Enantioselective, Organocatalytic Morita-Baylis-Hillman and Aza-Morita-Baylis-Hillman Reactions: Stereochemical Issues

Departamento de Química, Universidad de las Islas Baleares, 07122, Palma de Mallorca, Spain
*
Author to whom correspondence should be addressed.
Molecules 2010, 15(2), 709-734; https://doi.org/10.3390/molecules15020709
Submission received: 19 January 2010
/
Revised: 25 January 2010
/
Accepted: 29 January 2010
/
Published: 1 February 2010
(This article belongs to the Special Issue Baylis-Hillman Reaction and Related Processes)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Conscious of the importance that stereochemical issues may have on the design of efficient organocatalyts for both Morita-Baylis-Hillman and aza-Morita-Baylis-Hillman reaction we have analyzed them in this minireview. The so-called standard reactions involve “naked” enolates which therefore should lead to the syn adducts as the major products, irrespective of the E, Z stereochemistry of the enolate. Accordingly, provided the second step is rate determining step, the design of successful bifunctional or polyfunctional catalysts has to consider the geometrical requirements imposed by the transition structures of the second step of these reactions. On the other hand, MBH and aza-MBH reactions co-catalyzed by (S)-proline and a secondary or tertiary amine (co-catalyst) involve the aldol-type condensation of either a 3-amino-substituted enamine, dienamine, or both, depending on the cases. A Zimmerman-Traxler mechanism defines the stereochemical issues regarding these co-catalyzed condensations which parallel those of the well established (S)-proline catalyzed aldol-like reactions.
1. Introduction
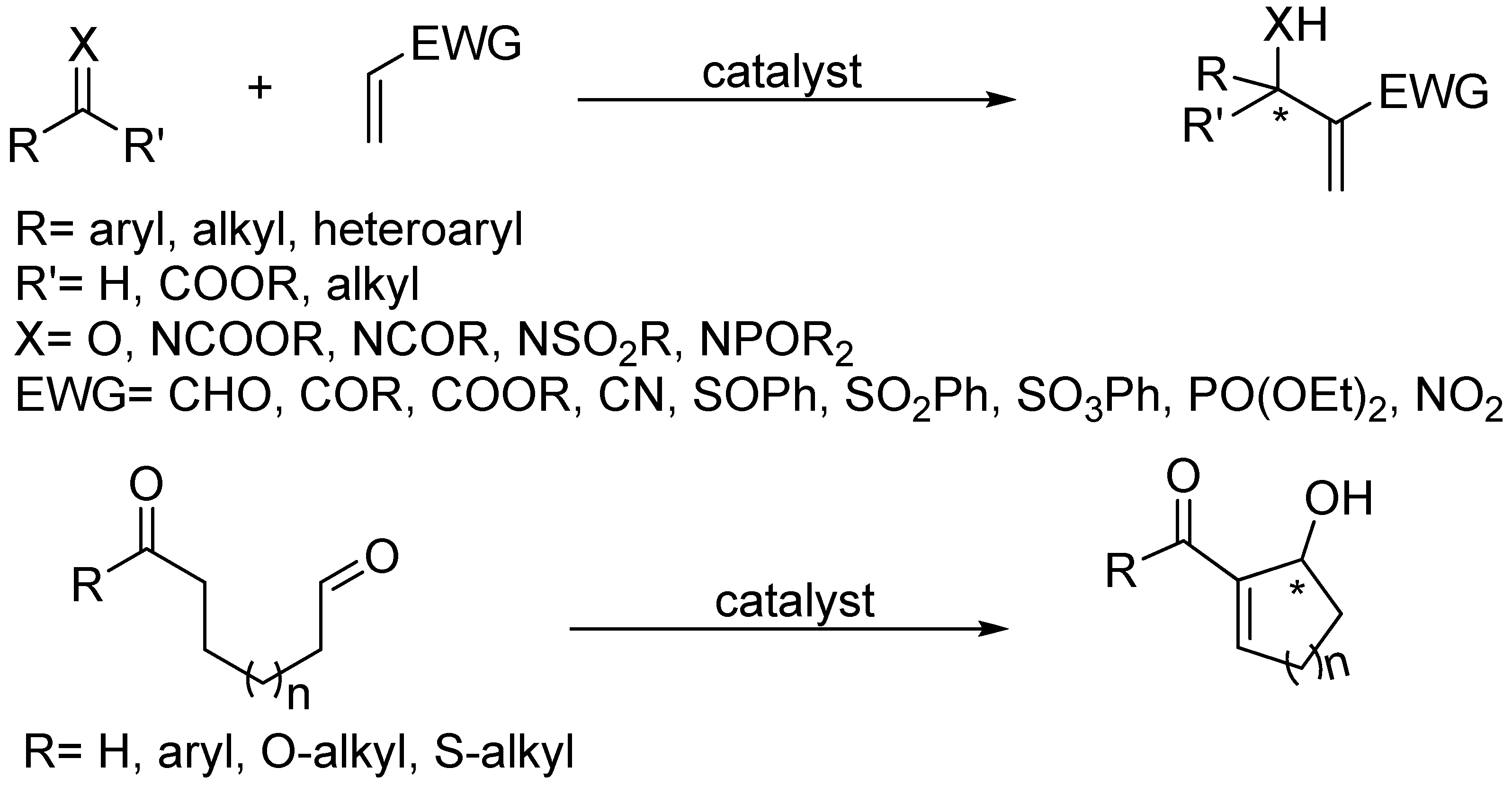
The original Morita-Baylis-Hillman (MBH) reaction [1,2] and its aza analogue (aza-MBH) [3] are unique reactions in many respects, the most relevant being perhaps its atom-economic and organocatalytic nature [4,5,6,7,8,9,10]. The standard reactions typically require bulky, conformationally rigid, basic tertiary amines such as quinuclidines [11,12,13], though DBU [14], DMAP [15], imidazoles [16], guanidine [17], or even heterocyclic carbenes [18,19] or nucleophilic phosphines [20,21] acting as Lewis base catalysts [22] have been used as well for promoting the condensation of an؟ α,β-unsaturated systems (aldehydes, ketones, esters, nitriles, amides, phosphonates, sulphonates, sulfones, sulfoxides or nitro compounds have been employed) with either the C=O functionality present in aldehydes, ketones or α-keto esters for the case of MBH reactions, or with the C=N moiety of N-sulfonyl, N-acyl, N-phosphinoyl, and N-alkoxycarbonyl imines in the case of aza-MBH reactions. The products are densely functionalized small molecules whose basic skeleton is that of a chiral, cyclic, or acyclic, α-methylene-β-hydroxycarbonyl, or α-methylene-β-aminocarbonyl, compound for MBH [23,24,25,26,27,28,29,30,31], or aza-MBH reactions [32,33,34,35], respectively. Both skeletons have attracted great synthetic interest, especially when derived from prochiral C=O or C=N funcionalities since enantioselective versions could then be designed (Scheme 1).
Scheme 1.
Morita-Baylis-Hillman (MBH) and aza-Morita-Baylis-Hilman (aza-MBH) reactions.

Needless to say, great efforts have been made by researchers all over the world to find efficient catalysts for achieving highly enantioselective MBH and aza-MBH reactions. The challenge was and still is huge, as investigators need to overcome many reaction hurdles such as low conversions, long reaction times and poor enantioselection. Impulse for the research race has come from extensive mechanistic studies.
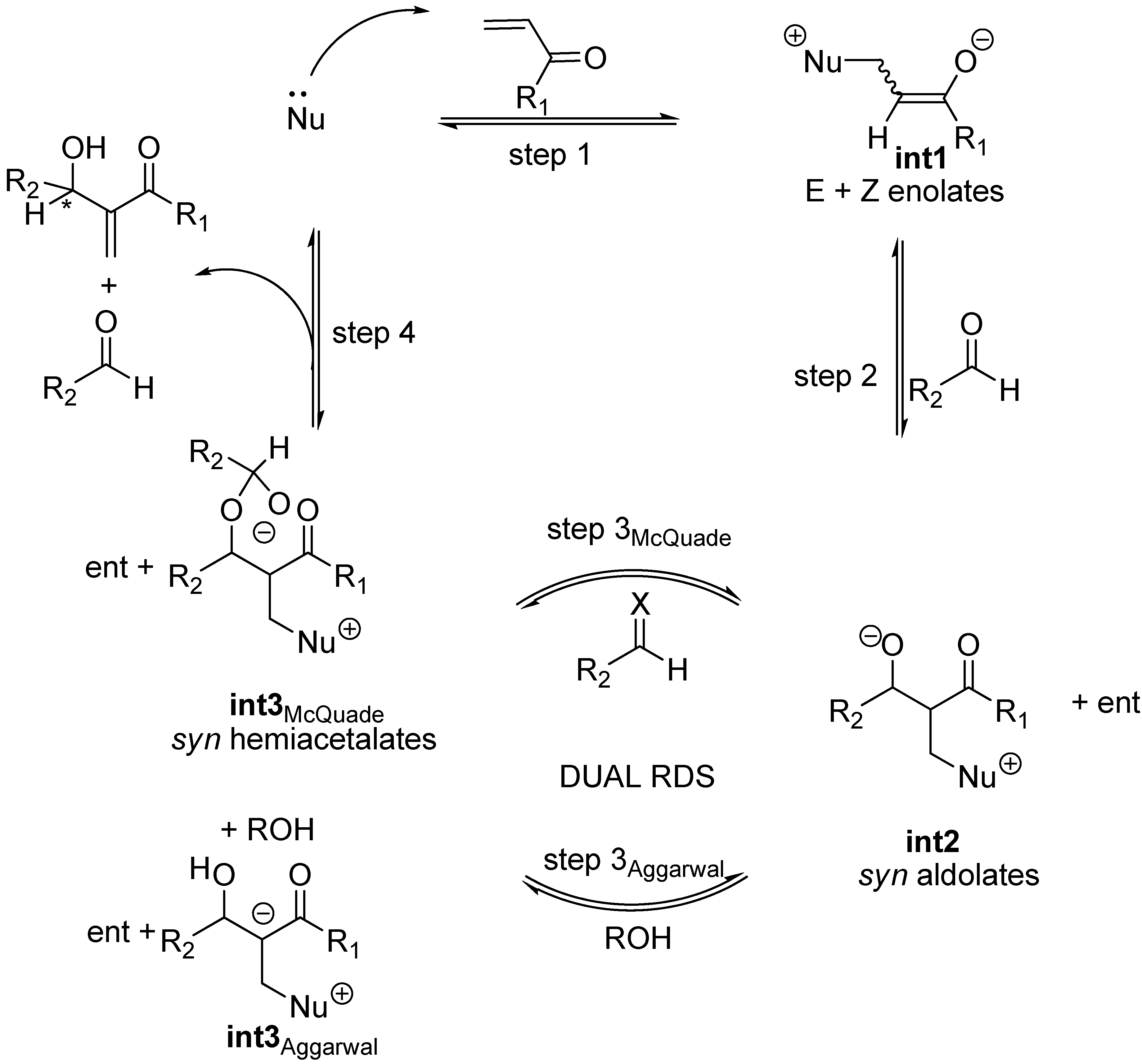
The original mechanism for the standard MBH reaction cycle, proposed by Hill and Isaacs [36,37], invoked a series of four transition structures giving rise to three highly dipolar, zwitterionic intermediates eventually collapsing to the final adduct together with the catalyst, then ready for a new cycle (Scheme 2). Strong support for the original MBH mechanism has come in the past from kinetic studies [38,39,40,41], and most recently by NMR studies [42] and ESI-MS data [43], according to which the aldol addition step (step 2 in Scheme 2) should be the rate determining step. Being highly dipolar means that the zwitterionic intermediates, as well as their preceding transition structures, should be high energy species impossible to detect or isolate in most cases [44], even if a specific, internal stabilization could be provided. Actually, it was soon recognized that Brönsted acid additives (water, methanol, ureas, thioureas, etc.) accelerated MBH reactions [45,46,47,48,49,50,51,15], thus spurring the search for suitable chiral, bifuntional molecules having a Lewis base (usually a rigid, tertiary amine or phosphine) and a Brönsted acid appropriately located for stabilizing those zwitterionic species and their precursory transition structures, i.e., LBBA catalysts [52,53,54,55,56,57,58,42].
Scheme 2.
Original mechanistic proposal for the MBH reaction.
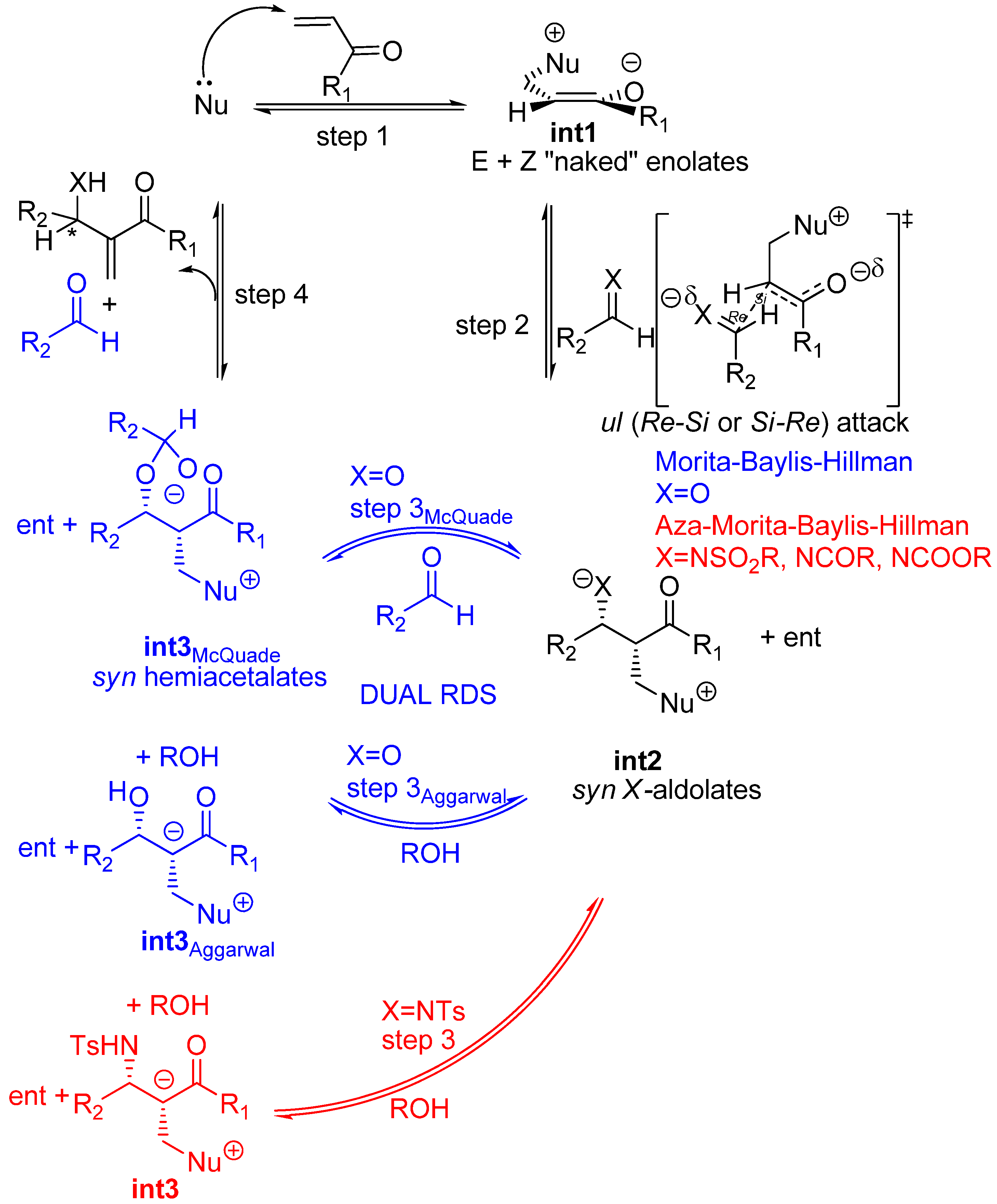
Recent experimental results and theoretical studies regarding the third step of the standard MBH and aza-MBH reactions (step 3 in Scheme 2) suggested a dualistic nature for it, thus significantly modifying the original mechanism. On the one hand McQuade has presented two key observations for quinuclidine-catalyzed MBH reaction of acrylate and benzaldehyde catalyzed by DABCO in non-polar, polar or even protic solvents, namely: 1) the rate equation is first order in DABCO and acrylate and second order in aldehyde, and 2) a large kinetic isotope effect was observed when α-deuterioacrylate was employed [59,60]. Altogether these facts are strong support for McQuade’s claim for a new mechanism for the standard MBH racemic reaction, according to which the rate determining step must be the proton shift occurring in the third step upon a hemiacetalate species. On the other hand, kinetic studies by Aggarwal et al. showed that the standard MBH reaction (ethyl acrylate with benzaldehyde catalyzed by quinuclidine) is autocatalytic at short reaction times, the implications being that in the presence of a proton donor molecule (alcohol, water, etc.) the rate determining step should be the protonation of the intermediate ammonium aldolate (int2) with the concomitant removal of the carbonyl α-hydrogen [61]. This proposal was further supported by computational data which showed that the energy barrier for this ROH-promoted proton shift was even somewhat lower than that envisioned in McQuade’s mechanism [62]. Experimental support for this dual option-mechanism (Scheme 3) has been recently provided by ESI-MS [63,64,65]. One of the key issues that remain unanswered is the stereochemical outcome of MBH and aza-MBH reactions. In particular, one notes the lack of a unified proposal for the stereochemical outcome of the aldol addition step (step 2) and, in addition, there is uncertainty upon whether or not the original kinetic outcome of the aldol reaction suffers any modification due to reaction reversal driven by thermodynamics (step 3). Needless to say, these issues hamper future development of novel and more efficient catalysts. These stereochemical issues appear to be more complex for the case of MBH reactions where the third step is dual and minor details surely play stereochemically relevant roles that can affect the diastereomeric composition of the so-called aldolate int2, and consequently erode the final enantioselectivity.
Scheme 3.
Dual mechanism for the standard MBH reactions according to recent physico-chemical studies.
Scheme 3.
Dual mechanism for the standard MBH reactions according to recent physico-chemical studies.

