Synthesis, Characterization and Spectral Properties of Substituted Tetraphenylporphyrin Iron Chloride Complexes

Institute of Green Chemistry and Fine Chemicals, Beijing University of Technology, Beijing 100124, China
*
Author to whom correspondence should be addressed.
Molecules 2011, 16(4), 2960-2970; https://doi.org/10.3390/molecules16042960
Submission received: 22 February 2011
/
Revised: 23 March 2011
/
Accepted: 31 March 2011
/
Published: 6 April 2011
Abstract
:A series of substituted tetraphenylporphyrin iron chloride complexes [RTPPFe(III)Cl, R=o/p-NO2, o/p-Cl, H, o/p-CH3, o/p-OCH3] were synthesized by a novel universal mixed-solvent method and the spectral properties of free base porphyrins and iron porphyrin compounds were compared with each other. The experimental results showed that the one-pot mixed solvent method was superior to the two-step method in the yields, reaction time and workup of reaction mixtures for the synthesis of iron porphyrin compounds. The highest yields (28.7%–40.4%) of RTPPFe(III)Cl were obtained in the mixed solvents propionic acid, glacial acetic acid and m-nitrotoluene under reflux for 2 h. A detailed analysis of ultraviolet-visible (UV-vis), infrared (IR) and far-infrared (FIR) spectra suggested the transformation from free base porphyrins to iron porphyrins. The red shift of the Soret band in ultraviolet-visible spectra due to the presence of p-nitrophenyl substituents and the blue shift of Fe-Cl bond of TPPFeCl in far-infrared spectra were further explained by the electron transfer and molecular planarity in the porphyrin ring.
1. Introduction
In recent years substituted porphyrin-like complexes with conjugated macrocycles have been essential to the study of biomimetic chemistry [1,2], iatrology [3], analytical chemistry [4] and molecular electronic devices [5]. One of the most important applications of the porphyrin-like complexes is as a model for the natural enzyme peroxidase, in which the dioxygen has been activated by metalloporphyrins under mild conditions [6]. Especially, the iron porphyrin complexes are widely used as model compounds to simulate the catalytic behavior of cytochrome P450 enzymes in life processes [7]. They can be used as biomimetic catalysts to catalyze the selective oxidation of saturated hydrocarbons, aromatic hydrocarbons and their side chains with dioxygen [8,9,10]. The importance of iron porphyrin complexes in biological systems and biomimetic catalytic reactions have prompted extensive studies on the synthesis of iron porphyrin compounds [11]. Many new synthetic methods of metalloporphyrins have been developed, including the tetramerization of pyrrole [12,13,14,15,16] and the self-condensation of dipyrromethene [17,18]. Thereinto, the synthesis of substituted tetraphenylporphyrin compounds via the tetramerization of benzaldehyde and pyrrole has also been improved by using different organic oxidants [19,20], carboxylic acids [21] and solvents [22].
The tetraarylporphyrins were obtained for the first time by Rothemund [23,24]. Up to now, there exist two simple and practical methods of synthesizing porphyrin compounds. Adler and Longo firstly converted aromatic aldehyde and pyrrole to corresponding porphyrin complexes in a single refluxing carboxylic acid with air oxidation [25]. The separation of products was relatively simple using the Adler-Longo method [13], but the yields of porphyrins rarely exceeded 20%. Lindsey’s group subsequently developed another synthetic strategy to form substituted tetraphenylporphyrin compounds in CH2Cl2 solvent with BF3 etherate as catalyst and p-chloranil as oxidant [14]. The Lindsey synthetic approach was more feasible for the larger scale synthesis [26] and obtained much higher yields of substituted tetraphenylporphyrins with the addition of salts [27]. However, large diluent agents and expensive quinones were involved in the Lindsey method, which brought about the high cost of porphyrin synthesis and restricted their applications at present. Therefore, the Adler method are of the advantages of simple manipulation and ease of workup with low yields, while the Lindsey method can obviously improve the yields of porphyrins with costly and environmental impacts. Meanwhile, the synthetic methods are different for the metalloporphyrins with various structures in the porphyrin rings, which lead to the universal synthetic method impossible to be used in the porphyrin complexes with various substituents. Therefore, the study on synthetic methods for conveniently improving the yields of metalloporphyrin complexes is of great significance. In this work, a series of substituted tetraphenylporphyrin iron chloride complexes were synthesized with two different methods. Simultaneously, the structures of above porphyrin-like complexes were characterized by UV-vis, IR, FIR and elemental analysis.
