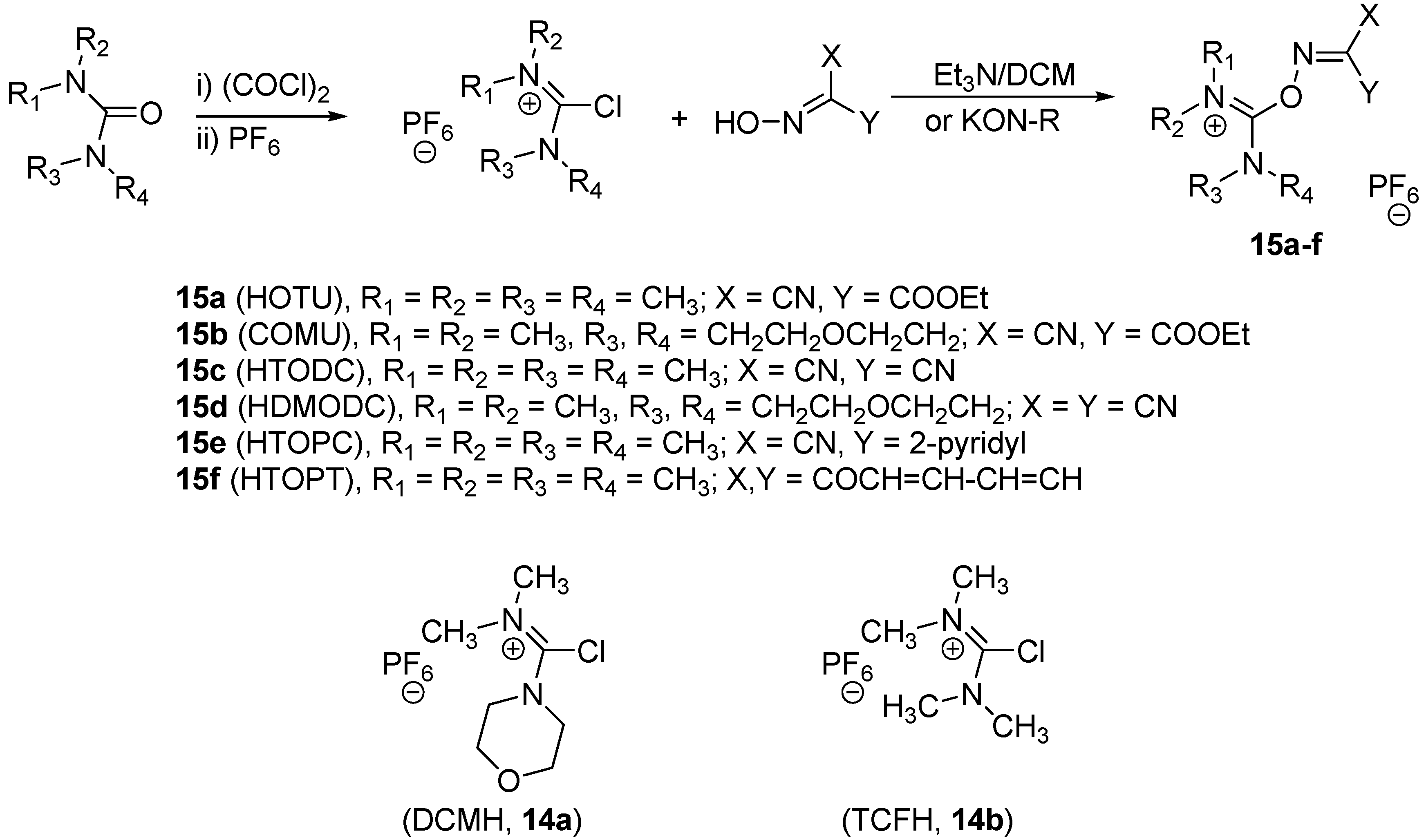{kind=link}
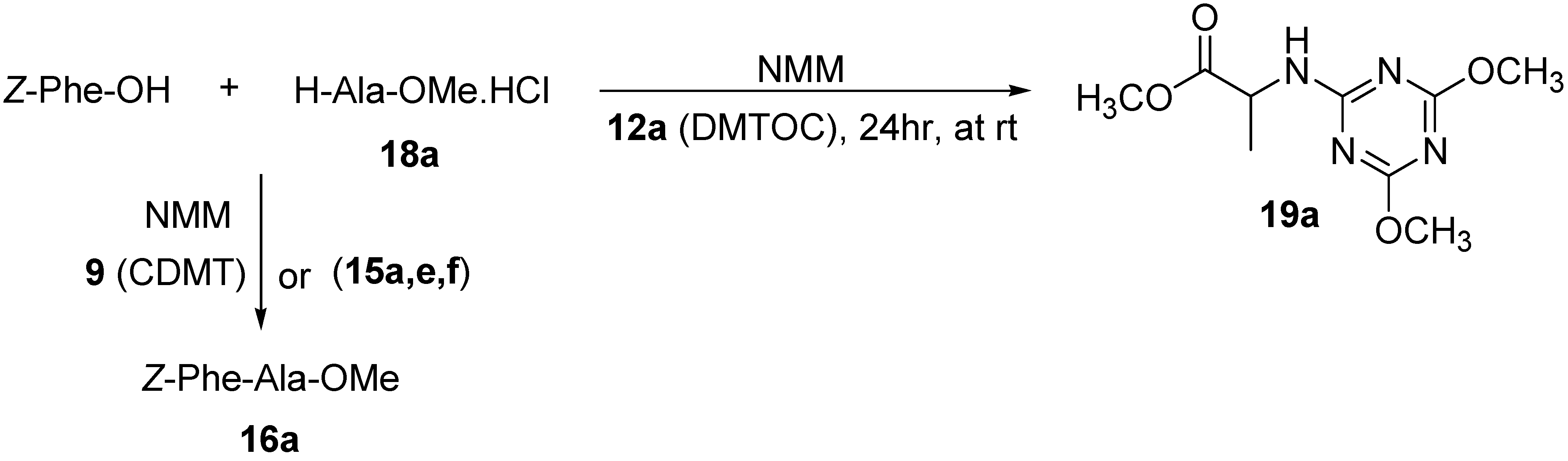{kind=link}
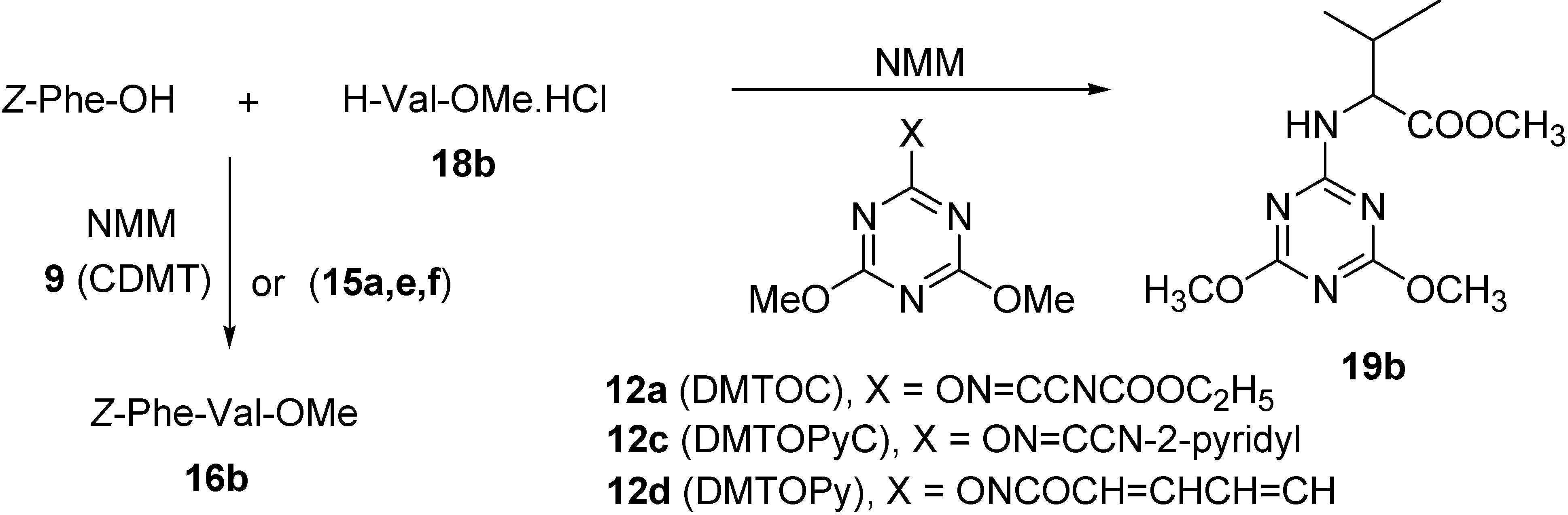{kind=link}
{kind=link}
| Coupling Reagent | Yield % | DL % |
|---|---|---|
| (HOTU, 15a) | 100 | <1 |
| (HTOPC, 15e) | 54 | <1 |
| (HTOPT, 15f) | 66 | <1 |
| (CDMT, 9) | 55 | 15 |


| Coupling Reagent | Yield % | DL % |
|---|---|---|
| (HOTU, 15a) | 83 | <1 |
| (HTOPC, 15e) | 89 | <1 |
| (HTOPT, 15f) | 68 | <1 |
| (CDMT, 9) | 69 | 2.0 |

| Coupling Reagent | Yield % | DL % |
|---|---|---|
| (HOTU, 15a) | 63 | <1 |
| (HTOPC, 15e) | 19 | 19.0 |
| (HTOPT, 15f) | 76 | 12.8 |
| (CDMT, 9) | gummy | 30 |
| Coupling Reagent | Yield % | DL % |
|---|---|---|
| (HOTU, 15a) | 80 | < 0.1 |
| (HTOPC, 15e) | 81 | 26.19 |
| (HTOPT, 15f) | 78 | 6.15 |
| (CDMT, 9) | 63 | 50.0 |
| Coupling Reagent | Yield % | DL % |
|---|---|---|
| (HOTU, 15a) | 81 | < 0.1 |
| (HTOPC, 15e) | ≈100 | 17.5 |
| (HTOPT, 15f) | 95 | 6.66 |
| (CDMT, 9) | 60 | 47.3 |
3. Experimental
3.1. General
3.2. General Synthesis of Triazine Coupling Reagents 12a-d from Oxime Derivatives
3.3. General Synthesis of Oximo-Uronium Type Coupling Reagents 15a-f
3.4. Synthesis of 16a,b (1+1)
3.5. General Method for Synthesis of 17a-c (2+1) Using Different Coupling Reagents
4. Conclusions
Acknowledgements
References
- Rich, D.H.; Singh, J. The Peptides: Analysis, Biology; Gross, E., Meienhofer, J., Eds.; Academic Press: New York, NY, USA, 1979; Volume 1, pp. 241–261. [Google Scholar]
- Knorr, R.; Trzeciak, A.; Bannwarth, W.; Gillessen, D. New coupling reagents in peptide chemistry. Tetrahedron Lett. 1989, 30, 1927–1930. [Google Scholar]
- König, W.; Geiger, R. A new method for the synthesis of peptides: activation of the carboxyl group with dicyclohexylcarbodiimide and 3-hydroxy-4-oxo-3,4-dihydro-1,2,3-benzotriazine. Chem. Ber. 1970, 103, 2034–2040. [Google Scholar] [CrossRef]
- König, W.; Geiger, R. A new method for the synthesis of peptides: activation of the carboxyl group with dicyclohexylcarbodiimide using 1-hydroxy-benzotriazoles as additives. Chem. Ber. 1970, 103, 788–789. [Google Scholar] [CrossRef]