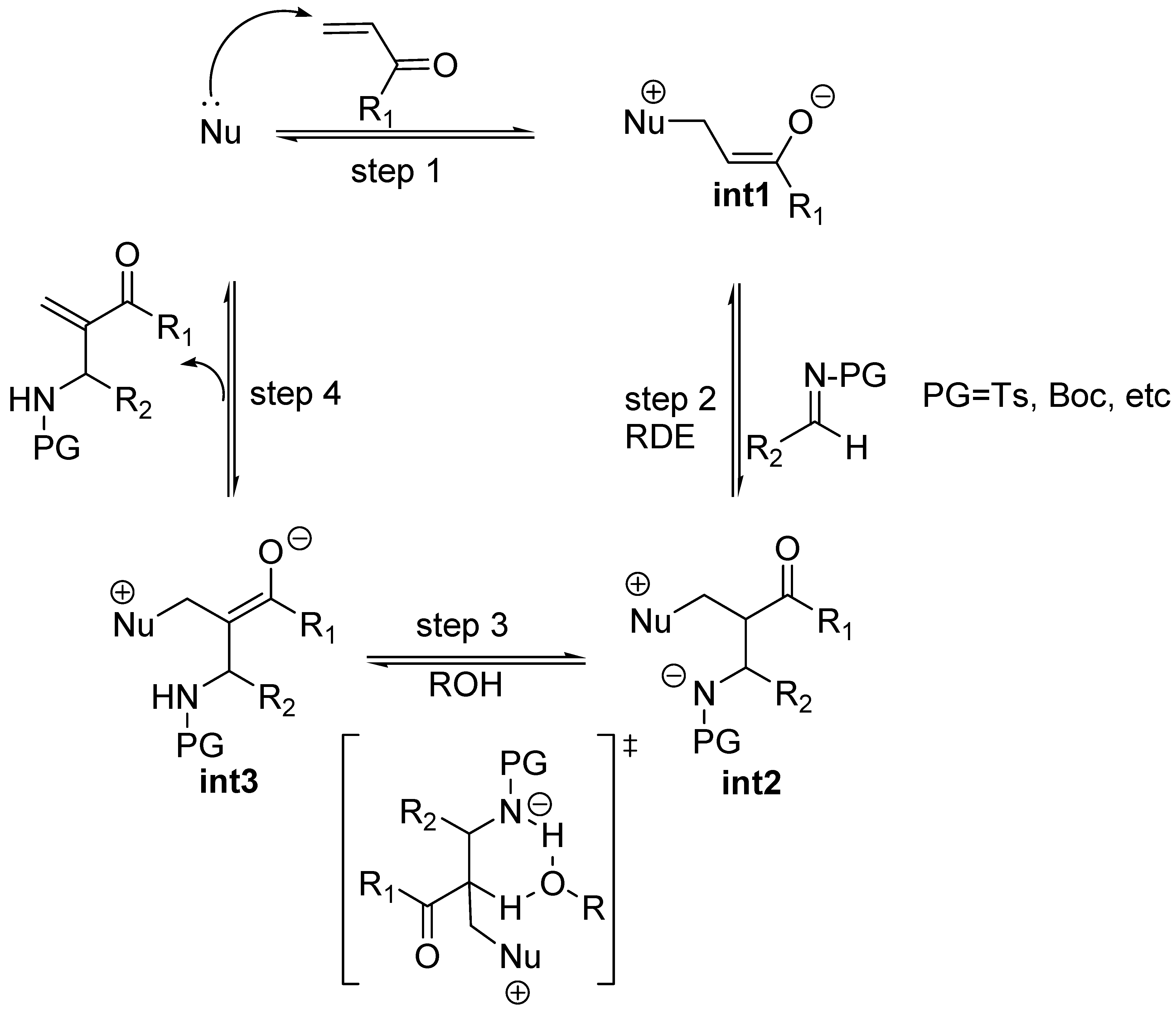
In spite of the strong mechanistic analogies between MBH and aza-MBH reactions there are some relevant dissimilarites worth noting here. Both are accelerated by Brönsted acid additives. However, at difference with the McQuade’s findings for MBH reactions, the kinetic studies run by Leitner et al. showed that the rate expression for the aza-MBH reaction, carried out in THF, between methyl vinyl ketone with an N-tosyl imine in the presence of triphenylphosphine as catalyst, and a Brönsted acid as cocatalyst whose pKa was in the range 16–8, was first order in both imine, ketone and triphenylphosphine, but independent of the acid cocatalyst [66]. The implication was evident: the elimination step of aza-MBH reactions, in which the acid cocatalyst operates, should not be the rate-determining step (Scheme 4). Instead, one could state, provided this behaviour could be extended to all kinds of aza-MBH reactions promoted by bifunctional LABA catalysts, that the rate-determining step should be the Mannich-type addition step also responsible for the eventual stereochemical results, provided racemization of the final product is avoided [66]. The consequence is evident: the race for the development of enantioselective, bifunctional catalysts for MBH and aza-MBH is being won by the latter, no doubt due to the fact that in this case the rate determining step is the addition step (second step) and the third step is fast, which derives in less stereochemical difficulties.
Scheme 4.
Mechanistic proposal for the aza-MBH reaction co-catalyzed with a Brönsted acid ROH.

From a mechanistic point of view there is a second major category of MBH and aza-MBH reactions, namely that which involves the aldol-like addition of an intermediate enamine species, instead of the enolates characteristic of the standard reactions. In particular, secondary α-aminoacid organocatalysts such as (S)-proline [67], or (S)-pipecolinic acid [68], often assisted with cocatalysts (N-methyl imidazole, DABCO, peptides, etc.), have recently been added to the above armoury of bifunctional organocatalysts [69,70,71,72]. Obviously, from a mechanistic viewpoint these α-amino acid-organocatalyzed MBH reactions differ completely to those categorized as standard MBH and aza-MBH reactions which involve tertiary amine or phosphine catalysts [73,74,75].
In addition, it is worth mentioning at this point that one can also reach MBH or aza-MBH products by means of non-organocatalytic protocols. These methods either employ Lewis acid catalysts such as BF3 or TiCl4 with an added halide as cocatalyst [76,77,78,79], or Ti(IV) tetraisopropoxide with, or without a chiral amine [80], phosphine or sulphur compound as co-catalyst [81]. Alternatively, a metallic salt can be used as catalyst or stoichiometric promoter. These miscellaneous methods which employ non-organocatalytic will not be examined in the present minireview.
In spite of the quite large number of reviews dedicated to the standard MBH and aza-MBH reactions no attempt has been made to outline their most prominent stereochemical aspects in a systematic manner, likely because of the still unanswered questions regarding their mechanism. The aim of this minireview is to analyze the stereochemical issues of the two main types of MBH reactions, namely the standard MBH reactions, and the α-aminoacid-cocatalyzed MBH reactions.
2. The Standard MBH and Aza-MBH Reactions
For the purpose of examining the stereochemical relevant details of the standard MBH and aza-MBH reactions one needs to consider the general mechanism illustrated in Scheme 3, and keep in mind that those steps taking place before the rate determining step involve reversible reactions and that, according to recent physico-chemical observations, the rate determining step is dual (in other words, it can be one or the other for the same reaction, as a function of time and/or solvent) as a function of the actual experimental conditions, as discussed below.
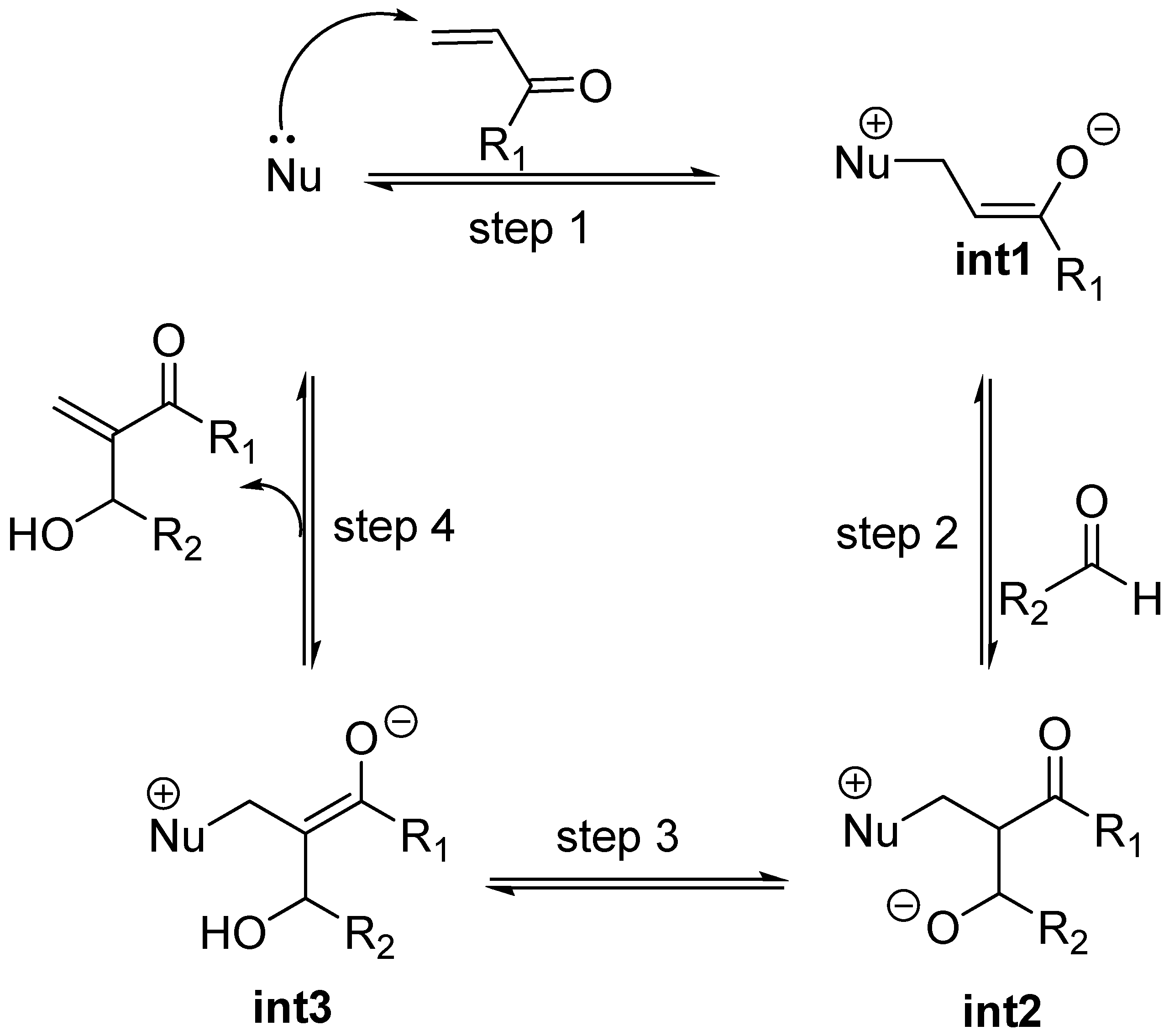
It is our contention that standard MBH and aza-MBH reactions should be considered as involving “naked” enolates i.e., fully separated ammonium, or phosphonium, enolates holding specific properties and reactivity [82]. A revealing experiment regarding reversibility of aldol reactions involving fully separated ion pairs (“naked” enolates), was reported in the nineties by Noyori et al. for tris(dimethylamino) sulfonium (TAS) enolates. What they found is that TAS enolates do not yield aldol products unless the reaction is driven by an O-silylation quench of the aldolate adducts which, accordingly, must be considered high energy species on the reaction profile [83,84]. This behaviour is reminiscent to that of standard MBH and aza-MBH reactions which usually require a protic solvent, a protic cosolvent, or a Lewis Base-Brönsted Acid (LBBA) catalyst for driving the reaction to an efficient level of conversion.
The first step MBH and aza-MBH reactions (step 1 in Scheme 3) involves the reversible nucleophillic attack upon the β carbon of an α,β-unsaturated system, thereby giving rise, for acyclic systems, to a zwitterionic ammonium or phosphonium, enolate (int1) [23,24,25,26,27,28], which can undergo aldol condensation with a C=O or C=N moiety, as reported by Noyori et al. for other systems [85,86]. Being this first step reversible, we can expect the formation of a thermodynamically controlled mixture of zwitterionic enolates, the most stable being the Z-configured ammonium enolate (thermodynamic enolate) by virtue of the fact that being the O- and CH2NR3+ moieties cis to each other [87], strong attractive interactions between the charged oxygen atom and the nearby α-hydrogens of the onium moiety are being developed, as reported by both Houk [88], and Leahy [89]. In this regard it is worth mentioning that Aggarwal, Harvey et al. have estimated by means of computation at the B3LYP/6-31+G*/THF level that the thermodynamic (Z)-enolate is stabilized by 1,1 kcal/mol relative to the kinetic enolate [87,90]. According to these authors this stabilization of the (Z)-enolate is due to the existence of stronger electrostatic interactions in the thermodynamic (Z)-enolate, rather than other specific bonding interactions. A recent experimental study has concluded that these specific bonding interactions are not present in the case of a phosphine-catalyzed MBH-like alkylation reaction involving the kinetic zwitterionic phosphonium enolates [91]. The MBH and aza-MBH reaction can thus be appropriately defined as a unique aldol condensation in which the enolate involved is a “naked”, zwitterionic, ammonium, or phosphonium, equilibrating enolate [92,93,94]. Since its formation is highly reversible it can be safely assessed that MBH reactions should involve the above mentioned thermodynamic (Z)-enolates as the major species [87]. Curiously enough, the rate of intramolecular MBH reactions has been shown to be highly dependant on the stereochemistry of the Michael acceptor, with the Z-stereoisomer being more reactive than the E-isomer [95].
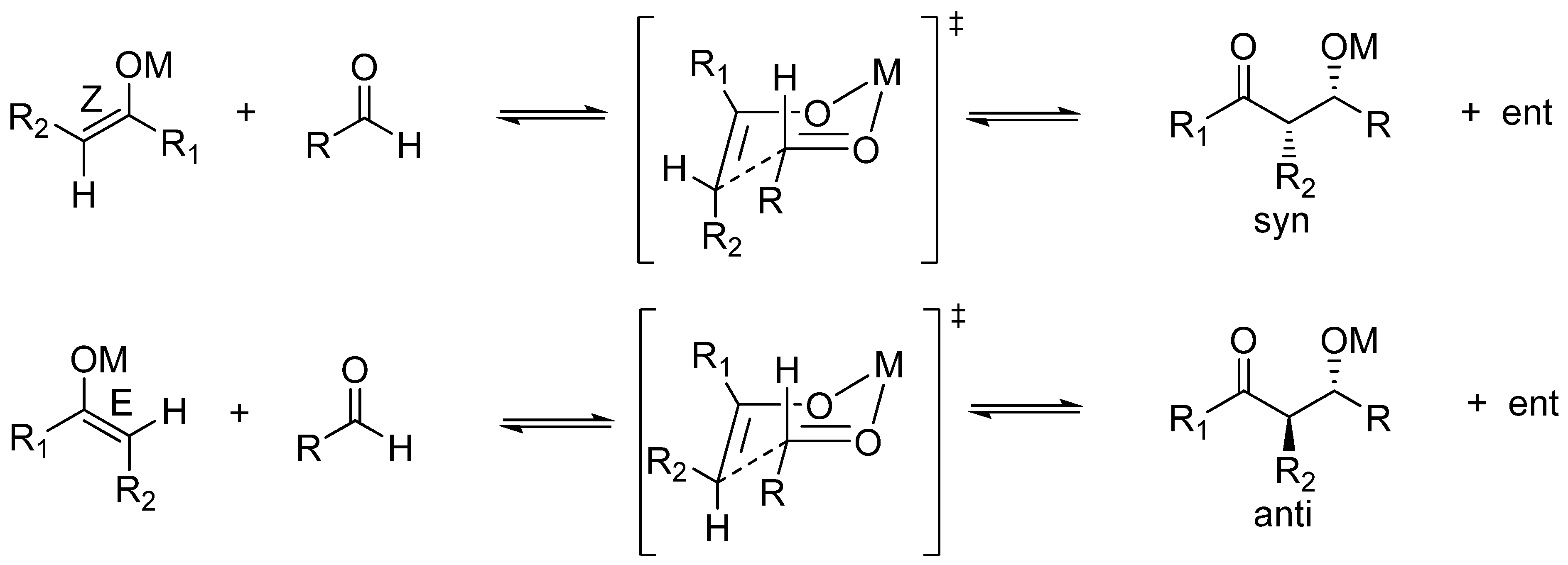
Common metal enolates and “naked” enolates are quite dissimilar in reactivity [96,97]. As mentioned previously, the most revealing aspect for the present analysis is that “naked” enolates do not react with aldehydes unless in the presence of trialkylsilyl fluoride which acts as a quencher of the resulting “naked” aldolate, as reported by Noyori et al. for TAS enolates [98,99]. A further revealing issue is that of the diastereoselection of aldol condensations. Kinetic diastereoselection when common metal enolates (Li, Na, K, Mg, Zn, B, Ti) are employed is rather well established as a consequence of the running of the so-called Zimmerman-Traxler mechanism which invokes cyclic, chair-like transition states where the metal plays a significant role as it is an integral part of this cyclic array [100]. According to the general ruling for the reactions of these metal enolates, thermodynamic (Z)-enolates give rise preferentially to syn aldols, whereas (E)-enolates lead to anti aldols (Scheme 5).
Scheme 5.
Kinetic diastereoselection for metal enolates according to the Zimmerman-Traxler mechanism of aldol condensations.
Scheme 5.
Kinetic diastereoselection for metal enolates according to the Zimmerman-Traxler mechanism of aldol condensations.