2. Results and Discussion
2.1. Synthesis with One-pot and Two-step Methods
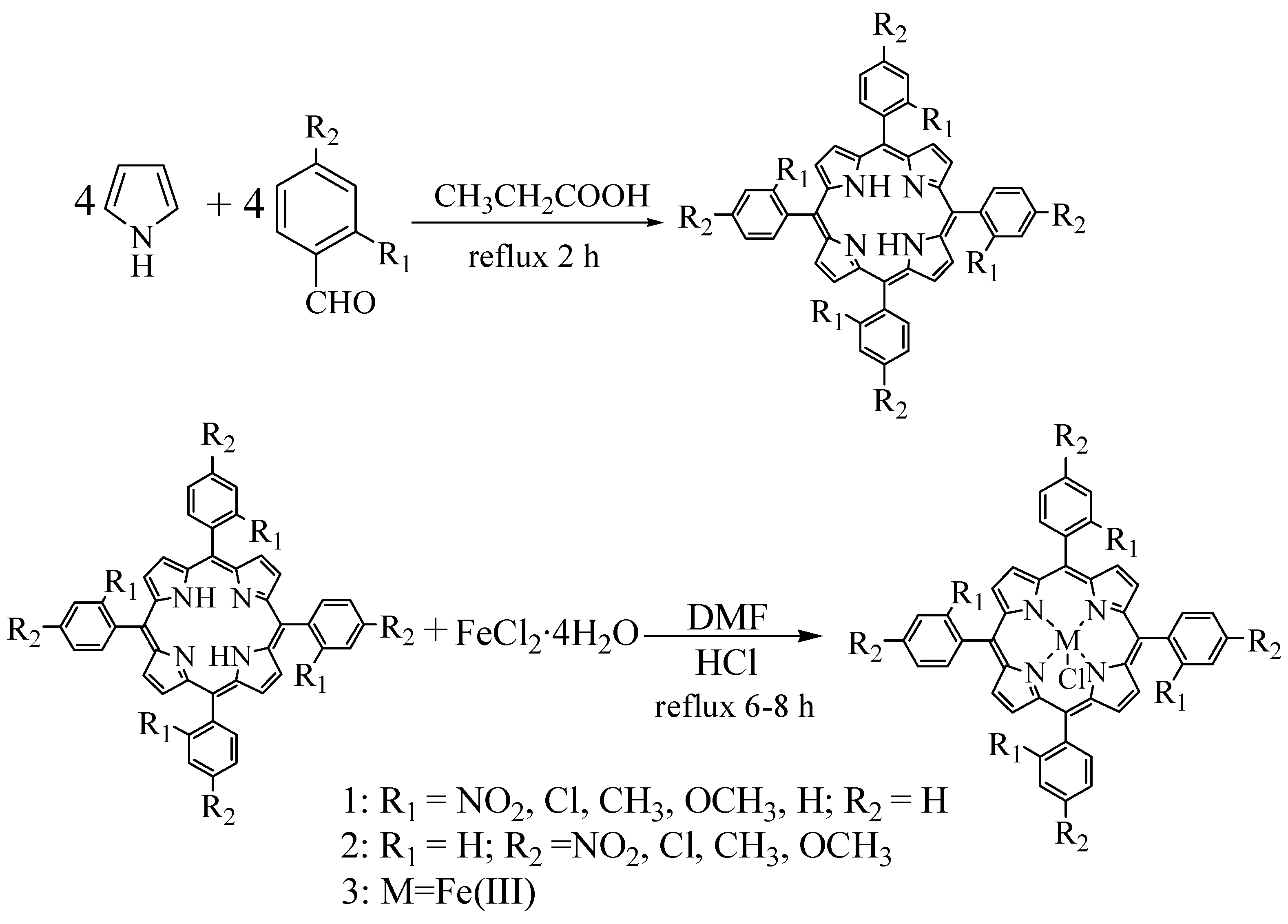
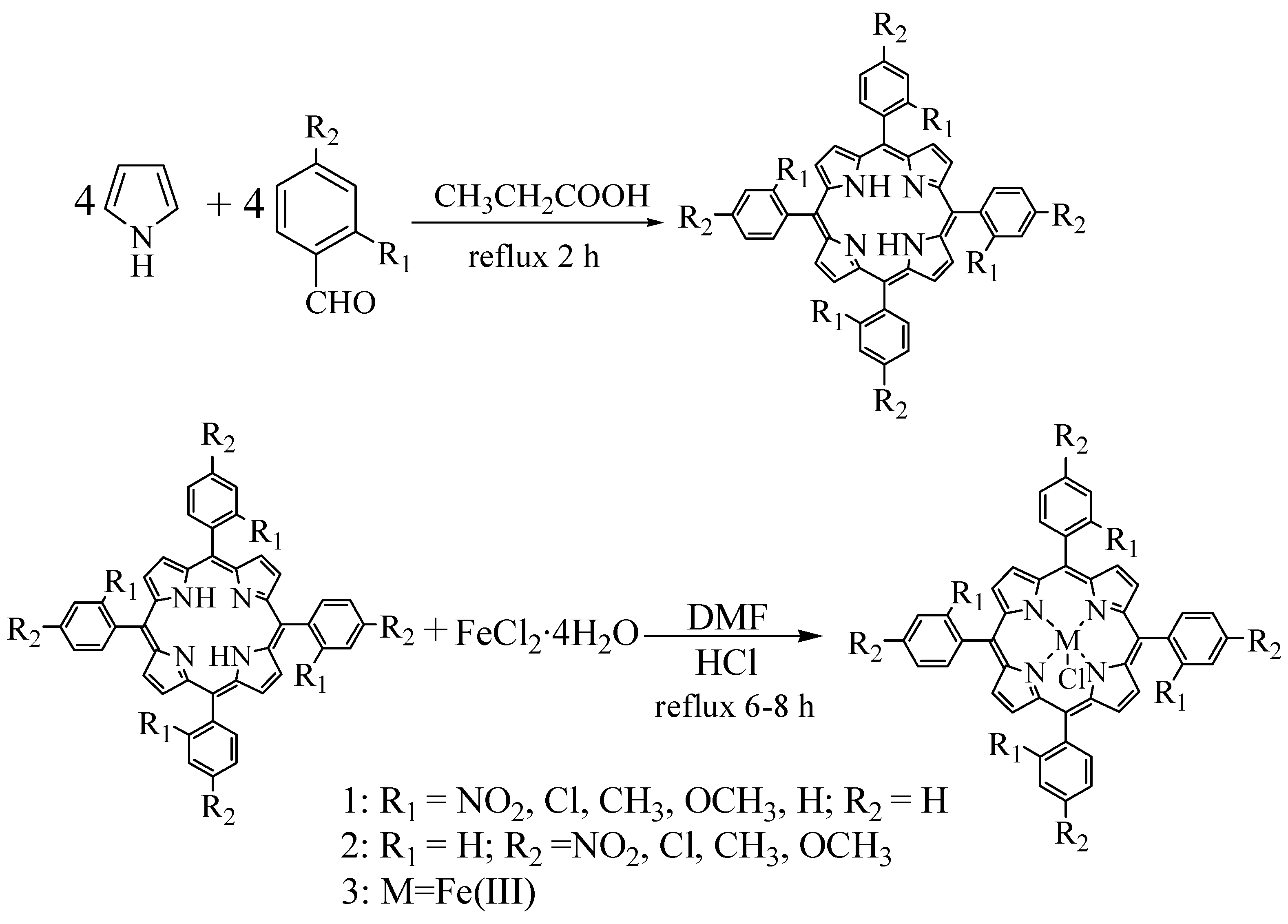
Nine substituted tetraphenylporphyrin iron chloride compounds were prepared using the one-pot mixed solvent method and the Adler two-step method. The synthetic reactions are shown in Scheme 1 and Scheme 2.
It was well known that the classic Adler two-step method of synthesizing metalloporphyrins is carried out by reacting free base porphyrins with metal salts in refluxing dimethylformamide (DMF) for a long time [28]. This method had advantages in the manipulation of synthesis and purities of products, but the yields of synthesized free base porphyrins were much lower (< 20%) and the reaction time for synthesizing metalloporphyrins was too long. Therefore, the prominent advantages of synthesizing metalloporphyrins by using one-pot mixed solvent method were that much higher yields of metalloporphyrin complexes could be obtained after shorter reaction times with simple workup of the reaction mixtures. A comparison of yields between the one-pot and two-step synthetic methods is given in Table 1.
From Table 1, it could be found that the synthetic yields of porphyrins were improved by the one-pot method, and the yields of substituted tetraphenylporphyrin iron chlorides were much higher (from 28.7% to 40.4%) than those (from 5.6% to 18.4%) in two-step method. For the synthesis of porphyrins in a single carboxylic acid (e.g. propionic acid) with air oxidation, it was difficult to turn up the appropriately corresponding conditions, especially such as polarity (dipole moment), acidity (pKa) and reaction temperature (reflux temperature), therefore, the synthetic yields of porphyrins were relatively low in the single carboxylic acid and this