- Carpino, L.A. 1-Hydroxy-7-azabenzotriazole. An efficient peptide coupling additive. J. Am. Chem. Soc. 1993, 115, 4397–4398. [Google Scholar] [CrossRef]
- Carpino, L.A.; Imazumi, H.; El-Faham, A.; Ferrer, F.J.; Zhang, C.; Lee, Y.; Foxman, B.M.; Henklein, P.; Hanay, C.; Gge, C.M.; Wenschuh, H.; Klose, J.; Beyermann, M.; Bienert, M. The Uronium/ Guanidinium Peptide Coupling Reagents: Finally the True Uronium Salts. Angew. Chem. Int. Ed. 2002, 41, 441–445. [Google Scholar]
- Albericio, F.; Bofill, J.M.; El-Faham, A.; Kates, S.A. Use of Onium Salt-Based Coupling Reagents in Peptide Synthesis. J. Org. Chem. 1998, 63, 9678–9683. [Google Scholar] [CrossRef]
- Wijkmans, J.C.H.M.; Kruijtzer, J.A.W.; van der Marel, G.A.; van Boom, J.H.; Bloemhoff, W. CF3-NO2-PyBOP: A new and highly efficient coupling reagent for N-methyl amino acids. Tetrahedron Lett. 1995, 36, 4643–4646. [Google Scholar]
- Kehler, J.; Püsch, A.; Dahl, O. Improved Syntheses of 1-Hydroxy-4-nitro-6-trifluoromethyl- benzotriazole and 1-Hydroxy-4,6-dinitrobenzotriazole. Acta Chem. Scan. 1996, 50, 1171–1173. [Google Scholar] [CrossRef]
- Carpino, L.A.; El-Faham, A.; Albericio, F. Racemization Studies During Solid Phase Peptide Synthesis Using Azabenzotriazole-Based Coupling Reagents. Tetrahedron Lett. 1994, 35, 2279–2282. [Google Scholar] [CrossRef]
- Carpino, L.A.; El-Faham, A.; Minor, C.H.; Albericio, F. Advantageous applications of azabenzotriazole (triazolopyridine)-based coupling reagents to solid-phase peptide synthesis. J. Chem. Soc., Chem. Commun 1994, 201–203. [Google Scholar]
- Quibell, M.; Packman, L.C.; Johnson, T. Identification of coupling conditions proceeding with low C-terminal epimerization during the assembly of fully protected backbone-substituted peptide segments. J. Chem. Soc. Perkin. Trans. 1996, 1, 1219–1225. [Google Scholar]
- Carpino, L.A.; El-Faham, A. The Diisopropylcarbodiimide/1-Hydroxy-7-azabenzotriazole System: Segment Coupling and Stepwise Peptide Assembly. Tetrahedron 1999, 55, 6813–6830. [Google Scholar] [CrossRef]