In contrast, a number of aldol reactions do not follow this general rule because a different mechanism is in effect. In particular, as put forward by Noyori and coworkers, “naked” enolates such as TAS enolates give rise to syn aldol derivatives regardless of the E-Z configuration of the enolate (Scheme 6) [101]. The reason for this is that an acyclic, extended transition state is at work and the energetically most favoured one is that which avoids electrostatic repulsion between the negatively charged oxygens, as illustrated in Scheme 6 [98,99,102,103].
Scheme 6.
Kinetic diastereoselection of aldol condensations undergone by “naked” enolates as according to Noyori et al.
Scheme 6.
Kinetic diastereoselection of aldol condensations undergone by “naked” enolates as according to Noyori et al.

Curiously enough, Aggarwal, Harvey et al. in their computational study (at the B3LYP/6-31+G*/THF level of theory) of the MBH reaction reported that the lowest transition estate of the C-C bond forming step i.e., that corresponding to the addition of the (Z)-configured enolate, was governed by the strength of the dipole-dipole interactions required for maximum electrostatic stabilization [104,105,106], even when an explicit methanol molecule was included for activation [98,99]. Therefore the product stereochemistry should be that of syn aldolates, as a consequence of the preferred face selective ul (Re-Si or Si-Re) condensation of either (Z)- or (E)-configured enolates, as reported by Noyori et al. for aldol condensation of TAS “naked” enolates [98,99]. Two additional flag features regarding those aldol reactions undergone by “naked” enolates are worth being mentioned because they perfectly match those of MBH reactions: 1) the final aldolate can react with a second molecule of aldehyde thereby giving rise to dioxane byproducts, and 2) the reaction shows autocatalysis [98,99]. The diastereoselectivity of the addition step to (E)-N-substituted imines i.e, the rate determining step of aza-MBH reactions, will also be governed by identical principles, though achiral, Brönsted acid additives can actually invert the stereochemical course of the reactions, as shown recently by Masson, Zhu et al [107,108]. To sum up, predictions for the standard MBH and aza-MBH reactions are that the ammonium or phosphonium syn aldolates should be the kinetic products of the second step (Scheme 7).
Scheme 7.
Stereochemical issues for the standard MBH ( ![Molecules 15 00709 i001]() ) and aza-MBH (
) and aza-MBH ( ![Molecules 15 00709 i002]() ) reactions.
) reactions.
 ) and aza-MBH (
) and aza-MBH (  ) reactions.
) reactions.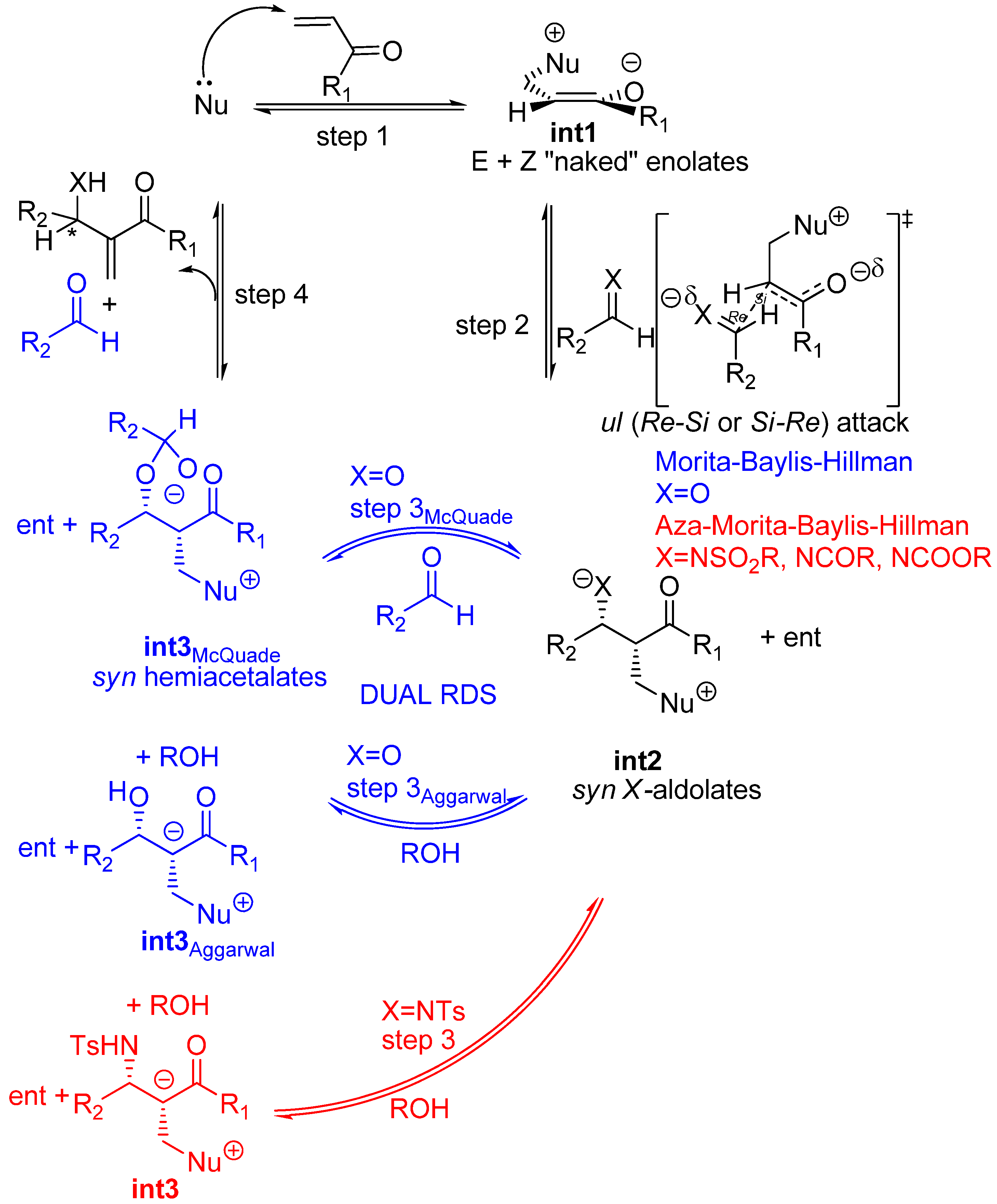
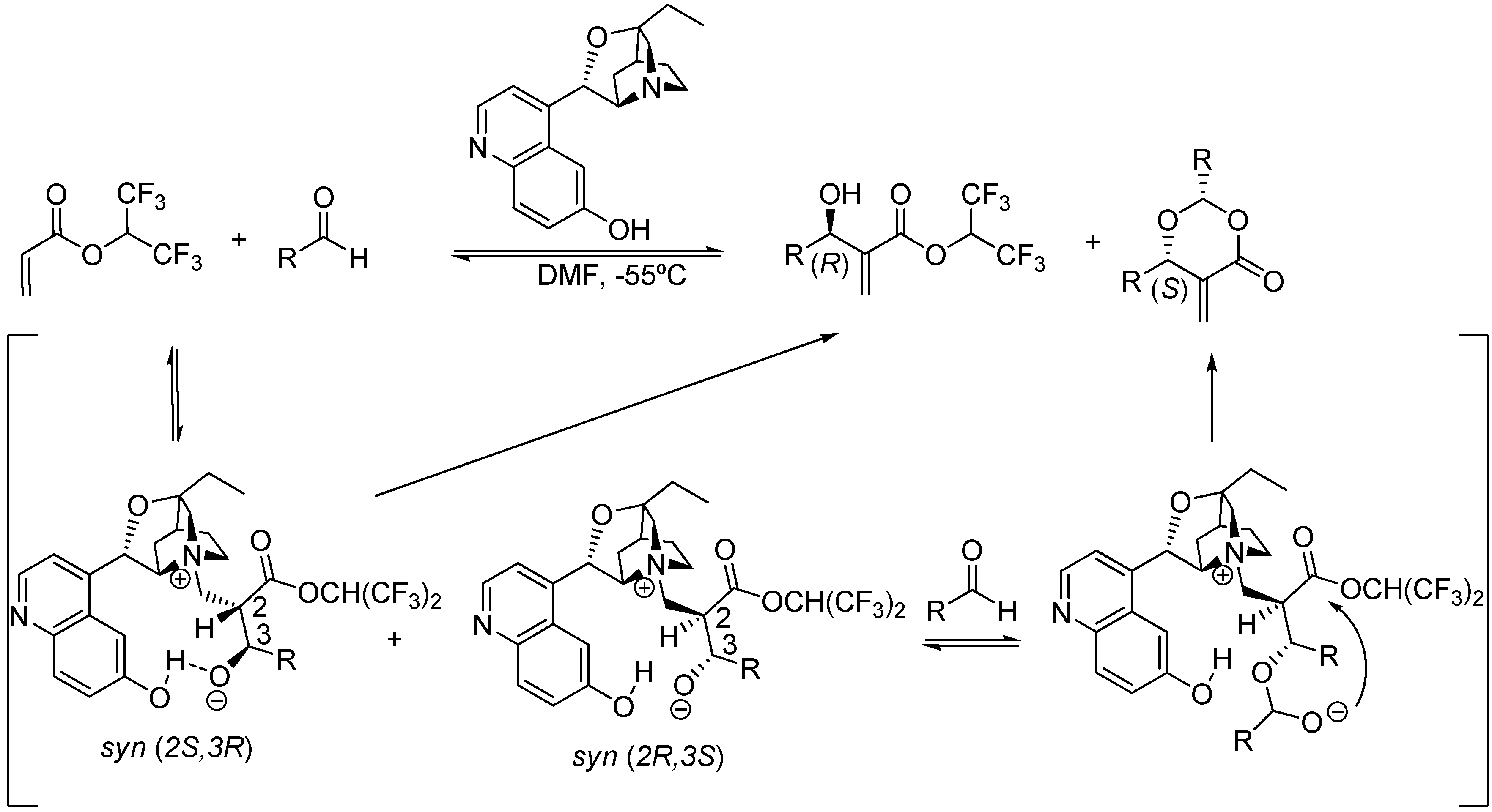
Thus far only scattered information about the intrinsic diastereoselectivity of the MBH and aza-MBH reactions had been advanced in the published literature due to the nature of the third step in which one of the two centers of chirality is eventually destroyed. A particular case is the mechanistic rationale proposed by Hatakeyama et al. for explaining the intriguing opposite enantioselectivity observed for the common MBH product and for the unexpected dioxanone adduct obtained in the β-isocupreidine-catalyzed MBH reaction between 1,1,1,3,3,3-hexafluoroisopropyl acrylate with aliphatic or aromatic aldehydes in DMF at -55 °C [109]. The authors called for the formation of the two diastereoisomeric syn adducts that evolved, presumably at similar rates, to the divergent products. In particular, the syn (2S,3R) adduct undergoes direct β-elimination thereby yielding the (R)-MBH product, whereas the syn (2R,3S) reacted with a second aldehyde molecule, as predicted by the McQuade’s dual mechanism, thereby giving rise to the (S)-dioxanone byproduct which has the opposite configuration, as shown in Scheme 8.
Scheme 8.
Hatakeyama’s stereochemically divergent β-isocupreidine-catalyzed MBH reaction.

Fortunately, a recent work by Xu et al. has provided clear-cut proof for the intrinsic diastereoselectivity, as well as the enantioselectivity, of the aza-MBH reaction of nitroalkenes with N-tosylimines catalyzed by several chiral amino thioureas derived either from quinine or 1,2-trans-diaminociclohexanes [110], which supports the above analysis. Instead of the prototypical proton shift and β-elimination, this reaction evolved through direct β-elimination thus resulting in the formation β-nitro-γ-enamines containing the two contiguous centers of chirality previously generated in the addition step. The results shown in Scheme 9 for the reaction catalyzed by (1R,2R)-diaminocyclohexane thiourea clearly provide sound support of the above reasoning as all reactions run in apolar, aprotic media yielded syn β-nitro-γ-enamines in high diastereomeric ratio, whereas those carried out in polar and protic solvents led to much lower diastereoselection.
Scheme 9.
Diastereo and enantioselective aza-MBH reaction of nitroalkenes with N-Ts protected imines catalyzed by bifunctional amino thioureas.
Scheme 9.
Diastereo and enantioselective aza-MBH reaction of nitroalkenes with N-Ts protected imines catalyzed by bifunctional amino thioureas.