single-solvent method was not universal for the synthesis of different substituted porphyrins. By mixing specific carboxylic acids and m-nitrotoluene in some proportion, the polarity and acidity of the reaction system may be conveniently improved, further promoting the deprotonation ability of α-H in pyrrole and the protonation strength of C=O in the aromatic aldehyde. Simultaneously, the reflux temperature (120 oC) in mixed solvents is lower than that in the single propionic acid (141 oC) or DMF (153 oC), which decreases the side effects caused by high reaction temperature. In addition, besides changing the polarity of the mixed solvent systems, the m-nitrotoluene in the mixed solvents played a role of oxidant [29], since m-nitrotoluene is a weak organic oxidant that can effectively promote the formation of free base porphyrins in contrast to atmospheric O2 as oxidant and correspondingly increased the yields of metalloporphyrins. The ratio of mixed solvents also had an important influence on the synthesis of substituted tetraphenylporphyrins. By changing the ratio of mixed solvents, the polarity of mixed solvents could be better adjusted to increase the collision frequencies of reactant molecules and accelerate the formation of porphyrins in the one-pot mixed solvent system. Moreover, the one-pot mixed solvent method was valid for different substituted benzaldehydes and the reaction time was shorter than that in the two-pot method. Therefore, the one-pot mixed solvent method was an efficient one to synthesize tetraphenylporphyrin iron compounds with different substituents.
2.2. Spectral Analysis of RTPPFe(III)Cl
The structures of all free base porphyrins and substituted tetraphenylporphyrin iron(III) chlorides were characterized through UV-vis and the spectral data are listed in Table 2.