- Carpino, L.A.; Imazumi, H.; Foxman, B.M.; Vela, M.J.; Henklein, P.; El-Faham, A.; Klose, J.; Bienert, M. Comparison of the Effects of 5- and 6-HOAt on Model Peptide Coupling Reactions Relative to the Cases for the 4- and 7-Isomers. Org. Lett. 2000, 2, 2253–2256. [Google Scholar] [CrossRef]
- Angell, Y.M.; Gacia, C.E.; Rich, D.H. Comparative studies of the coupling of N-methylated, sterically hindered amino acids during solid-phase peptide synthesis. Tetrahedron Lett. 1994, 35, 5981–5984. [Google Scholar]
- Gawne, G.; Kenner, G.; Sheppard, R.C. Acyloxyphosphonium salts as acylating agents. Synthesis of peptides. J. Am. Chem. Soc. 1969, 91, 5669–5671. [Google Scholar]
- Castro, B.; Dormoy, J.R. Azidotris(dimethylamino)phosphonium hexafluorophosphate. Excellent reagent for peptide coupling. Bull. Soc. Chim. Fr. 1973, 3359–3361. [Google Scholar]
- Castro, B.; Dormoy, J.R.; Evin, G.; Selve, C. Peptide coupling reagents. IV. N-[Oxytris(dimethylamino)phosphonium]benzotriazole hexafluorophosphate. Tetrahedron Lett. 1975, 14, 1219–1222. [Google Scholar]
- Castro, B.; Dormoy, J.R.; Evin, G.; Selve, C. Peptide coupling reagents. Part VII. Mechanism of the formation of active esters of hydroxybenzotriazole in the reaction of carboxylate ions on the BOP reagent for peptide coupling. A comparison with Itoh's reagent. J. Chem. Res. (S) 1977, 182–185. [Google Scholar]
- Coste, J.; Le-Nguyen, D.; Castro, B. PyBOP: A new peptide coupling reagent devoid of toxic by-product. Tetrahedron Lett. 1990, 31, 205–208. [Google Scholar]
- Dykstra, R.R. Encyclopedia of Reagents for Organic Synthesis; Paquette, L.A., Ed.; John Wiley & Sons Ltd.: Chichester, UK, 1995; Volume 4, pp. 2668–2675. [Google Scholar]
- Carpino, L.A.; El-Faham, A. Effect of Tertiary Bases on O-Benzotriazolyluronium Salt-Induced Peptide Segment Coupling. J. Org. Chem. 1994, 59, 695–698. [Google Scholar] [CrossRef]
- Carpino, L.A.; Ionescu, D.; El-Faham, A. Peptide Coupling in the Presence of Highly Hindered Tertiary Amines. J. Org. Chem. 1996, 61, 2460–2465. [Google Scholar] [CrossRef]
- Kates, S.A.; Diekmann, E.; El-Faham, A.; Herman, L.W.; Ionescu, D.; McGuinness, B.F.; Triolo, S.A.; Albericio, F.; Carpino, L.A. Techniques in Protein Chemistry VII; Marshak, D.R., Ed.; Academic Press: New York, NY, USA, 1996; pp. 515–523. [Google Scholar]