Nonetheless, a cautionary note is needed. Even though Noyori reported that the syn or antiO-silylated aldols are configurationally stable in the presence of fluoride in an aprotic medium, one should not forget that stereochemically defined lithium aldolates can suffer stereochemical isomerization in protic media [111]. Accordingly, at the time of devising new enantioselective catalysts for the MBH reaction, best candidates ought to be those that allow for a rapid protonation of the syn aldolates (int2) formed in the second step of MBH aldol condensations. In other words, the role of a bifunctional LBBA catalyst may be that of providing for a rapid protonation of the syn aldolate (int2) thereby giving rise to a basic site appropriately located for promoting a rapid β-elimination, i.e, promoting a concerted protonation-deprotonation sequence. Recent theoretical studies have provided evidences for declaring the second step the rate determining step for MBH reactions carried out in the presence of protic solvents or dual Lewis base-Brönsted acid catalysts [112,113,114], or the final β-elimination when in the absence of protic solvents or catalysts [56,60,115]. Trifunctional or multifunctional catalysts are conceivable and, in fact, some has already been described [116,117].
The lesson to be learned for the design of catalysts is clear: bifunctional LBBA molecules appear to be useful candidates for enantioselective catalysts for the standard MBH and aza-MBH reactions. Obviously, not all bifunctional LBBA catalysts will work, and in fact many of the so-called privileged catalysts have failed. In our view, in order to reach efficiency, catalysts will need to consider the spatial disposition of the dipoles in the stereochemically relevant transition state, as recently shown by Clarke, Philp et al. [118].
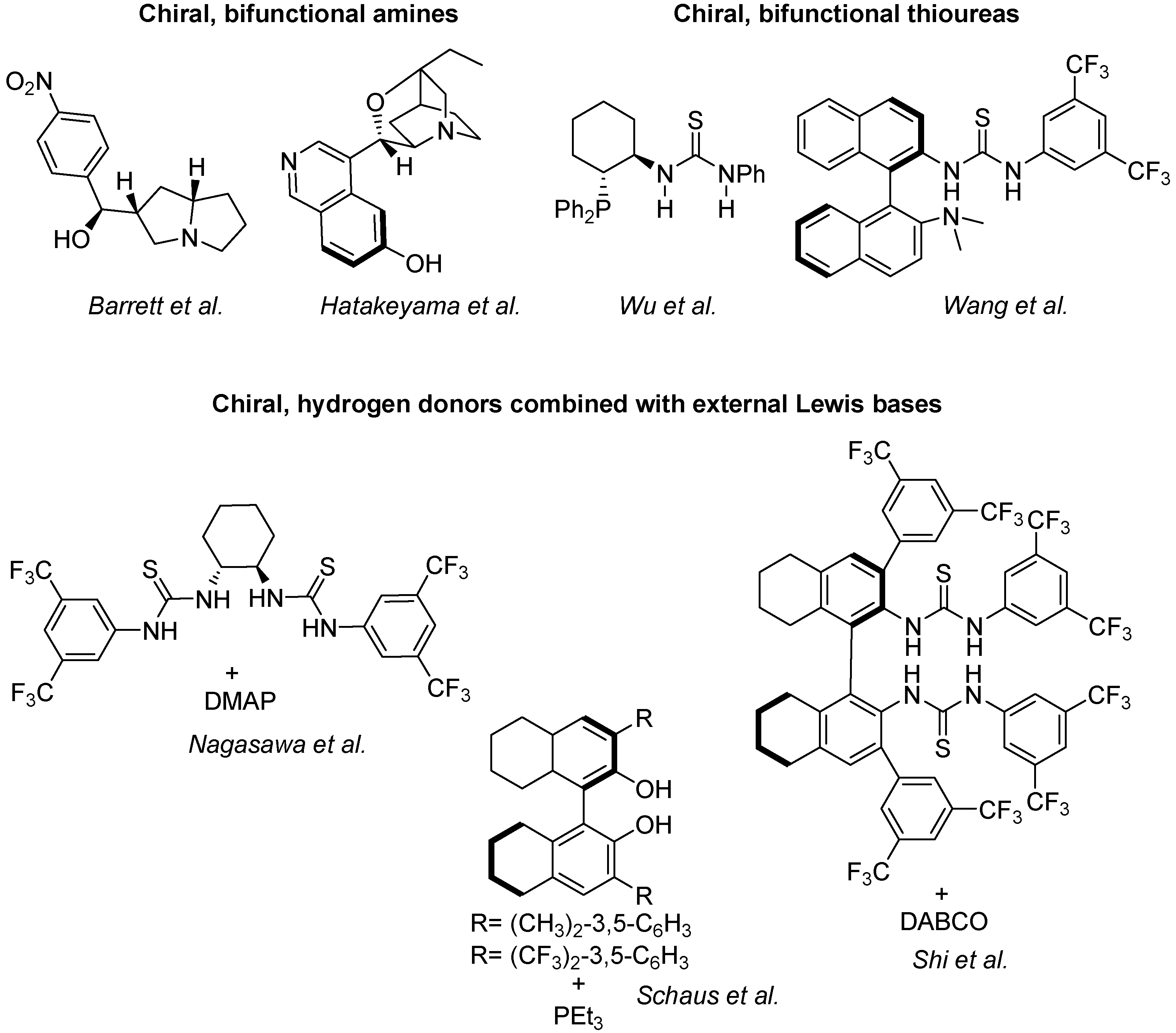
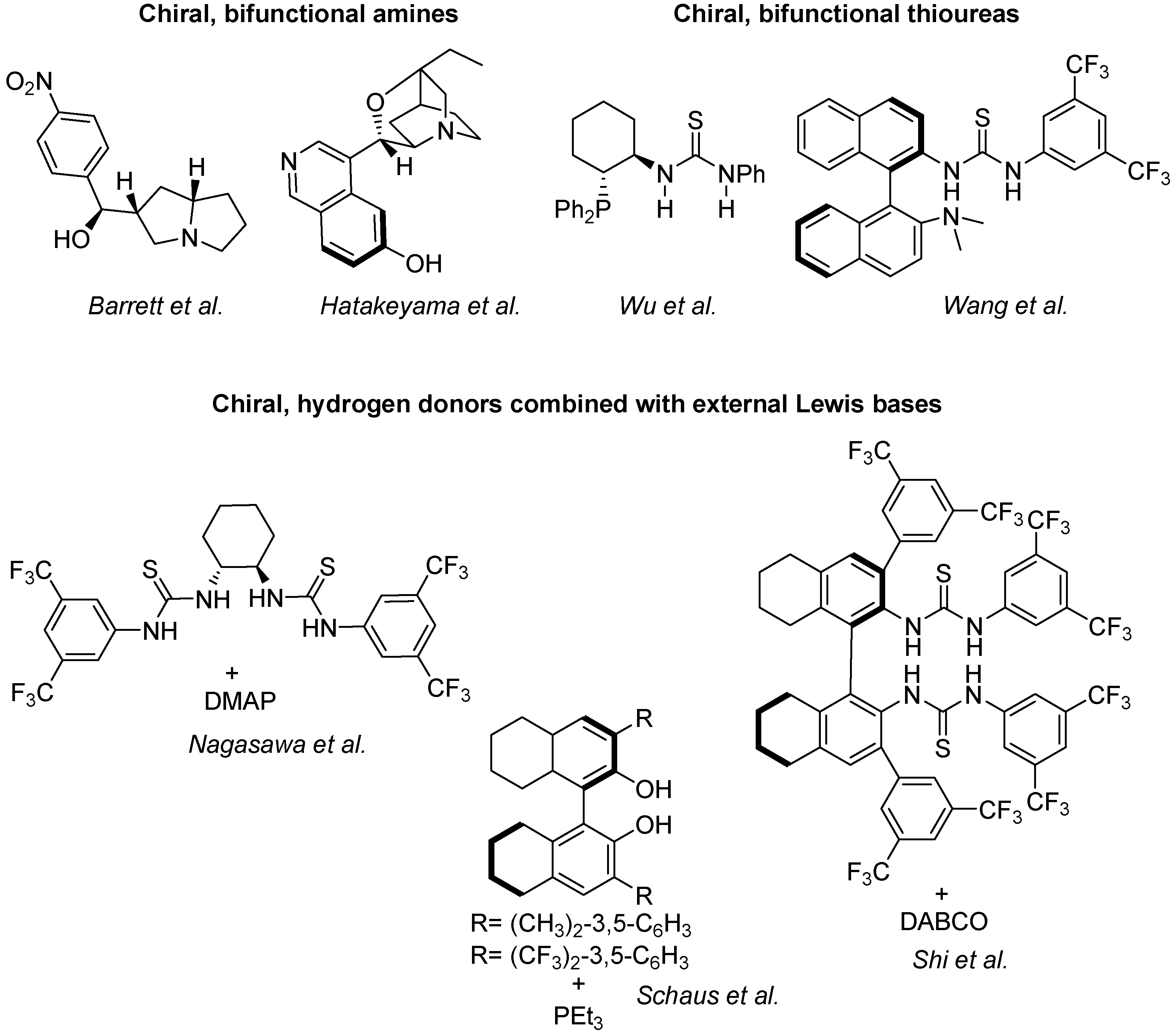
Quite a large armoury of highly successful enantioselective catalysts for standard MBH reactions is already available. Some of them, illustrated in Scheme 10, are classified in three main groups, namely the chiral bifunctional amines of Barrett [52] and Hatakeyama [109], the chiral, bifunctional thioureas of Wu [119] and Wang [120], and the chiral hydrogen donors combined with Lewis bases of Nagasawa [121], Schaus [122] and Shi [123].
Scheme 10.
Catalytic systems for the standard, enantioselective MBH reactions.
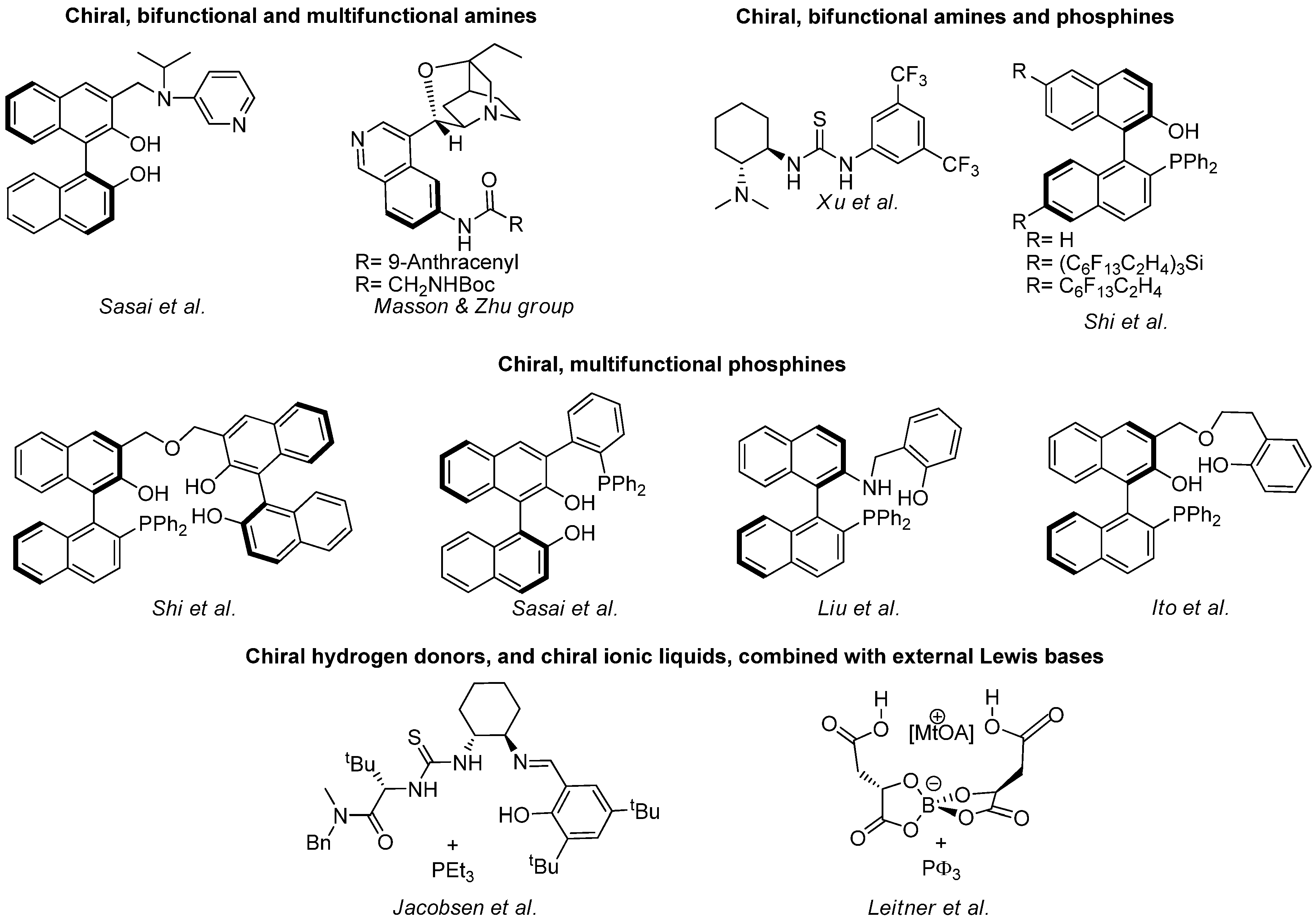
The number of enantioselective catalysts for the aza-MBH reaction has grown exponentially in recent years. Some of them, illustrated in Scheme 11, can be classified in four main groups, namely the chiral bifunctional and multifunctional amines of Sasai [124,125], and Mason and Zu [107,108], the chiral bifunctional amines and phosphines of Xu [110] and Shi [126,127,128,42], the chiral multifunctional phosphines of Shi [129,130], Sasai [58], Liu [116,117], and Ito [131], and the chiral hydrogen donors, as well as chiral ionic liquids, combined with external Lewis base of Jacobsen [44], and Leitner et al. [156].
Scheme 11.
Enantioselective catalysts for the standard aza-MBH reaction.

3. The α-Aminoacid Catalyzed and α-Aminoacid-Amine Cocatalyzed Mbh and Aza-Mbh Reactions
Secondary amines were recognized to promote intramolecular cyclizations of enonealdehydes thereby yielding MBH products. The reaction was interpreted, however, as involving a tandem Michael/aldol condensation [132,133,134]. To the best of our knowledge the first reported attempt at employing a secondary amine such as (S)-proline as enantioselective catalyst for MBH reactions is due to Shi and coworkers [135]. Their discovery was quite simple but fundamental: even though (S)-proline itself failed to promote the MBH reaction of arylaldehydes with a β-unsubstituted α,β-unsaturated ketone such as methyl vinyl ketone (MVK), the presence of an equimolar amount of a Lewis base such as imidazole, benzimidazole or DABCO acting as co-catalysts led to the corresponding MBH adducts in high yield, though in very low enantioselectivity (5–10% ee), the use of such chiral tertiary amines as Hatakeyama’s β-isocupreidine leading only to a somewhat marginal improvement in enantioselectivity [136].
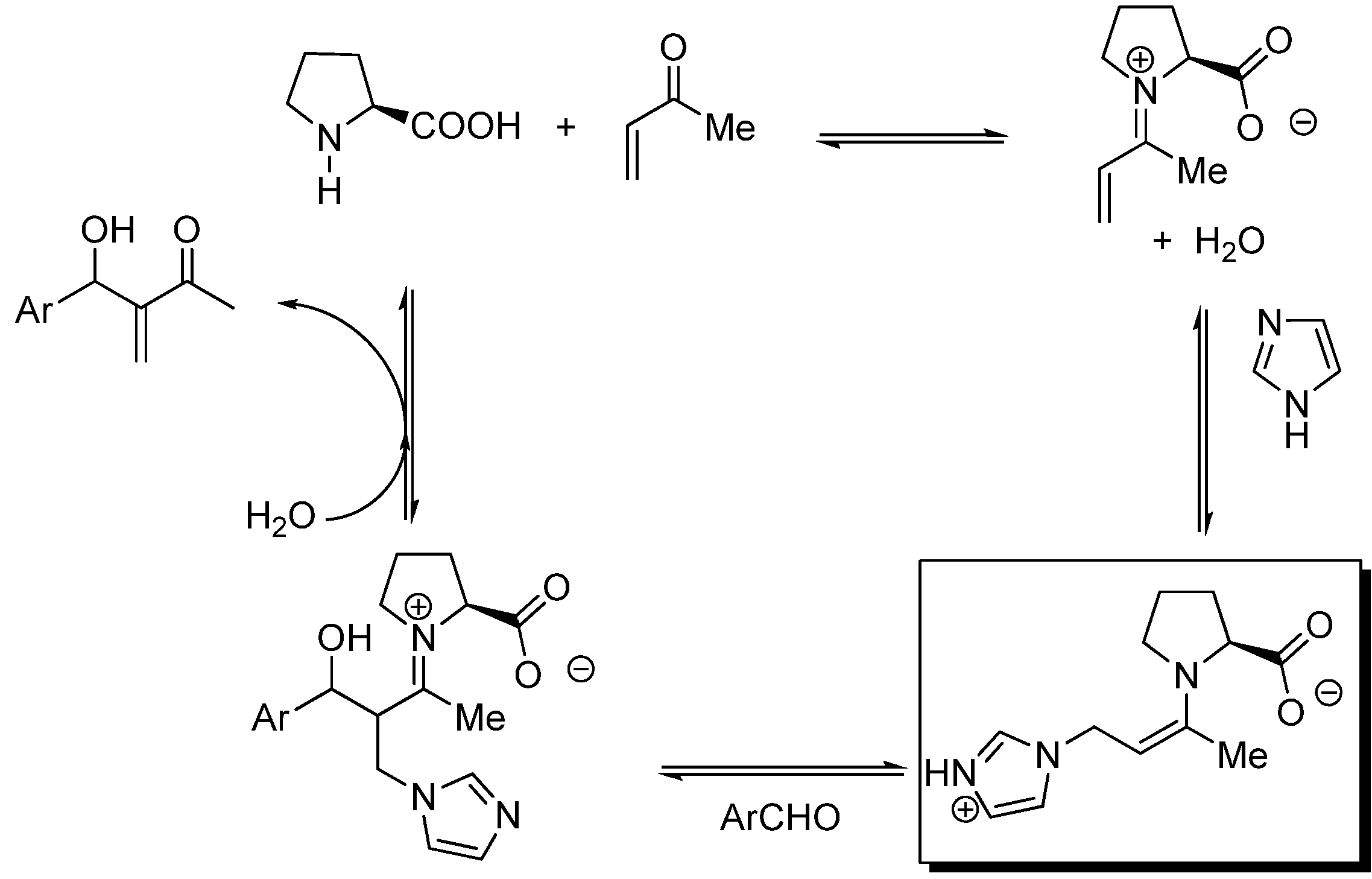
Scheme 12.
General mechanism for the MBH (S)-proline-Lewis base co-catalyzed reactions.