By comparing the UV-vis data in Table 2, it was noticed that four absorption bands presented at the Q band and the maximum wavelength absorption (λmax) presented at the Soret band along with the formation of free base porphyrins. When the metal ion was inserted into the porphyrin ring and then coordinated with four N atoms, the iron ion located in the center of the porphyrin ring to form the iron porphyrin compounds. Then the number and intensity of the Q bands decreased and the Soret band occurred slightly red shift (e.g. o-ClTPP to o-ClTPPFeCl, from 412 nm red shift to 418 nm), which was the characteristics of iron porphyrin compounds formed. The reason might be that the structure symmetry of iron porphyrin compounds with C4v point groups was improved and the energy gap decreased comparing with free base porphyrins with D2h point groups. Therefore, the UV-vis spectra of iron porphyrin compounds were obviously different from those of free base porphyrins.
It also could be observed that the absorption band in the UV-vis region of iron porphyrin compounds with –NO2 group located at ~422 nm, which revealed the red shift compared with other iron porphyrin compounds. The reason might be that the strong electron-withdrawing –NO2 group decreased the electronic density of the porphyrin ring. Thus, the energy levels of π1 and π2 orbits were increased and the energy gap between HOMO and LUMO of the porphyrin ring became smaller. The π-π* electron excitation of the porphyrin ring required absorbing the light of smaller energy (longer wavelength), accordingly the absorption band (Soret band) occurred red shift and located in the long wavelength region. Infrared and far-infrared spectra data of above porphyrin compounds were listed in Table 3.
As shown in Table 3, the IR absorption frequencies were different for free base porphyrins and iron porphyrin complexes with different functional groups. It was found that the N-H bond stretching and bending frequencies of free base porphyrins located at ~3,300 cm−1 and ~960 cm−1. When the iron ion was inserted into the porphyrin ring, the N-H bond vibration frequency of free base porphyrins disappeared and the characteristic functional groups of Fe-N bond formed at ~1,000 cm−1, which indicated the formation of iron porphyrin compounds [30]. The bands at 2,923~3,133 cm−1 were assigned to the C-H bond of the benzene ring and pyrrole ring. The bands at 1,494~1,682 cm−1 and 1,334~1,352 cm−1 were assigned to the C=C stretching mode and the C=N stretching vibration respectively. The bands at ~800 cm−1 and ~750 cm−1 were respectively assigned to the C-H bond bending vibration of para-substituted and ortho-substituted phenyl ring. The FIR characterization of Fe-Cl bond vibration located at 359~379 cm−1. The above results were in good agreement with the substituted tetraphenylporphyriniron chlorides as expected. Moreover, it could be well observed that the Fe-Cl bond vibration frequency (379 cm−1) of TPPFeCl was obviously higher than that in other substituted tetraphenylporphyrin iron chlorides. Generally, the vibration frequency shifted to the higher frequency region (blue shift) as the bond energy increased. Owing to the fact TPPFeCl has a MOOP structure [30] it was roughly planar and this led to better resonance effects than RTPPFeCl with a ruffling structure. Accordingly, the bond energy of Fe-Cl bond in TPPFeCl increased and the vibrational frequency greatly shifted to the higher region.
3. Experimental
3.1. Materials and Instruments
All chemicals were obtained commercially and used as received unless otherwise noted. Pyrrole was redistilled before use. CH2Cl2 was dehydrated. Neutral Al2O3 was baked at 100 °C for 5 h. Chromatography was performed on neutral Al2O3. UV-Vis spectra were recorded in CH2Cl2 with a HITACHI U-3010 spectrophotometer. IR spectra were recorded as KBr pellets via a Nicolet AVATAR-360 spectrophotometer. FIR spectra were recorded on a Brucker FT-IR VERTEX 70 spectrometer. The data of elemental analysis were obtained with an EURO EA3000 elemental analyzer.
3.2. Synthesis of RTPPFe(III)Cl
3.2.1. Synthesis–Adler Two-step Method
RTPPH2 (R=o/p-NO2, o/p-Cl, H, o/p-CH3, o/p-OCH3) were synthesized by the direct condensation of pyrrole with substituted benzaldehydes according to the documented procedure [13]. The free base porphyrins were purified through a column of Al2O3 (grade III) and eluted with CH2Cl2. The solvent was then removed under vacuum and the purified porphyrin complexes were obtained in yields of less than 20%.
o-NO2TPPFeCl. o-NO2TPPH2 (0.2 g, 0.25 mmol) was dissolved in DMF (100 mL). The solution was heated to reflux with magnetic stirring. Upon dissolution of the o-NO2TPPH2, FeCl2·4H2O (0.3 g, 1.5 mmol) was added into the solution in three portions over 30 min, thin-layer chromatography (alumina, using CH2Cl2 as eluant) indicated no free base porphyrins at this point. After that, DMF (50 mL) was removed from the solution. The solution was cooled to 50–60 °C and 6 M HCl (40 mL) was added into it. The solid appeared from the solution and then was filtrated and washed with 3 M HCl until the filter cake no longer appeared green. The resulting solid was vacuum-dried to afford over 91.9% yield of o-NO2TPPFeCl. Mp > 300 °C; Anal. Calcd. for C44H24N8O8ClFe: C 59.78; H 2.74; N 12.68; Found: C 59.35, H 2.64, N 12.52; UV-vis (CH2Cl2) λmax: 422 nm (Soret band), 510 nm, 579 nm (Q band); IR (KBr) νmax: 999 cm−1 (νFe-N), 367 cm−1 (νFe-Cl).