- Ehrlich, A.; Heyne, H.U.; Winter, R.; Beyermann, M.; Haber, H.; Carpino, L.A.; Bienert, M. Cyclization of all-L-Pentapeptides by Means of 1-Hydroxy-7-azabenzotriazole-Derived Uronium and Phosphonium Reagents. J. Org. Chem. 1996, 61, 8831–8838. [Google Scholar] [CrossRef]
- Jou, G.; Gozalez, I.; Albericio, F.; Lloyd, P.W.; Giralt, E. Total Synthesis of Dehydrodidemnin B. Use of Uronium and Phosphonium Salt Coupling Reagents in Peptide Synthesis in Solution. J. Org. Chem. 1997, 62, 354–366. [Google Scholar] [CrossRef]
- Han, Y.; Albericio, F.; Barany, G. Occurrence and Minimization of Cysteine Racemization during Stepwise Solid-Phase Peptide Synthesis. J. Org. Chem. 1997, 62, 4307–4312. [Google Scholar] [CrossRef]
- Albericio, F.; Cases, M.; Alsina, J.; Triolo, S.A.; Carpino, L.A.; Kates, S.A. On the use of PyAOP, a phosphonium salt derived from HOAt, in solid-phase peptide synthesis. Tetrahedron Lett. 1997, 38, 4853–4856. [Google Scholar] [CrossRef]
- Kim, S.; Chang, A.; Ko, Y.K. Benzotriazol-1-yl diethyl phosphate. A new convenient coupling reagent for the synthesis of amides and peptides. Tetrahedron Lett. 1985, 26, 1341–1349. [Google Scholar] [CrossRef]
- Fan, C.X.; Hao, X.L.; Ye, Y.H. A novel organophosphorus compound as a coupling reagent for peptide synthesis. Synth. Commun 1996, 26, 1455–1460. [Google Scholar] [CrossRef]
- Carpino, L.A.; Xia, J.; El-Faham, A. 3-Hydroxy-4-oxo-3,4-dihydro-5-azabenzo-1,2,3-triazene. J. Org. Chem. 2004, 69, 54–61. [Google Scholar] [CrossRef]
- Carpino, L.A.; Xia, J.; Zhang, C.; El-Faham, A. Organophoshorus and Nitro-substituted Sulfone Esters of 1-Hydroxy-7-azabenzotriazole as Highly Efficient Fast-Acting Peptide Coupling Reagents. J. Org. Chem. 2004, 69, 69–71. [Google Scholar]
- Carpino, L.A.; El-Faham, A.; Albericio, F. Efficiency in Peptide Coupling: 1-Hydroxy-7-azabenzotriazole vs. 3,4-Dihydro-3-hydroxy-4-oxo-1,2,3-benzotriazine. J. Org. Chem. 1995, 60, 3561–3564. [Google Scholar] [CrossRef]