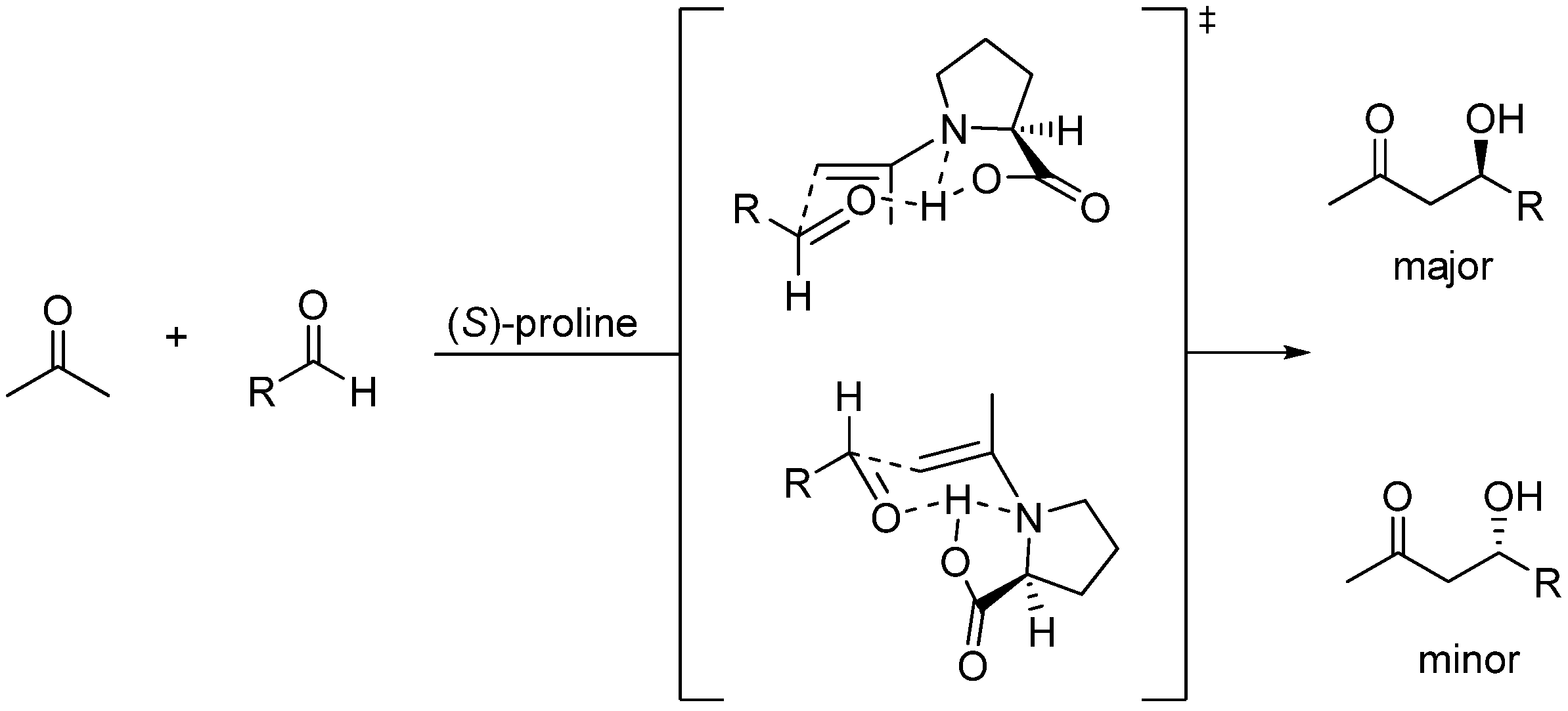
Inspired by the (S)-proline organocatalyzed aldol condensations reported by List [137,138], Barbas [139], and MacMillan [140], Shi et al. proposed a mechanism (Scheme 12) involving formation of an intermediate enamine species (highlighted in Scheme 12) derived from the trapping of the conjugated iminium species resulting from the condensation of (S)-proline with MVK by the co-catalyst which bears a clear-cut relationship with the zwitterionic enolates (int1) of the standard MBH reactions previously mentioned. The stereochemical issues regarding (S)-proline-catalized intermolecular aldol additions have been well established by Houk, List and coworkers [141]. Accordingly, Shi’s enamine intermediate should react with the aldehyde in keeping with the generalized mechanism based on the Zimmerman-Traxler six-membered ring chair-like model [100]. Eventually, recycling of the catalyst should take place as a consequence of hydrolysis of the final iminium condensate (Scheme 13). At the time of writting this review it is clear that there is much room for improvement of the enantioselectivity of MBH co-catalyzed reactions.
Scheme 13.
Generalized mechanism for (S)-proline-catalyzed aldol reactions based on the Zimmerman-Traxler model.
Scheme 13.
Generalized mechanism for (S)-proline-catalyzed aldol reactions based on the Zimmerman-Traxler model.

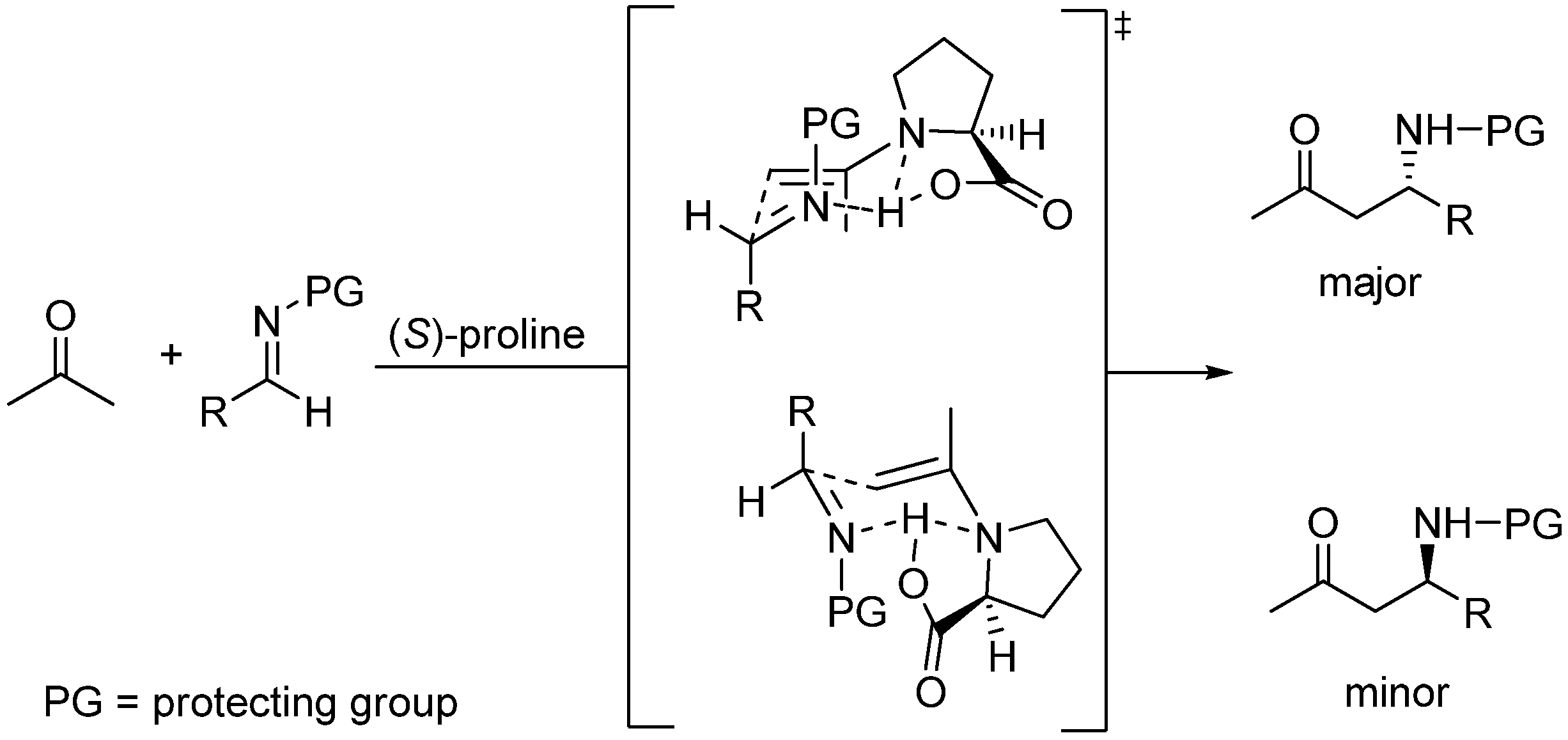
Other chiral, tertiary amino alcohols have also been explored for synergy with (S)-proline in catalyzing enantioselective MBH reactions by Zhou, He and coworkers with some improvement [142,143]. Miller’s approach to reach ideal synergy called for exploring π-(Me)His (Pmh)-containing peptides of various lengths and constitution [144]. The best enantioselection (78% ee) for the prototypical reaction of the o-nitrobenzaldehyde with an β-unsubstituted α,β-unsaturated (MVK) was obtained when using (S)-proline with the chiral octapeptide BOC-(π-Me)His-Aib-Chg-(trt)Gln-D-Phe-D-Pip-Cha-Phe-OMe as cocatalyst, the key observation being that the “unmatched” pair (R)-proline/ BOC-(π-Me)His-Aib-Chg-(trt)Gln-D-Phe-D-Pip-Cha-Phe-OMe yielded the MBH adduct with opposite configuration in only 33% ee. This observation led the authors to conclude that the stereochemical issues regarding proline/tertiary amine cocatalyzed MBH reactions are dominated by proline stereochemistry. The scope of the reaction has been examined [145].
Application of these ideas to an intramolecular MBH cyclization led Miller and coworkers to find that the (S)-pipecolinic acid/N-methylimidazole pair gave better enantioselectivities than the (S)-proline/N-methylimidazole pair [146]. Simultaneously, Hong and coworkers not only observed that (S)-proline itself behaved as an effective catalyst but also reported the unexpected observation that the Lewis base co-catalyst employed (e.g., imidazole) gave rise to inversion of the enantioselectivity, an event that required a new reactive intermediate, as suggested by the authors [147]. From a mechanistic viewpoint, it was proposed that the reaction catalyzed by (S)-proline involved (E,E)-dienamine species which followed the widely accepted enamine mechanism as applied to an intramolecular aldol condensation, whereas that co-catalyzed by imidazole involved imidazole-substituted (E)-enamines having two centers of chirality [148,149].
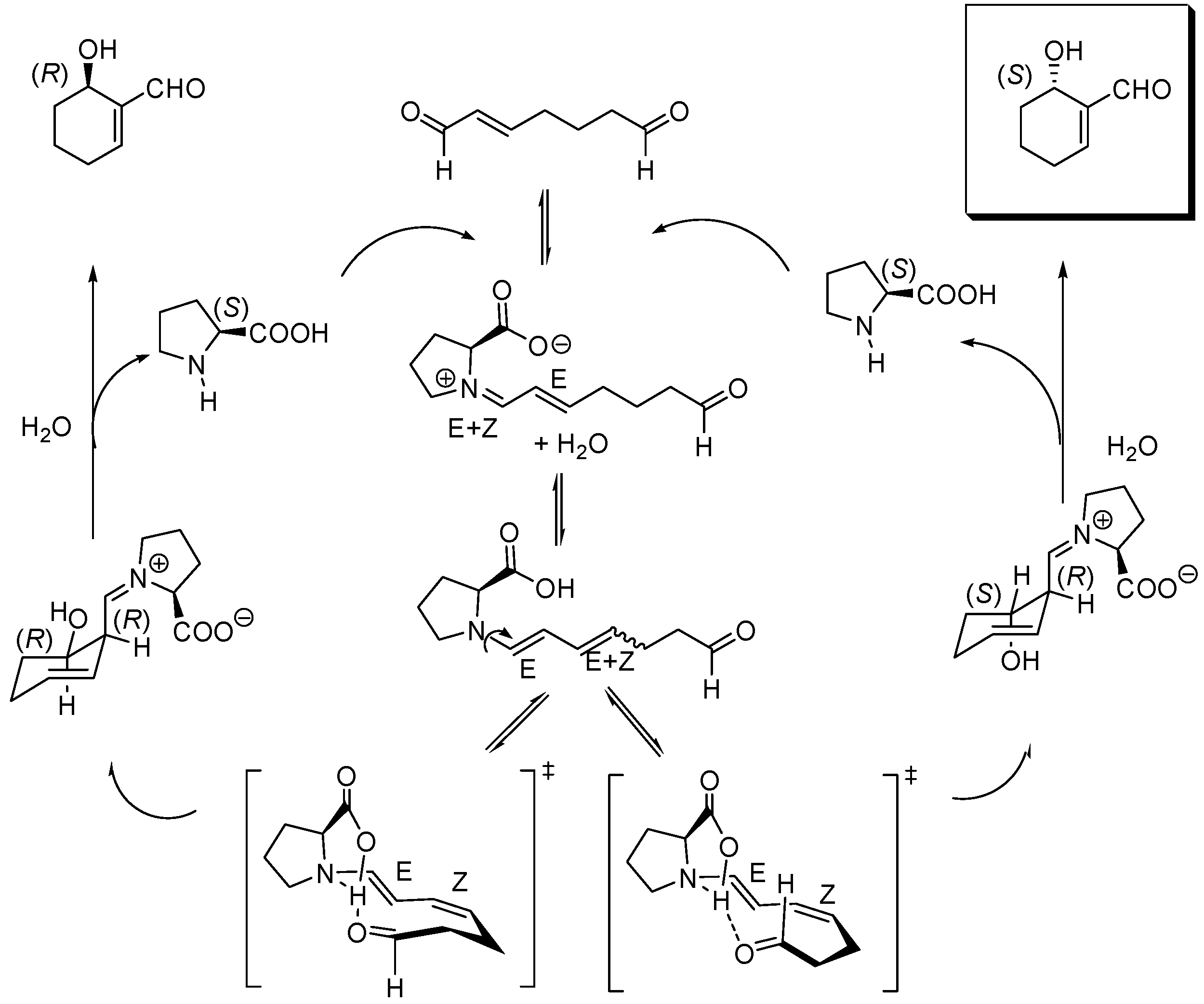
Scheme 14.
Mechanism for the (S)-proline-catalyzed intramolecular cyclization of hept-2-enedial to (S)-6-hydroxy-cyclohex-1-enecarbaldehyde.
Scheme 14.
Mechanism for the (S)-proline-catalyzed intramolecular cyclization of hept-2-enedial to (S)-6-hydroxy-cyclohex-1-enecarbaldehyde.

Subtle details of this mechanism have come to light as a consequence of an extensive density functional analysis at the B3LYP/6-31-G(d,p) level which included the use of the polarized continuum model (PCM B3LYP/6-31++G(d,p)//B3LYP/6-31G(d,p)) to describe solvent effects, the most important being the role played by water to give rise to the required syn and anti, (E,Z)-dienamine key intermediates in equilibrium, as the theoretical calculations demonstrated that (E,E)-enamines could not undergo cyclization [150]. According to these PCM calculations, the cyclization of the anti, E,Z-dienamine is biased towards the formation of the (S)-configured product, as illustrated in Scheme 14 which displays, in a simplified manner, the modifications introduced by the computational work by Gil Santos et al., in close analogy with the model proposed and by List and coworkers [151].
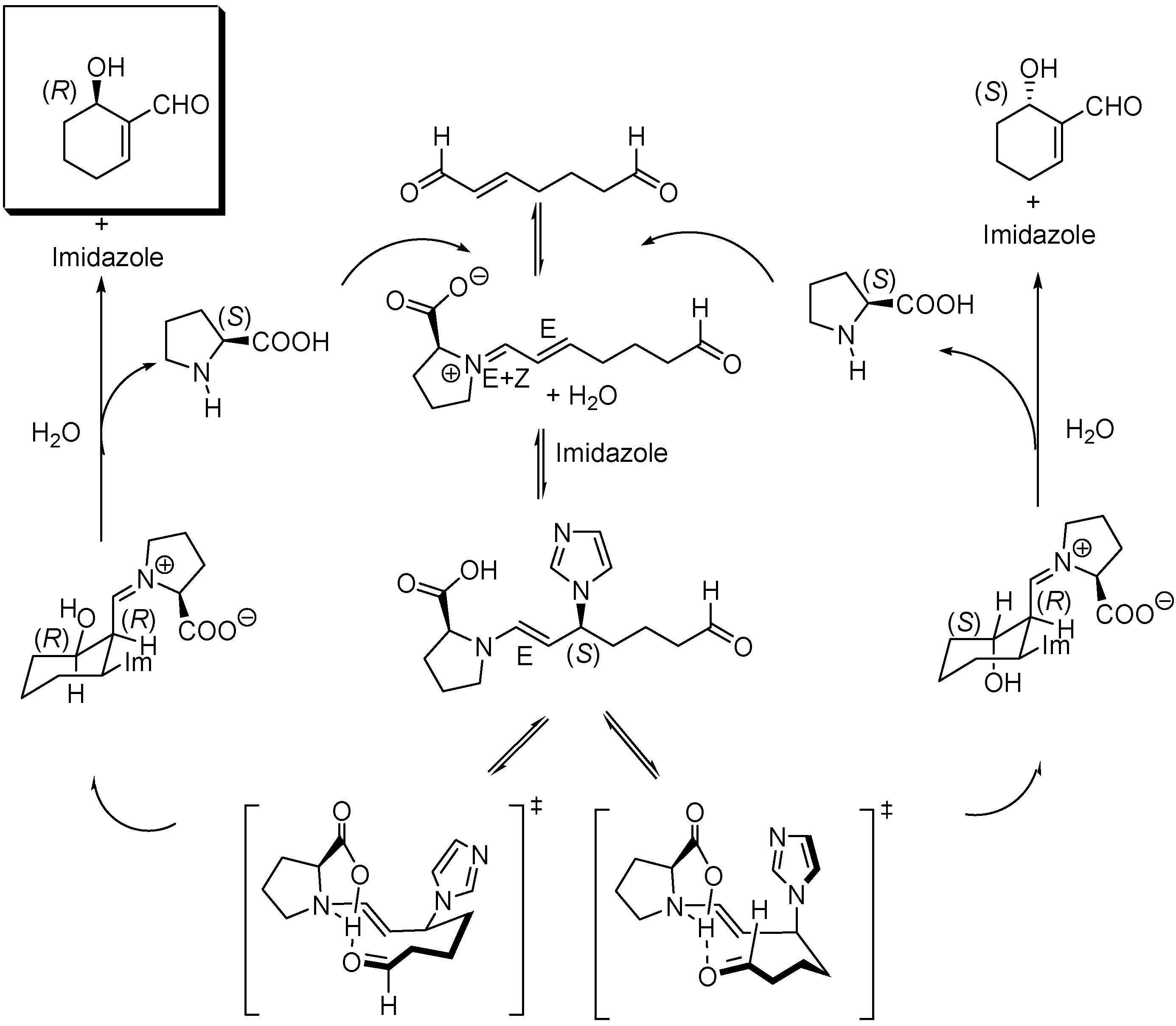
The original mechanistic proposal of Hong et al. for the imidazole co-catalyzed reaction has also been modified as a consequence of the theoretical studies of Gil Santos et al., which predict the formation of the (S,S)-diastereoisomer of the 3-(1-imidazolyl)-substituted enamine by attack of imidazole to the (E)-iminium ion assisted by water in the rate limiting step. Cyclization of this intermediate followed by hydrolysis yields the (R)-6-hydroxycyclohex-1-enecarbaldehyde and (S)-proline, as illustrated in Scheme 15. Calculations also provide satisfactory data for explaining the temperature and solvent polarity dependence of the cyclization.
Scheme 15.
Mechanism for the (S)-proline, imidazole co-catalyzed intramolecular cyclization of hept-2-enedial to (R)-6-hydroxycyclohex-1-enecarbaldehyde.
Scheme 15.
Mechanism for the (S)-proline, imidazole co-catalyzed intramolecular cyclization of hept-2-enedial to (R)-6-hydroxycyclohex-1-enecarbaldehyde.