o-ClTPPFeCl was prepared using the same procedure as described for o-NO2TPPFeCl and the final solid was recrystallized from CH2Cl2 to yield 94.7% of the title compound. Mp > 300 °C; Anal. Calcd. for C44H24N4Cl5Fe: C 62.78, H 2.87, N 6.66; Found: C 62.44, H 3.16, N 7.01; UV-vis (CH2Cl2) λmax: 418 nm (Soret band), 505 nm, 576 nm (Q-band); IR (KBr) νmax: 999 cm−1 (νFe-N), 367 cm−1 (νFe-Cl).
o-CH3TPPFeCl was prepared using the same procedure as described for o-NO2TPPFeCl and the final solid was recrystallized from CH2Cl2 in 91.5% yield. Mp > 300 °C; Anal. Calcd. for C48H36N4ClFe: C 59.78, H 2.64, N 12.61; Found: C 59.35, H 2.74, N 12.68; UV-vis (CH2Cl2) λmax: 416 nm (Soret band), 511 nm, 585 nm (Q-band); IR (KBr) νmax: 998 cm−1 (νFe-N), 361 cm−1 (νFe-Cl).
o-OCH3TPPFeCl was prepared in 91.5% yield using the same procedure as described for o-NO2TPPFeCl and the final solid was recrystallized from CH2Cl2. Mp > 300 °C; Anal. Calcd. for: C48H36N4ClFeO4: C 69.96, H 4.40, N 6.80; Found: C 69.36, H 4.73, N 6.78; UV-vis (CH2Cl2) λmax: 418 nm (Soret band), 513 nm (Q-band); IR (KBr) νmax: 998 cm−1 (νFe-N), 360 cm−1 (νFe-Cl).
TPPFeCl was prepared using the same procedure as described for o-NO2TPPFeCl and recrystallized from CH2Cl2/CH3OH to yield up to 97.4% of the target compound. Mp > 300 °C; Anal. Calcd. for C44H28N4ClFe: C 75.07, H 4.01, N 7.96; Found: C 74.29, H 4.07, N 7.92; UV-vis (CH2Cl2) λmax: 418 nm (Soret band), 507 nm, 572 nm (Q-band); IR (KBr) νmax: 991 cm−1 (νFe-N), 379 cm−1 (νFe-Cl).
p-NO2TPPFeCl was prepared using the same procedure as described for o-NO2TPPFeCl and the final solid was vacuum-dried and afforded above 92.0% yield of product. Mp > 300 °C; Anal. Calcd. for C44H24N8O8ClFe: C 59.78; H 2.74; N 12.68; Found: C 59.35, H 2.74, N 12.68; UV-vis (CH2Cl2) λmax: 421 nm (Soret band), 514 nm, 584 nm (Q-band); IR (KBr) νmax: 999 cm−1 (νFe-N), 368 cm−1 (νFe-Cl).
p-ClTPPFeCl was prepared using the same procedure as described for o-NO2TPPFeCl and recrystallized from CH2Cl2/CH3OH to yield up to 99.4% of product. Mp > 300 °C; Anal. Calcd. for C44H24N4Cl5Fe: C 62.78, H 2.87, N 7.04; Found: C 62.32, H 3.31, N 6.66; UV-vis (CH2Cl2) λmax: 417 nm (Soret band), 509 nm, 573 nm (Q-band); IR (KBr) νmax: 999 cm−1 (νFe-N), 359 cm−1 (νFe-Cl).
p-CH3TPPFeCl was prepared in up to 90.4% yield using the same procedure as described for o-NO2TPPFeCl and the final solid was recrystallized from CH2Cl2. Mp > 300 °C; Anal. Calcd. for C48H36N4ClFe: C 59.78, H 2.64, N 12.61; Found: C 59.43, H 2.89, N 12.96; UV-vis (CH2Cl2) λmax: 418 nm (Soret band), 452 nm, 511 nm (Q-band); IR (KBr) νmax: 999 cm−1 (νFe-N), 360 cm−1 (νFe-Cl).
p-OCH3TPPFeCl was prepared using the same procedure as described for o-NO2TPPFeCl. The resulting solid was washed twice with water (50 mL) and recrystallized from CH2Cl2/CH3OH to yield up to 95.2% of product. Mp > 300 °C; Anal. Calcd. for C48H36N4ClFeO4: C 69.96, H 4.40, N 6.80; Found: C 69.30, H 4.68, N 6.86; UV-vis (CH2Cl2): λmax: 420 nm (Soret band), 509 nm, 572 nm (Q-band); IR (KBr) νmax: 998 cm−1 (νFe-N), 359 cm−1 (νFe-Cl).