- El-Faham, A.; Khattab, Sh.N.; Abdul-Ghani, M.; Albericio, F. Design and Synthesis of New Immonium-Type Coupling Reagents. Eur. J. Org. Chem. 2006, 6, 1563–1573. [Google Scholar]
- El-Faham, A.; Albericio, F. Novel Proton Acceptor Immonium-Type Coupling Reagents: Application in Solution and Solid Phase Peptide Synthesis. Org. Lett. 2007, 9, 4475–4477. [Google Scholar] [CrossRef]
- El-Faham, A.; Albericio, F. Morpholine-Based Immonium and Halogeno amidinium Salts as Coupling Reagents in Peptide Synthesis. J. Org. Chem. 2008, 73, 2731–2737. [Google Scholar] [CrossRef]
- Kamiński, Z.J. Triazine-based condensing reagents. Biopolymers (Peptide Science) 2000, 55, 140–164. [Google Scholar] [CrossRef]
- Kamiński, Z.J.; Paneth, P.; Rudzinski, J. A Study on the Activation of Carboxylic Acids by Means of 2-Chloro-4,6-dimethoxy-1,3,5-triazine and 2-Chloro-4,6-diphenoxy-1,3,5-triazine. J. Org. Chem. 1998, 63, 4248–4255. [Google Scholar] [CrossRef]
- Kunishima, M.; Kawachi, C.; Iwasaki, F.; Terao, K.; Tani, S. Synthesis and characterization of 4-(4,6-dimethoxy-1,3,5-triazin-2-yl)-4-methylmorpholinium chloride. Tetrahedron Lett. 1999, 40, 5327–5330. [Google Scholar] [CrossRef]
- Kunishima, M.; Kawachi, C.; Morita, J.; Hioki, K.; Terao, K.; Tani, S. Formation of carboxamides by direct condensation of carboxylic acids and amines in alcohols using a new alcohol- and water-soluble condensing agent: DMT-MM. Tetrahedron 2001, 57, 1551–1558. [Google Scholar] [CrossRef]
- Kjell, D.P.; Hallberg, D.W.; Kalbfleisch, J.M.; McCurry, C.K.; Semo, M.J.; Sheldo, E.M.; Spitler, J.T.; Wang, M. Determination of the Source of the N-Methyl Impurity in the Synthesis of Pemetrexed Disodium Heptahydrate. Org. Proc. Res. Dev. 2005, 9, 738–742. [Google Scholar] [CrossRef]
- Kunishima, M.; Kawachi, C.; Morita, J.; Terao, K.; Iwasaki, F.; Tani, S. 4-(4,6-dimethoxy-1,3,5-triazin-2-yl)-4-methyl-morpholinium chloride: an efficient condensing agent leading to the formation of amides and esters. Tetrahedron 1999, 55, 13159–13170. [Google Scholar] [CrossRef]
- Tanaka, T.; Noguchi, M.; Kobayashi, A.; Shoda, S.I. A novel glycosyl donor for chemo-enzymatic oligosaccharide synthesis: 4,6-dimethoxy-1,3,5-triazin-2-yl glycoside. Chem. Commun 2008, 2016–2018. [Google Scholar]