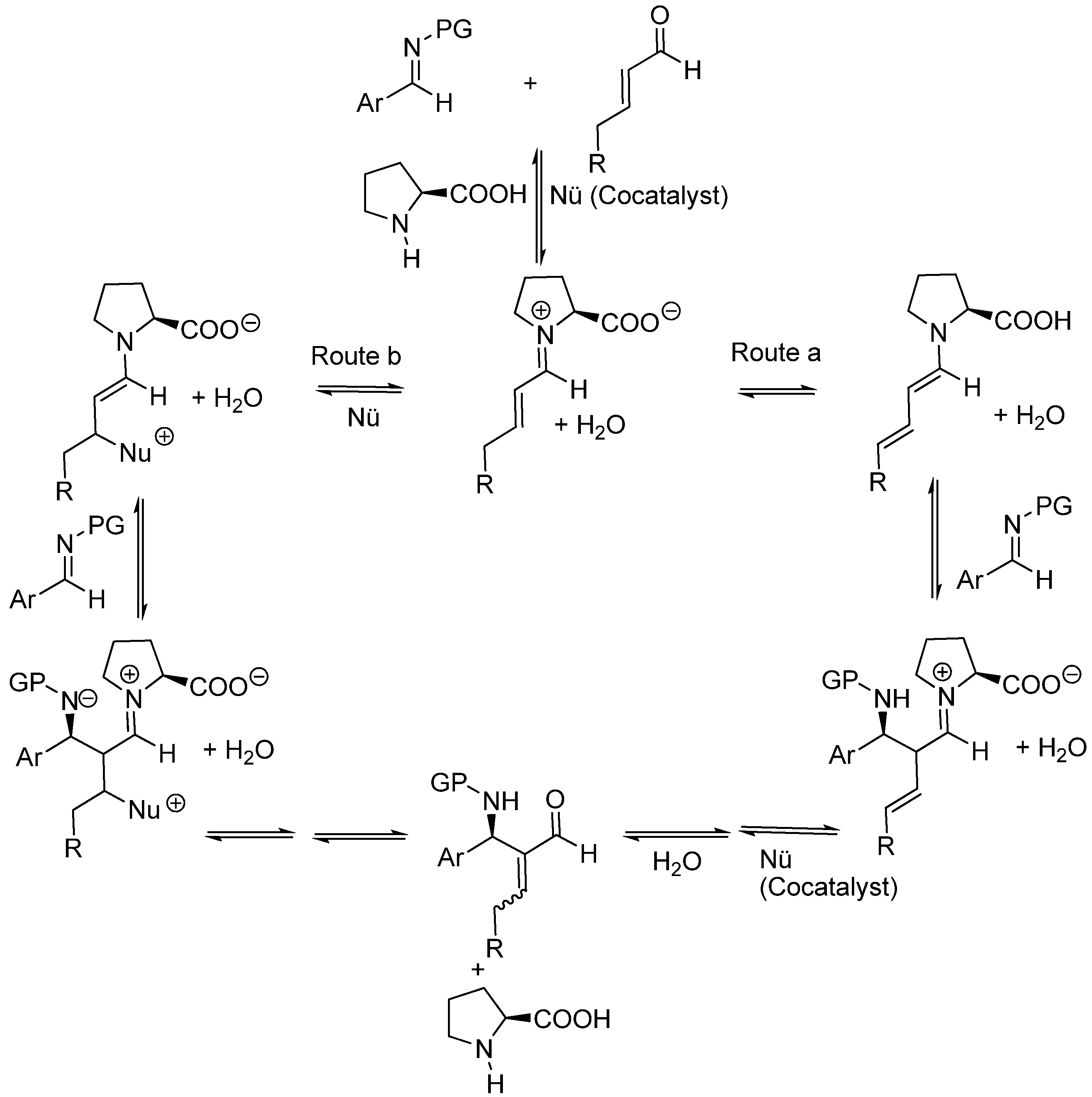
First examples of aza-MBH reactions catalyzed by (S)-proline, as well as co-catalyzed with added bases, have come to light quite recently first published by Barbas, Tanaka and coworkers [152], and then by Córdova and coworkers [153]. From a stereochemical viewpoint, intermolecular reactions of β-alkyl substituted-α,β-unsaturated aldehydes with either N-PMP (Barbas and Tanaka) or N-BOC protected imines (Córdova) catalyzed by (S)-proline, can be understood as Mannich condensations involving either a (E,E)-dienamine, or the 3-substituted enamine resulting from the trapping of the precursory conjugated iminium ion by the cocatalyst. Whichever the case, a Zimmerman-Traxler six-membered ring chair-like model as applied to N-substituted imines (Scheme 16) should apply [154,155].
Scheme 16.
Generalized mechanism for (S)-proline-catalyzed Mannich reactions based on the Zimmerman-Traxler model.
Scheme 16.
Generalized mechanism for (S)-proline-catalyzed Mannich reactions based on the Zimmerman-Traxler model.

The available evidence found by Barbas, Tanaka and coworkers suggests that the role of the co-catalyst (imidazole) was only that of increasing the rate of the reaction and improving the chemical yield, but had no influence on the enantioselectivity and absolute configuration of the final adducts, therefore implying that the reaction actually involves the condensation of a (E,E)-dienamine with the protected imine (route a in Scheme 17). Córdova and coworkers did not find positive evidences for distinguishing between the dienamine route (route a) and the 3-substituted enamine route (route b).
Scheme 17.
Alternative mechanisms for (S)-proline-amine co-catalyzed Mannich reactions of β-substituted aldehydes and N-protected imines (N-PG).
Scheme 17.
Alternative mechanisms for (S)-proline-amine co-catalyzed Mannich reactions of β-substituted aldehydes and N-protected imines (N-PG).