3.2.2. Synthesis–One-pot Mixed Solvent Method
o-NO2TPPFeCl: Propionic acid (50 mL), glacial acetic acid (10 mL) and m-nitrotoluene (10 mL) were added into a 250 mL three-neck round-bottom flask equipped with stirrer, reflux condenser and dropping funnel. The mixture was stirred under reflux for 30 min. After that, o-nitrobenzaldehyde (1.51 g, 10 mmol) dissolved in propionic acid (20 mL) and freshly distilled pyrrole (0.7 mL, 10 mmol) dissolved in m-nitrotoluene (10 mL) were simultaneously added into the flask through two dropping funnels in 15 min. After 10 min, the mixture was stopped heating and cooled to 90-100 °C, FeCl2·4H2O (2 g, 10 mmol) was added into the reaction solution and was again heated to reflux with magnetic stirring for about 50 min. When the thin-layer chromatography (alumina) indicated no free base porphyrins at this point, the reaction was stopped. The crude mixture was filtered and washed with methanol three times. The further purification by column chromatography on alumina using 100% CH2Cl2 removed the porphyrin isomers and use of acetone/ethyl acetate (1:1) as eluent gave the final products. The iron porphyrin was eluted as the second band, leading to the pure compound obtained after solvent elimination (yield up to 28.7%). Other iron porphyrin complexes (RTPPFeCl) were prepared with the same procedure as o-NO2TPPFeCl. The ratios of mixed solvents and the yields of porphyrins were listed in Table1. The spectra data of iron porphyrins were in accordance with the results of two-step method.
4. Conclusions
Two different methods were applied to synthesize substituted tetraphenylporphyrin iron chloride complexes and the structures of above porphyrin compounds were characterized by UV-vis, IR, FIR and elemental analysis. Compared with the two-step method, the one-pot mixed solvent method offered the advantages of higher yields, shorter reaction time and convenient workup of reaction mixtures. The yields of iron porphyrins may be improved effectively by mixing different carboxylic acids and m-nitrotoluene in some proportion. The highest yields (28.7%-40.4%) of RTPPFe(III)Cl were obtained by using the one-pot mixed solvent method. Additionally, the characteristics of electronic spectra and molecular vibration spectra of above porphyrin compounds were analyzed respectively. Meanwhile, the red shift of the Soret band due to the presence of p-nitrophenyl substituents and the blue shift of Fe-Cl bond of TPPFeCl in the vibration frequency were explained by the electron transfer and molecular planarity in the porphyrin ring.
Acknowledgements
This work was supported by the Project of the National Natural Science Foundation of China (Grant No. 20776003, 21036009 and 1076004) and the Funding Project for Academic Human Resources Development in Institutions of Higher Learning under the Jurisdiction of Beijing Municipality (Grant No. PHR201107104).
References and Notes
- Haber, J.; Matachowski, L.; Pamin, K.; Poltowicz, J. The effect of peripheral substituents in metalloporphyrins on their catalytic activity in Lyons system. J. Mol. Catal. A: Chem. 2003, 198, 215–221. [Google Scholar] [CrossRef]
- Zakharieva, O.; Trautwein, A.X.; Veeger, C. Porphyrin-Fe(III)-hydroperoxide and porphyrin-Fe(III)-peroxide anion as catalytic intermediates in cytochrome P450-catalyzed hydroxylation reactions: a molecular orbital study. Biophys. Chem. 2000, 88, 11–34. [Google Scholar] [CrossRef]
- Bourre, L.; Simonneaux, G.; Ferrand, Y.; Thibaut, S.; Lajat, Y.; Patrice, T. Synthesis, and in vitro and in vivo evaluation of a diphenylchlorin sensitizer for photodynamic therapy. J. Photochem. Photobiol. B Biol. 2003, 69, 179–192. [Google Scholar] [CrossRef]
- Biesaga, M.; Orska, J.; Fiertek, D.; Izdebski, J.; Trojanowicz, M. Immobilized metal-ion affinity chromatography of peptides on metalloporphyrin stationary phases. Fresenius J. Anal. Chem. 1999, 364, 160–164. [Google Scholar] [CrossRef]
- Drain, C.M.; Russell, K.C.; Lehn, J.M. Self-assembly of a multi-porphyrin supramolecular macrocycle by hydrogen bond molecular recognition. Chem. Commun. 1996, 3, 337–338. [Google Scholar] [CrossRef]
- Adachi, S.; Nagano, S.; Ishimori, K.; Watanabe, Y.; Morishima, I.; Egawa, T.; Kitagawa, T.; Makino, R. Roles of proximal ligand in heme proteins: replacement of proximal histidine of human myoglobin with cysteine and tyrosine by site-directed mutagenesis as models for P-450, chloroperoxidase, and catalase. Biochemistry 1993, 32, 241–252. [Google Scholar] [CrossRef] [PubMed]