- Paoline, I.; Nuti, F.; de la Cruz Pozo Carrero, M.; Barbetti, F.; Kolesinska, B.; Kamiński, Z.J.; Chelli, M.; Papini, A.M. A convenient microwave-assisted synthesis of N-glycosyl amino acids. Tetrahedron Lett. 2007, 48, 2901–2904. [Google Scholar]
- Wozniak, L.A.; Gora, M.; Stec, W.J. Chemoselective Activation of Nucleoside 3'-O-Methylphosphonothioates with 1,3,5-Triazinyl Morpholinium Salts. J. Org. Chem. 2007, 72, 8584–8587. [Google Scholar] [CrossRef]
- El-Faham, A.; Subirós-Funosas, R.; Prohens, R.; Albericio, F. COMU, a Safer and More Effective Replacement for Benzotriazole-based Uronium Coupling Reagents. Chem. Eur. J. 2009, 15, 9404–9416. [Google Scholar] [CrossRef]
- Kitamura, M.; Chiba, S.; Narasaka, K. Synthesis of Primary Amines and N-Methylamines by the Electrophilic Amination of Grignard Reagents with 2-Imidazolidinone O-Sulfonyloxime. Bull. Chem. Soc. Jpn. 2003, 76, 1063–1070. [Google Scholar] [CrossRef]
- Izdebski, J. New Reagents Suppressing Racemization in Peptide Synthesis by the DCC Method. Polish J. Chem. 1979, 53, 1049–1057. [Google Scholar]
- Sample Availability: Samples of the compounds triazinyloximino derivatives are available from the authors.
© 2010 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Al-Warhi, T.I.; AL-Hazimi, H.M.A.; El-Faham, A.; Albericio, F. Synthesis of 2-(4,6-Dimethoxy-1,3,5-triazin-2-yloxyimino) Derivatives: Application in Solution Peptide Synthesis. Molecules 2010, 15, 9403-9417. https://doi.org/10.3390/molecules15129403
Al-Warhi TI, AL-Hazimi HMA, El-Faham A, Albericio F. Synthesis of 2-(4,6-Dimethoxy-1,3,5-triazin-2-yloxyimino) Derivatives: Application in Solution Peptide Synthesis. Molecules. 2010; 15(12):9403-9417. https://doi.org/10.3390/molecules15129403
Chicago/Turabian StyleAl-Warhi, Tarfah I., Hassan M.A. AL-Hazimi, Ayman El-Faham, and Fernando Albericio. 2010. "Synthesis of 2-(4,6-Dimethoxy-1,3,5-triazin-2-yloxyimino) Derivatives: Application in Solution Peptide Synthesis" Molecules 15, no. 12: 9403-9417. https://doi.org/10.3390/molecules15129403