4. Conclusions
Major efforts are being dedicated to the search of chemically efficient, enantioselective, organocatalytic Morita-Baylis-Hillman (MBH) and aza-Morita-Baylis-Hillman (aza-MBH) reactions. These reactions provide enantiomerically-enriched, densely-functionalized molecules of interest to synthetic organic chemists. The development of efficient reactions has been plagued with difficulties derived from low conversions, meagre chemical yields and poor enantioselectivities. Fortunately, along the years, chemists in their search for better oganocatalysts have identified a number of key mechanistic issues by means of fundamental, physical organic chemistry studies, and otherwise. Breakthroughs focussing on the nature of the rate determining step of both the MBH and aza-MBH reactions, and of course those that reveal the nature of the species actually involved paved the way for the actual development of the two major avenues that lead to efficient, enantioselective, organocatalytic MBH and aza-MBH methodologies.
The so-called standard MBH and aza-MBH reactions involve Lewis base catalysts (typically tertiary amines or phosphines). Efficient catalytic systems for them either entail an external Lewis base and a chiral hydrogen donor, or instead the catalyst usually is a single chiral, bifunctional or multifunctional molecule having both a Lewis base and one or several hydrogen donors appropriately located in space. However, we believe that there is still room for improvement. This minireview has examined the stereochemical issues regarding these reactions in a coherent manner. The most relevant conclusion of this analysis is that standard MBH and aza-MBH reactions involve the aldol-type condensation of “naked” enolates, thereby leading to syn adducts irrespective of the configuration of the enolate. Several pieces of evidence already available in the published literature support this conclusion. Accordingly, provided the second step is rate determining step, the design of successful bifunctional or polyfunctional catalysts has to consider the geometrical requirements imposed by the transition structures of the second step of these reactions.
On the other hand, all MBH and aza-MBH reactions promoted by both (S)-proline and a co-catalyst (a secondary or tertiary amine) invoke the aldol-type condensation of either a 3-amino substituted enamine, dienamine, or both, depending on cases. The stereochemical issues regarding these co-catalyzed condensations appear to mirror those of the well established (S)-proline catalyzed aldol-like reactions.
Acknowledgements
Thanks are due to the MICINN (Ministerio de Ciencia e Innovación, Spain) for financial support through project CTQ2007-62952.
References and Notes
- Morita, K.; Suzuki, Z.; Hirose, H. A Tertiary Phosphine-catalyzed Reaction of Acrylic Compounds with Aldehydes. Bull. Chem Soc. Jpn. 1968, 41, 2815–2815. [Google Scholar] [CrossRef]
- Baylis, A.B.; Hillman, M.E.D. Ger. Offen. 2, 155,133 (1972). Acrylic compounds. US Pat. 3,743,669, 1973. Chem. Abstr. 1972, 77, 34174. [Google Scholar]
- Perlmutter, P.; Teo, C.C. A simple synthesis of 2-methyledene-3-aminopropanoates. Tetrahedron Lett. 1984, 25, 5951–5952. [Google Scholar] [CrossRef]
- Dalko, P.I.; Moisan, L. Enantioselective Organocatalysis. Angew. Chem. Int. Ed. 2001, 40, 3726–3748. [Google Scholar] [CrossRef]
- Jarvo, E.R.; Miller, S.J. Amino acids and peptides as asymmetric organocatalysts. Tetrahedron 2002, 58, 2481–2495. [Google Scholar] [CrossRef]
- Benaglia, M.; Puglisi, A.; Cozzi, F. Polymer-Supported Organic Catalysts. Chem. Rev. 2003, 103, 3401–3430. [Google Scholar] [CrossRef]
- Berkessel, A.; Gröger, H. Metal-Free Organic Catalysts in Asymmetric Synthesis; Wiley-VCH: Weinheim, Germany, 2004. [Google Scholar]
- Dalko, P.I.; Moisan, L. In the Golden Age of Organocatalysis. Angew. Chem. Int. Ed. 2004, 43, 5138–5175. [Google Scholar] [CrossRef]
- Seayad, J.; List, B. Asymmetric Organocatalysis. Org. Biomol. Chem. 2005, 3, 719–724. [Google Scholar] [CrossRef]
- Marcelli, T.; vanMaarseveen, J.H.; Hiemstra, H. Cupreines and Cupreidines: An Emerging Class of Bifunctional Cinchona Organocatalysts. Angew. Chem. Int. Ed. 2006, 45, 7496–7504. [Google Scholar] [CrossRef]
- Shi, M.; Xu, Y.-M.; Zhao, G.-L.; Wu, X.-F. Lewis Base Effects in the Baylis-Hillman Reaction of Arenecarbaldehydes and N-Aryliden-4-methylbenzenesolfonamides with α-β-Unsaturated Cyclic Ketones. Eur. J. Org. Chem. 2002, 3666–3679. [Google Scholar]
- Shi, M.; Xu, Y.-M. Lewis base effects in the Baylis-Hillman Reaction of imines with methyl vinyl ketone. Eur. J. Org. Chem. 2002, 4, 696–701. [Google Scholar]
- Aggarwal, V.K.; Emme, S.Y.; Fulford, S.Y. Correlation between pKa and reactivity of quinuclidine-based catalysts in the Baylis-Hillman Reaction: discovery of quinuclidine as optimum catalyst leading to substantial enhancement of scope. J. Org. Chem. 2003, 68, 692–700. [Google Scholar]
- Aggarwal, V.K.; Mereu, A. Superior amine catalysts for the Baylis-Hillman reaction: the use of DBU and its implications. Chem. Commun. 1999, 2311–2312. [Google Scholar] [CrossRef]
- Rezgui, F.; El Gaied, M.M. DMAP-catalyzed hydroxymethylation of 2-cyclohexenone in aqueous medium through Baylis-Hillman reaction. Tetrahedron Lett. 1998, 39, 5965–5966. [Google Scholar] [CrossRef]
- Luo, S.; Zhang, B.; He, J.; Janczuk, A.; Wang, P.G.; Cheng, J. Aqueous Baylis-Hillman reaction of cyclopent-2-enone using imidazole as catalyst. Tetrahedron Lett. 2002, 43, 7369–7371. [Google Scholar] [CrossRef]
- Leadbeater, N.E.; Van der Pol, C. Development of catalysts for the Baylis-Hillman reaction: the application of tetramethylguanidine and attempts to use a supported analogue. J. Chem. Soc. Perkin Trans. 2001, 1, 2831–2835. [Google Scholar] [CrossRef]
- He, L.; Jian, T.-Y.; Ye, S. N-Heterocyclic Carbene Catalyzed aza-Morita-Baylis-Hillman Reaction of Cyclic Enones with N-Tosylarylimines. J. Org. Chem. 2007, 72, 7466–7468. [Google Scholar] [CrossRef]
- He, L.; Zhang, Y.-R.; Huang, X.-L.; Ye, S. Chiral Bifunctional N-Heterocyclic Carbenes: Synthesis and Application in the aza-Morita-Baylis-Hillman Reaction. Synthesis 2008, 2825–2829. [Google Scholar]
- Oda, R.; Kawabata, T.; Tanimoto, S. Entstehung von P-Ylid aus Triphenylphosphin und Acrylsäurederivaten. Tetrahedron Lett. 1964, 5, 1653–1657. [Google Scholar] [CrossRef]
- For a recent review: Methot, J.L.; Roush, W.R. Nucleophilic phosphine organocatalysis. Adv. Synth. Catal. 2004, 346, 1035–1050. [Google Scholar] [CrossRef]
- Denmark, S.E.; Gregory, L.B. Lewis Base Catalysis in Organic Synthesis. Angew. Chem. Int. Ed. 2008, 47, 1560–1638. [Google Scholar] [CrossRef]
- Drewes, S.E.; Roos, G.H.P. Synthetic potential of the tertiary-amine-catalyzed reaction of activated vinyl carbanions with aldehydes. Tetrahedron 1988, 44, 4653–4670. [Google Scholar] [CrossRef]
- Basavaiah, P.D. Rao; Hyma, R.S. The Baylis−Hillman reaction: a novel carbon-carbon forming reaction. Tetrahedron 1996, 52, 8001–8062. [Google Scholar] [CrossRef]
- Ciganek, E. Organic Reactions; John Wiley & sons, Inc.: New York, NY, USA, 1997; Volume 51, p. 201. [Google Scholar]
- Langer, P. New strategies for the development of an asymmetric version of the Baylis-Hillman reaction. Angew. Chem. Int. Ed. 2000, 39, 3049–3052. [Google Scholar] [CrossRef]
- Basavaiah, D.; Rao, A.J.; Satyanarayana, T.T. Recent advances in the Baylis−Hillman Reaction and Applications. Chem. Rev. 2003, 103, 811–892. [Google Scholar] [CrossRef]
- Basavaiah, D.; Rao, K.V.; Reddy, R.J. The Baylis–Hillman reaction: a novel source of attraction, opportunities, and challenges in synthetic chemistry. Chem. Soc. Rev. 2007, 36, 1581–1588. [Google Scholar] [CrossRef]
- Singh, V.; Batra, S. Advances in the Baylis-Hillman reaction-assisted synthesis of cyclic frameworks. Tetrahedron 2008, 64, 4511–4574. [Google Scholar] [CrossRef]
- Keck, G.E.; Welch, D.S. Intramolecular Baylis-Hillman and Morita Reactions Using Unsaturated Thiol Ester Substrates Containing Enolizable Aldehydes. Org. Lett. 2002, 4, 3687–3690. [Google Scholar] [CrossRef]
- Yagi, K.; Turitani, T.; Shinokubo, H.; Oshima, K. Intramolecular Tamdem Michael-Type Addition/Aldol Cyclization Induced by TiCl4/R4NX Combinations. Org. Lett. 2002, 4, 3111–3114. [Google Scholar] [CrossRef]
- Shi, Y.-L.; Shi, M. Aza-Baylis-Hillman reactions and their synthetic applications. Eur. J. Org. Chem. 2007, 2905–2916. [Google Scholar]
- Masson, G.; Housseman, C.; Zhu, J. The enantioselective Morita−Baylis−Hillman reaction and its aza counterpart. Angew. Chem. Int. Ed. 2007, 46, 4614–4628. [Google Scholar] [CrossRef]
- Declerck, V.; Martinez, J.; Lamaty, F. Aza-Baylis−Hillman Reaction. Chem. Rev. 2009, 109, 1–48. [Google Scholar] [CrossRef]
- Ma, G.-N.; Jiang, J.-J.; Shi, M.; Wei, Y. Recent extensions of the Morita-Baylis-Hillman reaction. Chem. Commun. 2009, 5496–5514. [Google Scholar]
- Hill, J.S.; Isaacs, N.S. Mechanism of substitution reactions of acrylic derivatives. J. Phys. Org. Chem. 1990, 3, 285–288. [Google Scholar]
- Menozzi, C.; Dalko, P.I. Enantioselective Organocatalysis: Reactions and Experimental Procedures; Wiley-VCH: Weinheim, Germany, 2004; pp. 151–187. [Google Scholar]
- Hoffman, H.R.M.; Rabe, J. A new, efficient and stereocontrolled synthesis of trisubstituted alkenes via functionalized acrylic esters. Angew. Chem. Int. Ed. 1983, 22, 796–797. [Google Scholar] [CrossRef]
- Bode, M.L.; Kaye, P.T. A kinetic and mechanistic study of the Baylis−Hillman reaction. Tetrahedron Lett. 1991, 32, 5611–5614. [Google Scholar] [CrossRef]
- Fort, Y.; Berthe, M.C.; Caubère, P. The Baylis−Hillman reaction-Mechanism and applications revisited. Tetrahedron 1992, 48, 6371–6384. [Google Scholar] [CrossRef]
- Rozendaal, E.M.L.; Voss, B.M.W.; Scheeren, H.W. Effect of solvent, pressure and catalyst on the E/Z selectivity in the Baylis-Hillman reaction between crotononitrile and benzaldehyde. Tetrahedron 1993, 49, 6931–6936. [Google Scholar] [CrossRef]
- Shi, M.; Chen, L.-H.; Li, C.-Q. Chiral phosphine Lewis bases catalyzed asymmetric aza-Baylis-Hillman reaction of N-sulfonated imines with activated olefins. J. Am. Chem. Soc. 2005, 127, 3790–3800. [Google Scholar] [CrossRef]
- Santos, L.S.; Pavam, C.H.; Almeida, W.P.; Coelho, F.; Eberlin, M.N. Probing the mechanism of the Baylis-Hillman reaction by electrospray ionization mass and tandem mass spectrometry. Angew. Chem. Int. Ed. 2004, 43, 4330–4333. [Google Scholar] [CrossRef]
- Raheem, I.T.; Jacobsen, E.N. Highly Enantioselective Aza-Baylis-Hillman Reactions Catalyzed by Chiral Thiourea Derivatives. Adv. Synth. Catal. 2005, 347, 1701–1708. [Google Scholar] [CrossRef]
- Byun, H.S.; Reddy, K.C.; Bittman, R. Improved syntheses of ethyl(α-bromomethyl)acrylate and 2-methylene-1,3-propanediol via ethyl α-(hydroxymethyl) acrylate. Tetrahedron Lett. 1994, 35, 1371–1374. [Google Scholar]
- Auge, J.; Lubin, N.; Lubineau, A. Acceleration in water of the Baylis-Hillman reaction. Tetrahedron Lett. 1994, 35, 7947–7948. [Google Scholar]
- Basavaiah, D.; Krishnamacharyulu, M.; Rao, A.J. The Aqueous Trimethylamine Mediated Baylis-Hillman Reaction. Synth. Commun. 2000, 30, 2061–2069. [Google Scholar] [CrossRef]
- Yu, C.Z.; Liu, B.; Hu, L.Q. Efficient Baylis-Hillman Reaction Using Stoichiometric Base Catalyst and an Aqueous Medium. J. Org. Chem. 2001, 66, 5413–5418. [Google Scholar] [CrossRef]
- Cai, J.; Zhou, Z.; Zhao, G.; Tang, C. Dramatic Rate Acceleration of the Baylis-Hillman Reaction in Homogeneous Medium in the Presence of Water. Org. Lett. 2002, 4, 4723–4725. [Google Scholar] [CrossRef]
- Park, K.S.; Kim, J.; Choo, H.; Chong, Y. Octanol-Accelerated Baylis-Hillman Reaction. Synlett. 2007, 395–398. [Google Scholar]
- Shi, M.; Liu, Y.H. Traditional Morita-Baylis-Hillman reaction of aldehydes with methyl vinyl ketone co-catalyzed with triphenylphosphine and nitrophenol. Org. Biomol. Chem. 2006, 4, 1468–1470. [Google Scholar] [CrossRef]
- Barrett, A.G.M.; Cook, A.S.; Kamimura, A. Asymmetric Baylis-Hillman reaction: catalysis using a chiral pirrolizidine base. Chem. Commun. 1998, 2533–2534. [Google Scholar]
- Drewes, S.E.; Freese, S.D.; Emslie, N.D.; Roos, G.H.P. Synthesis of 3-Hydroxy-2-Methylene Carbonyl Compounds. Synth. Commun. 1988, 18, 1565–1572. [Google Scholar] [CrossRef]
- Bailey, M. ; Markó, I.E.; Ollis, W.D.; Rasmussen, P.R. Stereoselective epoxidation of hydroxyenones. The synthesis of the sidechain of clerocidin. Tetrahedron Lett. 1990, 31, 4509–4512. [Google Scholar] [CrossRef]
- Kawahara, S.; Nakano, A.; Esumi, T.; Iwabuchi, Y.; Hatakeyama, S. Isocupreidine-catalyzed Asymmetric Baylis-Hillman Reaction of Imines. Org. Lett. 2003, 5, 3103. [Google Scholar] [CrossRef]
- Shi, M. Catalytic, Asymmetric Baylis-Hillman Reaction of Imines with Methyl Vinyl Ketone and Methyl Acrylate. Angew. Chem. Int. Ed. 2002, 41, 4507. [Google Scholar] [CrossRef]
- Meng, X.; Huang, Y.; Chen, R. A Novel Selective Aza-Morita-Baylis-Hillman (aza-MBH) Domino Reaction and aza-MBH Reaction of N-Sulfonyl Imines with Acrolein Catalyzed by Bifunctional Phosphine Organocatalyst. Chem. Eur. J. 2008, 14, 6852–6856. [Google Scholar] [CrossRef]
- Matsui, K.; Takizawa, S. A Brönsted acid and Lewis base organocatalyst for the aza-Morita–Baylis–Hillman reaction. Synlett. 2006, 761–765. [Google Scholar]
- Price, K.E.; Broadwater, S.J.; Jung, H.M.; McQuade, D.T. Baylis−Hillman mechanism: A New Interpretation in Aprotic Solvents. Org. Lett. 2005, 7, 147–150. [Google Scholar]
- Price, K.E.; Broadwater, S.J.; Walker, B.J.; McQuade, D.T. A new interpretation of the Baylis−Hillman mechanism. J. Org. Chem. 2005, 70, 3980–3987. [Google Scholar] [CrossRef]
- Aggarwal, V.K.; Fulford, F.Y.; Lloyd-Jones, G.C. Re-evaluation of the mechanism of the Baylis−Hillman reaction - implications for asymmetric catalysis. Angew. Chem. Int. Ed. 2005, 44, 1706–1708. [Google Scholar] [CrossRef]
- Robiette, R.; Aggarwal, V.K.; Harvey, J.N. Mechanism of the Morita−Baylis−Hillman reaction: A computational investigation. J. Am. Chem. Soc. 2007, 129, 15513–15525. [Google Scholar] [CrossRef]
- Amarante, G.W.; Milagre, H.M.S.; Vaz, B.G.; Ferreira, B.R.V.; Eberlin, M.C.; Coelho, F. Dualistic nature of the mechanism of the Morita-Baylis-Hillman reaction probed by electrospray ionization mass spectrometry. J. Org. Chem. 2009, 74, 3031–3037. [Google Scholar]
- Carrasco-Sanchez, V.; Simirgiotis, M.J.; Santos, L.S. The Morita-Baylis-Hillman reaction: insights into asymmetry and reaction mechanisms by electrospray ionization mass spectrometry. Molecules 2009, 14, 3989–4021. [Google Scholar] [CrossRef]
- Online mechanistic investigations of catalyzed reactions by electrospray ionization mass spectrometry: A tool to intercept transient species in solution. Eur. J. Org. Chem. 2008, 235–253.
- Buskens, P.; Klankermayer, J.; Leitner, W. Bifunctional Activation and Racemization in the Catalytic Asymmetric aza-Baylis-Hillman Reaction. J. Am. Chem. Soc. 2005, 127, 16762–16763. [Google Scholar] [CrossRef]
- Utsumi, N.; Zhang, H.; Tanaka, F.; Barbas, C.B., III. A Way to Highly Enantiomerically Enriched aza-Morita-Baylis-Hillman-Type Products. Angew. Chem. Int. Ed. 2007, 46, 1878–1880. [Google Scholar] [CrossRef]
- Aroyan, C.E.; Vasbinder, M.M.; Miller, S.J. Dual Catalyst Control in the Enantioselective Intramolecular Morita-Baylis-Hillman Reaction. Org. Lett. 2005, 7, 3849–3851. [Google Scholar] [CrossRef]
- Miller, S.J. In Search of Peptide-Based Catalysts for Asymmetric Organic Synthesis. Acc. Chem. Res. 2004, 37, 601–610. [Google Scholar] [CrossRef]
- Imbriglio, J.E.; Vasbinder, M.M.; Miller, S.J. Dual Catalyst Control in the Amino Acid-Peptide-Catalyzed Enantioselective Baylis-Hillman Reaction. Org. Lett. 2003, 5, 3741–3743. [Google Scholar] [CrossRef]
- Vasbinder, M.M.; Imbriglio, J.E.; Miller, S.J. Amino acid-peptide-catalyzed enantioselective Morita-Baylis-Hillman reactions. Tetrahedron 2006, 62, 11450–11459. [Google Scholar] [CrossRef]
- Vesely, J.; Dziedzic, P.; Córdova, A. Aza-Morita-Baylis-Hillman-type reactions: highly enantioselective addition of unmodified α,β-unsaturated aldehydes with N-Boc protected imines. Tetrahedron Lett. 2007, 48, 6900–6904. [Google Scholar] [CrossRef]
- Shi, M.; Jiang, J.-K.; Li, C.-Q. Lewis base and L-proline co-catalyzed Baylis-Hillman reaction of arylaldehydes and methyl vinyl ketone. Tetrahedron Lett. 2002, 43, 127–130. [Google Scholar] [CrossRef]
- Chen, S.-H.; Hong, B.-C.; Su, C.-F.; Sarshar, S. An unexpected inversion of enantioselectivity in the proline catalyzed intramolecular Baylis-Hillman reaction. Tetrahedron Lett. 2005, 46, 8899–8903. [Google Scholar] [CrossRef]
- Duarte, F.J.S.; Cabrita, E.J.; Frenking, G.; Santos, A.G. Density functional study of proline-catalyzed intramolecular Baylis-Hillman Reactions. Chem. Eur. J. 2009, 15, 1734–1746. [Google Scholar] [CrossRef]
- Taniguchi, M.; Hino, T.; Kishi, Y. Aldol reactions of allenolates generated by 1,4-addition of iodide anion or its equivalent to acetylenic ketones. Tetrahedron Lett. 1986, 27, 4767–4770. [Google Scholar] [CrossRef]
- Uehira, S.; Nan, Z.; Shinokubo, H.; Oshima, K. Highly Stereoselective coupling reaction of acrolein or vinyl ketone with aldehydes. Org. Lett. 1999, 1, 1383–1385. [Google Scholar] [CrossRef]