- Nebert, D.W.; Dalton, T.P. The role of cytochrome P450 enzymes in endogenous signalling pathways and environmental carcinogenesis. Nat. Rev. Cancer 2006, 6, 947–960. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Ji, H.B.; Chen, Y.X.; Han, Y.; Song, X.F.; She, Y.B.; Zhong, R.G. Oxidation of cyclohexane to adipic acid using Fe-porphyrin as a biomimetic catalyst. Org. Process Res. Dev. 2004, 8, 418–420. [Google Scholar] [CrossRef]
- Wang, L.Z.; She, Y.B.; Zhong, R.G.; Ji, H.B.; Zhang, Y.H.; Song, X.F. A green process for oxidation of p-nitrotoluene catalyzed by metalloporphyrins under mild conditions. Org. Process Res. Dev. 2006, 10, 757–761. [Google Scholar] [CrossRef]
- Rahimi, R.; Tehrani, A.A.; Fard, M.A.; Sadegh, B.M.M.; Khavasi, H.R. First catalytic application of metal complexes of porpholactone and dihydroxychlorin in the sulfoxidation reaction. Catal. Commun. 2009, 11, 232–235. [Google Scholar] [CrossRef]
- Meunier, B. Metalloporphyrins as versatile catalysts for oxidation reactions and oxidative DNA cleavage. Chem. Rev. 1992, 92, 1411–1456. [Google Scholar] [CrossRef]
- Rothemund, P. A new porphyrin synthesis. The synthesis of porphin. J. Am. Chem. Soc. 1936, 58, 625–627. [Google Scholar] [CrossRef]
- Adler, A.D.; Longo, F.R.; Finarelli, J.D.; Assour, J.; Korsakoff, L. A simplified synthesis for meso-tetraphenylporphine. J. Org. Chem. 1967, 32, 476. [Google Scholar] [CrossRef]
- Lindsey, J.S.; Schreiman, I.C.; Hsu, H.C.; Kearney, P.C.; Marguerettaz, A.M. Rothemund and Adler-Longo reactions revisited: synthesis of tetraphenylporphyrins under equilibrium conditions. J. Org. Chem. 1987, 52, 827–836. [Google Scholar] [CrossRef]
- Kumar, A.; Maji, S.; Abhilash, G.J.; Pandey, S.; Sarkar, S. One-pot general synthesis of metalloporphyrins. Tetrahedron Lett. 2007, 48, 7287–7290. [Google Scholar] [CrossRef]
- Sharghi, H.; Nejad, A.H. Phosphorus pentachloride (PCl5) mediated synthesis of tetraarylporphyrins. Helv. Chim. Acta 2003, 86, 408–414. [Google Scholar] [CrossRef]
- Nguyen, L.T.; Senge, M.O.; Smith, K.M. Simple methodology for syntheses of porphyrins possessing multiple peripheral substituents with an element of symmetry. J. Org. Chem. 1996, 61, 998–1003. [Google Scholar] [CrossRef]
- Johnston, M.R.; Latter, M.J.; Warrener, R.N. Porphyrin-containing molecular capsules: metal mediated dimerization of a bis-porphyrin cavity. Org. Lett. 2002, 4, 2165–2168. [Google Scholar] [CrossRef] [PubMed]
- Rocha Gonsalves, A.M.d’A.; Varejao, J.M.T.B.; Pereira, M.M. Some new aspects related to the synthesis of meso-substituted porphyrins. J. Heterocycl. Chem. 1991, 28, 635–640. [Google Scholar] [CrossRef]
- Serra, A.C.; Rocha Gonsalves, A.M.d’A. Controlled porphyrinogen oxidation for the selective synthesis of meso-tetraarylchlorins. Tetrahedron Lett. 2010, 51, 4192–4194. [Google Scholar] [CrossRef]
- Geier, G.R., III; Chick, J.F.B.; Callinan, J.B.; Reid, C.G.; Auguscinski, W.P. A survey of acid catalysis and oxidation conditions in the two-step, one-flask synthesis of meso-substituted corroles via dipyrromethanedicarbinols and pyrrole. J. Org. Chem. 2004, 69, 4159–4169. [Google Scholar] [CrossRef] [PubMed]
- Ono, N.; Kawamura, H.; Bougauchi, M.; Maruyama, K. Porphyrin synthesis from nitrocompounds. Tetrahedron 1990, 46, 7483–7496. [Google Scholar] [CrossRef]
- Rothemund, P. Formation of porphyrins from pyrroles and aldehydes. J. Am. Chem. Soc. 1935, 57, 2010–2011. [Google Scholar] [CrossRef]
- Rothemund, P.; Menotti, A.R. Porphyrin studies. IV. The synthesis of α,β,γ,δ-tetraphenylporphine. J. Am. Chem. Soc. 1941, 63, 267–270. [Google Scholar] [CrossRef]
- Adler, A.D.; Longo, F.R.; Shergalis, W. Mechanistic investigation of porphyrin syntheses. I. Preliminary studies on ms-tetraphenylporphin. J. Am. Chem. Soc. 1964, 86, 3145–3149. [Google Scholar] [CrossRef]
- Lindsey, J.S.; MacCrum, K.A.; Tyhonas, J.S.; Chuang, Y.Y. Investigation of a synthesis of meso-porphyrins employing high concentration conditions and an electron transport chain for aerobic oxidation. J. Org. Chem. 1994, 59, 579–587. [Google Scholar] [CrossRef]
- Li, F.R.; Yang, K.X.; Tyhonas, J.S.; Maccrum, K.A.; Lindsey, J.S. Beneficial effects of salts on an acid-catalyzed condensation leading to porphyrin formation. Tetrahedron 1997, 53, 12339–12360. [Google Scholar] [CrossRef]
- Adler, A.D.; Longo, F.R.; Kampas, F.; Kim, J. On the preparation of metalloporphyrins. J. Inorg. Nucl. Chem. 1970, 32, 2443–2445. [Google Scholar] [CrossRef]
- Johnstone, R.A.W.; Nunes, M.L.P.G.; Pereira, M.M.; Rocha Gonsalves, A.M.d’A.; Serra, A.C. Improved syntheses of 5,10,15,20-tetrakisaryl- and tetrakisalkylporphyrins. Heterocycles 1996, 43, 1423–1437. [Google Scholar] [CrossRef]
- Prendergast, K.; Spiro, T.G. Core expansion, ruffling, and doming effects on metalloporphyrin vibrational frequencies. J. Am. Chem. Soc. 1992, 114, 3793–3801. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are available from the authors. |
Scheme 1.