- Wei, H.X.; Gao, J.J.; Li, G. Substoichiometric TiCl4-mediated vicinal difunctionalization of α,β-acetylenic ketones for the synthesis of β-halo Baylis-Hillman olefins. Tetrahedron Lett. 2001, 42, 9119–9122. [Google Scholar] [CrossRef]
- Kataoka, T.; Kinoshita, H. Chalcogenide-lewis acid mediated tandem michael aldol reaction. An alternative to the morita-baylis-hillman reaction and a new development. Eur. J. Org. Chem. 2005, 45–48. [Google Scholar] [CrossRef]
- Balan, D.; Adolfsson, H. titanium isopropoxide as efficient catalyst for the Aza-Baylis-Hillman Reaction. selective formation of α-methylene-β-amino acid derivatives. J. Org. Chem. 2002, 67, 2329–2334. [Google Scholar] [CrossRef]
- Walsh, L.M.; Winn, C.L.; Goodman, J.M. Sulfide-BF3.Et2O mediated Baylis-Hillman reactions. Tetrahedron Lett. 2002, 43, 8219–8222. [Google Scholar] [CrossRef]
- Kuwajima, I.; Nakamura, E. Quaternary Ammonium Enolates as Synthetic Intermediates. Regiospecific Alkylation Reaction of Ketones. J. Am. Chem. Soc. 1975, 97, 3257–3258. [Google Scholar] [CrossRef]
- Noyori, R.; Nishida, I.; Sakata, J.; Nishizawa, M. Tris(dimethylamine)sulfonium Enolates. J. Am. Chem. Soc. 1980, 102, 1223–1225. [Google Scholar] [CrossRef]
- Noyori, R.; Nishida, I.; Sakata, J. Alkylation via tris(dialkylamino)sulfonium enolates. Tetrahedron Lett. 1980, 21, 2085–2088. [Google Scholar] [CrossRef]
- Noyori, R.; Yokoyama, K.; Sakata, J.; Kuwajima, I.; Nakamura, E. Fluoride ion catalyzed Aldol Reaction between enol silyl ethers and carbonyl compounds. J. Am. Chem. Soc. 1977, 99, 1265–1267. [Google Scholar] [CrossRef]
- Kleshick, W.A.; Buse, C.T.; Heathcock, C.H. Stereoselection in aldol condensation. J. Am. Chem. Soc. 1977, 99, 247–248. [Google Scholar] [CrossRef]
- Notice that for the case of unsaturated esters this intermediate is in fact the E enolate as OMe has precedence over the O- for nomenclature
- Cannizzaro, C.E.; Houk, K.N. Magnitudes and Chemical Consequences of R3N+-C-H…O=C Hydrogen Bonding. J. Am. Chem. Soc. 2002, 124, 7163–7169. [Google Scholar] [CrossRef]
- Rafel, S.; Leahy, J.W. An Unexpected rate acceleration-practical improvements in the Baylis-Hillman Reaction. J. Org. Chem. 1997, 62, 1521–1522. [Google Scholar] [CrossRef]
- Robiette, R.; Aggarwal, V.K.; Harvey, J.N. Mechanism of the Morita−Baylis−Hillman reaction: A computational investigation. J. Am. Chem. Soc. 2007, 129, 15513–15525. [Google Scholar] [CrossRef]
- Kraftt, M.E.; Haxell, T.F.N.; Seibert, K.A.; Abboud, K.A. Implications in the Morita−Baylis−Hillman Alkylation: Isolation and Characterization of an Intermediate. J. Am. Chem. Soc. 2006, 128, 4174–4175. [Google Scholar]
- Reviews on catalytic enantioselective condensations: Palomo, C.; Oiarbide, M.; García, J.M. The Aldol Addition Reaction : An Old Transformation at Constant Rebirth. Chem. Eur. J. 2002, 8, 36–44. [Google Scholar] [CrossRef]
- Alcaide, B.; Almendros, P. The Direct Catalytic Asymmetric Cross-Aldol Reaction of Aldehydes. Angew. Chem. Int. Ed. 2003, 42, 858. [Google Scholar] [CrossRef]
- Heathcock, C.H. Comprehensive Organic Synthesis; Trost, B.M., Fleming, I., Heathcock, C.H., Eds.; Pergamon: Oxford, UK, 1991; Volume 2, pp. 133–176. [Google Scholar]
- Teng, W.-D.; Huang, R.; Kwong, C.K.-W.; Shi, M.; Toy, P.H. Influence of Michael Acceptor Stereochemistry on Intramolecular Morita-Baylis-Hillman Reactions. J. Org. Chem. 2006, 71, 368–371. [Google Scholar]
- Heathcock, C.H. Asymmetric Syntheis; Morrison, J.D., Ed.; Academic Press, Inc.: New York, NY, USA, 1984; Volume 3, pp. 111–212. [Google Scholar]
- Trost, B.M; Fleming, I.; Heathcock, C.H. Comprehensive Organic Synthesis; Pergamon: Oxford, UK, 1991; Volume 2, pp. 173–179. [Google Scholar]
- Noyori, R.; Nishida, I.; Sakata, J. Erytro-selective aldol reaction via tris(dimethylamine)sulfonium enolates. J. Am. Chem. Soc. 1981, 103, 2106–2108. [Google Scholar] [CrossRef]
- Noyori, R.; Nishida, I.; Sakata, J.; Nishizawa, M. Tris(dialkylamine)sulfonium enolates. Synthesis, structure and reactions. J. Am. Chem. Soc. 1983, 105, 1598–1608. [Google Scholar] [CrossRef]
- Zimmerman, H.E.; Traxler, M.D. The Stereochemistry of the Ivanov and Reformatsky Reactions. J. Am. Chem. Soc. 1957, 79, 1920–1923. [Google Scholar] [CrossRef]
- According to nomenclature employed by Noyori’s group at the eighties syn aldols were named as erythro and anti aldols were named as threo
- Dubois, J-E.; Fort, J.-F. Dynamic stereochemistry of aldolization-XXI. Definition of “restoring energy” of a system of reversible competitive reactions. Tetrahedron 1972, 28, 1665–1675. [Google Scholar] [CrossRef]
- Murata, S.; Suzuki, M.; Noyori, R. Trialkylsilyl triflates. 5. A stereoselective aldol-type condensation of enol silyl ethers and acetals catalyzed by trimethylsilyl trifluoromethanesulfonate. J. Am. Chem. Soc. 1980, 102, 3248–3249. [Google Scholar] [CrossRef]
- Heathcock, C.H.; Davidsen, S.K.; Flippin, L.A. Acyclic stereoselection. 36. Simple diastereoselection in the Lewis acid mediated reactions of enol silanes with aldehydes. J. Org. Chem. 1986, 51, 3027–3037. [Google Scholar] [CrossRef]
- Denmark, S.E; Beutner, G.L.; Wynn, T.; Eastgate, M.D. Lewis base activation of lewis acids: catalytic, enantioselective addition of silyl ketene acetals to aldehydes. J. Am. Chem. Soc. 2005, 127, 3774–3789. [Google Scholar] [CrossRef]
- Denmark, S.; Chung, W.-j. Lewis Base activation of lewis acids: catalytic enantioselective glycolate aldol reactions. Angew. Chem. Int. Ed. 2008, 47, 1890–1892. [Google Scholar] [CrossRef]
- Abermil, M.; Masson, G.; Zhu, J. Highly Enantioselective aza Morita-Baylis-Hillman Reaction Catalyzed by Bifunctional β-isocupreidine Derivatives. J. Am. Chem. Soc. 2008, 130, 12596–12597. [Google Scholar]
- Abermil, M.; Masson, G.; Zhu, J. Invertible Enantioselectivity in 6’-Deoxy-6’-acylamino-β-Isocupreidine-catalyzed Aza-Morita-Baylis-Hillman Reaction: Key Role of an Achiral Additive. Org. Lett. 2009, 11, 4648–4651. [Google Scholar] [CrossRef]
- Ibawuchi, Y.; Nakatani, M.; Yokoyama, N.; Hatakeyama, S. Chiral amine-catalyzed asymmetric Baylis-Hillman reaction: A reliable route to highly enantiomerically-enriched to (α-Methylene-β-hydroxy)esters. J. Am. Chem. Soc. 1999, 121, 10219–10220. [Google Scholar] [CrossRef]
- Wang, X.; Chen, Y.-F.; Niu, L.-F.; Xu, P.-X. Diastereo- and enantioselective aza-mbh-type reaction of nitroalkenes to n-tosylimines catalyzed by bifunctional organocatalysts. Org. Lett. 2009, 11, 3310–3313. [Google Scholar] [CrossRef]
- Dubois, J.E.; Dubois, M. The aldol condensation: influence of the solvent and the cation on stereoselectivity. Chem Commun. 1968, 1567–1568. [Google Scholar]
- Roy, D.; Sunoj, R.B. Ab Initio and Density Functional Theory Evidence on the Rate-Limiting Step in the Morita−Baylis−Hillman Reaction. Org. Lett. 2007, 9, 4873–4876. [Google Scholar] [CrossRef]
- Roy, D.; Sunoj, R.B. Water Catalysis in the Morita-Baylis-Hillman Reaction: A Mechanistic Perspective. Chem. Eur. J. 2008, 14, 10530–10534. [Google Scholar] [CrossRef]
- Fan, J.-F.; Yang, C.-H.; He, L.-J. DFT Study on the Role of Methanol Solvent in Morita-Baylis-Hillman Reaction. J. Int. Q. Chem. 2009, 109, 1311–1321, See also [62]. [Google Scholar] [CrossRef]
- Xu, J. Probing the mechanism of Morita-Baylis-Hillman reaction in dichloromethane by density functional theory. J. Mol. Struct. (TeoChem.) 2006, 767, 61–66, See also [62]. [Google Scholar] [CrossRef]
- Garnier, J.-M.; Anstiss, C.; Liu, F. Enantioselective Trifunctional Organocatalysis for Rate-Enhanced aza-Morita-Baylis-Hillman Reactions at Room Temperature. Adv. Synth. Catal. 2009, 351, 331–338. [Google Scholar] [CrossRef]
- Garnier, J.-M.; Liu, F. Trifunctional organocatalyst-promoted counterion catalysis for fast and enantioselective aza-Morita-Baylis-Hillman reactions at ambient temperature. Org. Biomol. Chem. 2009, 7, 1272–1275. [Google Scholar] [CrossRef]
- Jones, C.E.S.; Turega, S.M.; Clarke, M.L.; Philp, D. A rationally designed cocatalyst for the Morita−Baylis−Hillman reaction. Tetrahedron Lett. 2008, 49, 4666–4669. [Google Scholar] [CrossRef]
- Yuan, K.; Zhang, L.; Song, H.-L.; Hu, Y.; Wu, X.-Y. Chiral phosphinothiourea organocatalyst in the enantioselective Morita–Baylis–Hillman reactions of aromatic aldehydes with methyl vinyl ketone. Tetrahedron Lett. 2008, 49, 6262–6264. [Google Scholar]
- Wang, J.; Li, H.; Yu, X.; Zu, L.; Wang, W. Chiral binaphthyl-derived amine-thiourea organocatalyst-promoted asymmetric Morita-Baylis-Hillman reaction. Org. Lett. 2005, 7, 4293–4296. [Google Scholar] [CrossRef]
- Sohtome, Y.; Takemura, N.; Takagi, R.; Hashimoto, Y.; Nagasawa, K. Thiourea-catalyzed Morita–Baylis–Hillman reaction. Tetrahedron 2008, 64, 9423–9429. [Google Scholar] [CrossRef]
- McDougal, N. T.; Schaus, S. E. Asymmetric Morita-Baylis-Hillman Reactions Catalyzed by Chiral Brønsted Acids. J. Am. Chem. Soc. 2003, 125, 12094–12095. [Google Scholar] [CrossRef]
- Shi, M.; Liu, X.-G. Asymmetric Morita-Baylis-Hillman Reaction of Arylaldehydes with 2-Cyclohexen-1-one Catalyzed by Chiral Bis(Thio)urea and DABCO. Org. Lett. 2008, 10, 1043–1046. [Google Scholar] [CrossRef]
- Matsui, K.; Takizawa, S.; Sasai, H. Bifunctional organocatalysts for enantioselective aza-Morita-Baylis-Hillman reaction. J. Am. Chem. Soc. 2005, 127, 3680–3681. [Google Scholar] [CrossRef]
- Matsui, K.; Tanaka, K.; Horii, A.; Takizawa, S.; Sasai, H. Conformational lock in a Brønsted acid–Lewis base organocatalyst for the aza-Morita–Baylis–Hillman reaction. Tetrahedron: Asymmetry 2006, 17, 578–583. [Google Scholar] [CrossRef]
- Shi, M.; Chen, L.-H. Chiral phosphine Lewis base catalyzed asymmetric aza-Baylis–Hillman reaction of N-sulfonated imines with methyl vinyl ketone and phenyl acrylate. Chem. Commun. 2003, 1310–1311. [Google Scholar]
- Shi, M.; Chen, L.-H.; Teng, W.D. Asymmetric aza-Morita-Baylis-Hillman reaction of N-sulfonated imines with methyl vinyl ketone catalyzed by chiral phosphine Lewis bases bearing perfluoroalkanes as pony tails. Adv. Synth. Catal. 2005, 347, 1781–1789. [Google Scholar] [CrossRef]
- Shi, M.; Li, C.Q. Catalytic, asymmetric aza-Baylis–Hillman reaction of N-sulfonated imines with 2-cyclohexen-1-one and 2-cyclopenten-1-one in the presence of a chiral phosphine Lewis base. Tetrahedron Asymmetry 2005, 16, 1385–1391. [Google Scholar] [CrossRef]
- Liu, Y.-H.; Chen, L.-C.; Shi, M. Asymmetric Aza-Morita–Baylis_Hillman Reaction of N-Sulfonated Imines with Activated Olefins Catalyzed by Chiral Phosphine Lewis Bases Bearing Multiple Phenol Groups. Adv. Synth. Catal. 2006, 348, 973–979. [Google Scholar] [CrossRef]
- Guan, X.- Y.; Jiang, Y.-Q.; Shi, M. Chiral Sterically Congested Phosphane-Amide Bifunctional Organocatalysts in Asymmetric Aza-Morita–Baylis–Hillman Reactions of N-Sulfonated Imines with Methyl and Ethyl Vinyl Ketones. Eur. J. Org. Chem. 2008, 2150–2155. [Google Scholar]
- Ito, K.; Nishida, K.; Gotanda, T. Highly enantioselective aza-Morita–Baylis–Hillman reaction with a bisphenol-based bifunctional organocatalyst. Tetrahedron Lett. 2007, 48, 6147–6149. [Google Scholar] [CrossRef]
- Richards, E.L.; Murphy, P.J.; Dinon, F.; Fratucello, S.; Brown, P.M.; Gelbrich, T.; Hurstouse, M.B. Assesing the scope of the tandem Michael/intramolecular aldol reaction mediated by secondary amines, thiols and phosphines. Tetrahedron 2001, 57, 7771–7784. [Google Scholar] [CrossRef]
- Dinon, F.; Richards, E.; Murphy, P.J.; Hibbs, D.E.; Hurtshouse, M.B.; Abdul Malik, K.M. Tandem intramolecular/aldol reactions mediated by secondary amines, thiols and phosphines. Tetrahedron Lett. 1999, 40, 3279–3282. [Google Scholar]
- Black, G.P.; Dinon, F.; Fratucello, S.; Murphy, P.J.; Nielsen, M.; Williams, H.L. Intramolecular Baylis-Hillman reaction catalyzed by secondary amines. Tetrahedron Lett. 1997, 38, 8561–8564. [Google Scholar]
- Shi, M.; Jiang, J.-K.; Li, C.-Q. Lewis base and L-proline co-catalyzed Baylis-Hillman reaction of arylaldehydes with metil vinyl ketone. Tetrahedron Lett. 2002, 43, 127–130. [Google Scholar] [CrossRef]
- Shi, M.; Jiang, J.-K. An exploration of asymmetric Baylis-Hillman reactions catalyzed by quinidine-derived chiral amines. Tetrahedron Asymmetry 2002, 13, 1941–1947. [Google Scholar] [CrossRef]
- List, B.; Lerner, R.; Barbas, C.F., III. Proline-catalyzed direct asymmetric aldol reactions. J. Am. Chem. Soc. 2000, 122, 2395–2396. [Google Scholar] [CrossRef]
- Notz, W.; List, B. Catalytic Asymmetric Synthesis of anti-1,2-diols. J. Am. Chem. Soc. 2000, 122, 7386–7387. [Google Scholar] [CrossRef]
- Bui, T.; Barbas, III, C.F. A proline-catalyzed asymmetric Robinson annulation reaction. Tetrahedron Lett. 2000, 41, 6951–6954. [Google Scholar] [CrossRef]
- Ahrendt, K.A.; Borths, C.J.; MacMillan, D.W.C. New strategies for organic catalysis: the First highly enantioselective organocatalytic Diels-Alder reaction. J. Am. Chem. Soc. 2000, 122, 4243–4244. [Google Scholar] [CrossRef]
- Bahmanyar, S.; Houk, K.N.; Martin, H.J.; List, B. Quantum-mechanical predictions of the stereoselectivities of proline-catalyzed asymmetric intermolecular Aldol Additions. J. Am. Chem. Soc. 2003, 125, 2475–2479. [Google Scholar] [CrossRef]
- Tang, H.; Gao, P.; Zhao, G.; Zhou, Z.; He, L.; Tang, C. (1R,2R)-(-)-2-Dimethylamino-1-(4-nitrophenyl)-1,3-propanediol/L-proline cocatalyzed enantioselective Morita-Baylis-Hillman reaction. Cat. Commun. 2007, 8, 1811–1814. [Google Scholar] [CrossRef]
- Tang, H.; Zhao, G.; Zhou, Z.; Gao, P.; He, L.; Tang, C. Chiral tertiary Amine/L-Proline Cocatalyzed Enantioselective Morita-Baylis-Hillman (MBH) Reaction. Eur. J. Org. Chem. 2008, 126–135. [Google Scholar]
- Imbriglio, J.E.; Vasbinder, M.M.; Miller, S.J. Dual catalyst control in the amino acid-peptide-catalyzed enantioselective Baylis-Hillman Reaction. Org. Lett. 2003, 5, 3741–3743. [Google Scholar] [CrossRef]
- Vasbinder, M.M.; Imbriglio, J.E.; Miller, S.J. Amino acid-peptide-catalyzed enantioselective Morita-Baylis-Hillman reactions. Tetrahedron 2006, 62, 11450–11459. [Google Scholar] [CrossRef]
- Aroyan, C.E.; Vasbinder, M.M.; Miller, S.J. Dual Catalyst Control in the Enantioselective Intramolecular Morita-Baylis-Hillman Reaction. Org. Lett. 2005, 7, 3849–3851. [Google Scholar] [CrossRef]
- Chen, S.-H.; Hong, B.-C.; Su, C.-F.; Sarshar, S. An unexpected inversion of enantioselectivity in the proline catalyzed intramolecular Baylis-Hillman reaction. Tetrahedron Lett. 2005, 46, 8899–8903. [Google Scholar] [CrossRef]
- Clemente, F.R.; Houk, K.N. Computational evidence for the enamine mechanism of intramolecular Aldol Reactions catalyzed by proline. Angew. Chem. Int. Ed. 2004, 43, 5766–5768. [Google Scholar] [CrossRef]
- Clemente, F.R.; Houk, K.N. Theoretical Studies of Stereoselectivities of Intramolecular Aldol Cyclizations Catalyzed by Amino Acids. J. Am. Chem. Soc. 2005, 127, 11294–11302. [Google Scholar] [CrossRef]
- Duarte, F.J.S.; Cabrita, E.J.; Frenking, G.; Gil Santos, A. Density Functional Study of Proline-Catalyzed Intramolecular Baylis-Hillman Reactions. Chem. Eur. J. 2009, 15, 1734–1746. [Google Scholar] [CrossRef]
- Pidathala, C.; Hoang, L.; Vignola, N.; List, B. Direct, Catalytic Asymmetric eno/exo Aldolizations. Angew. Chem. Int. Ed. 2003, 42, 2785–2788. [Google Scholar] [CrossRef]
- Utsumi, N.; Zhang, H.; Tanaka, F.; Barbas, III, C.F. A Way to highly enantiomerically enriched Aza-Morita-Baylis-Hillman-type products. Angew. Chem. Int. Ed. 2007, 46, 1878–1880. [Google Scholar]
- Vesely, J.; Dziedzic, P.; Córdova, A. Aza-Morita-Baylis-Hillman-type reactions: highly enantioselective organocatalytic addition of unmodified α,β-unsaturated aldehydes to N-BOC protected imines. Tetrahedron Lett. 2007, 48, 6900–6904. [Google Scholar] [CrossRef]
- List, B. the direct catalytic asymmetric three-component mannich reaction. J. Am. Chem. Soc. 2000, 126, 9336–9337. [Google Scholar] [CrossRef]
- List, B.; Pojarliev, P.; Biller, W.T.; Martin, H.J. The proline-catalized asymmetric three component Mannich reaction: Scope, optimization, and applications to the highly enbantioselective synthesis of 1,2-aminoalcohols. J. Am. Chem. Soc. 2002, 124, 827–833. [Google Scholar]
- Gausepohl, R.; Buskens, P.; Kleinen, J.; Bruckman, A.; Lehmann, C.W.; Klankermayer, J.; Leitner, W. Highly enantioselective Aza-Baylis-Hillman in a chiral reaction medium. Angew. Chem. Int. Ed. 2006, 45, 3689–3692. [Google Scholar] [CrossRef]
© 2010 by the authors;
Share and Cite
MDPI and ACS Style
Mansilla, J.; Saá, J.M. Enantioselective, Organocatalytic Morita-Baylis-Hillman and Aza-Morita-Baylis-Hillman Reactions: Stereochemical Issues. Molecules 2010, 15, 709-734. https://doi.org/10.3390/molecules15020709
AMA Style
Mansilla J, Saá JM. Enantioselective, Organocatalytic Morita-Baylis-Hillman and Aza-Morita-Baylis-Hillman Reactions: Stereochemical Issues. Molecules. 2010; 15(2):709-734. https://doi.org/10.3390/molecules15020709
Chicago/Turabian StyleMansilla, Javier, and José M. Saá. 2010. "Enantioselective, Organocatalytic Morita-Baylis-Hillman and Aza-Morita-Baylis-Hillman Reactions: Stereochemical Issues" Molecules 15, no. 2: 709-734. https://doi.org/10.3390/molecules15020709