Synthesis of iron porphyrin complexes by the one-pot mixed solvent method.

Scheme 2.
Synthesis of iron porphyrin complexes with the two-step method.

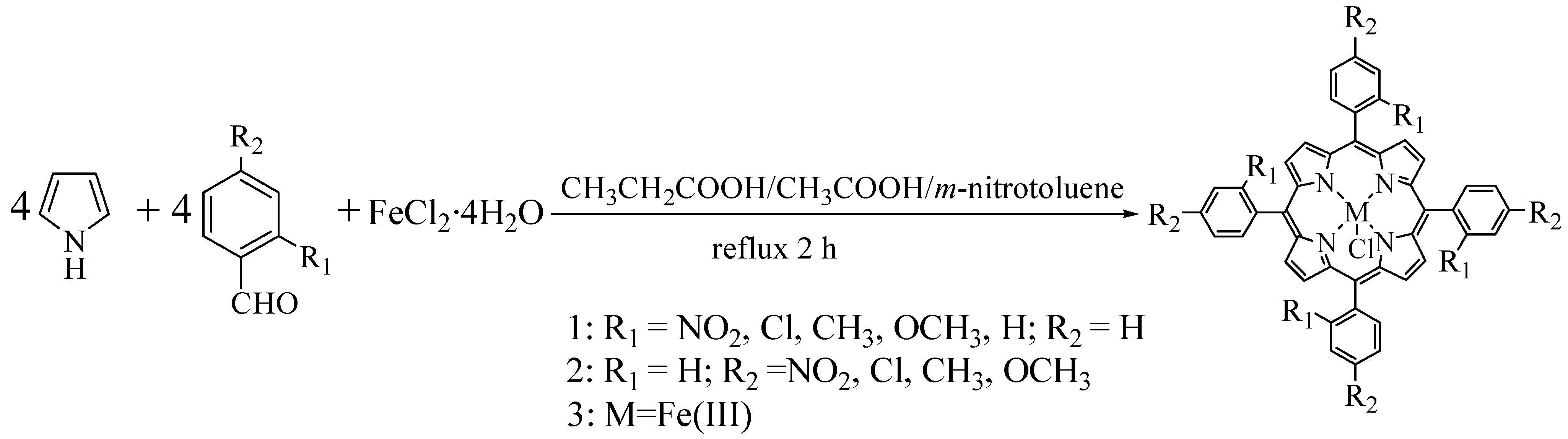{kind=link}
{kind=link}
Table 1.
Comparison of metalloporphyrin yields by the one-pot and two-step methods. Optimum ratios of solvents: the volume ratios of propionic acid, glacial acetic acid and m-nitrotoluene.
Table 1.
Comparison of metalloporphyrin yields by the one-pot and two-step methods. Optimum ratios of solvents: the volume ratios of propionic acid, glacial acetic acid and m-nitrotoluene.
| No. | Compounds | Optimum ratios of solvents (one-pot method) | Total yields (one-pot method) (%) | Total yields (two-step method) (%) |
|---|---|---|---|---|
| 1 | o-NO2TPPFeCl | 70:10:20 | 28.7 | 13.1 |
| 2 | o-ClTPPFeCl | 40:20:20 | 31.8 | 9.7 |
| 3 | o-CH3TPPFeCl | 40:20:20 | 33.6 | 16.7 |
| 4 | o-OCH3TPPFeCl | 70:10:20 | 32.5 | 5.7 |
| 5 | TPPFeCl | 70:10:20 | 38.3 | 18.4 |
| 6 | p-NO2TPPFeCl | 40:20:20 | 30.1 | 5.6 |
| 7 | p-ClTPPFeCl | 40:20:20 | 40.4 | 16.7 |
| 8 | p-CH3TPPFeCl | 70:10:20 | 32.7 | 15.4 |
| 9 | p-OCH3TPPFeCl | 40:20:20 | 35.9 | 13.0 |
Table 2.
UV-vis data of free base porphyrins and substituted tetraphenylporphyrin iron chlorides.
| No. | Compounds | λmax (nm) | ||||
|---|---|---|---|---|---|---|
| Soret bands | Q bands | |||||
| 1 | o-NO2TPP | 421 | 516 | 551 | 593 | 650 |
| 2 | o-ClTPP | 412 | 511 | 542 | 587 | 642 |
| 3 | o-CH3TPP | 416 | 512 | 544 | 589 | 645 |
| 4 | o-OCH3TPP | 417 | 512 | 545 | 589 | 643 |
| 5 | TPP | 417 | 514 | 538 | 585 | 620 |
| 6 | p-NO2TPP | 424 | 516 | 551 | 594 | 604 |
| 7 | p-ClTPP | 418 | 514 | 549 | 589 | 645 |
| 8 | p-CH3TPP | 419 | 516 | 551 | 592 | 647 |
| 9 | p-OCH3TPP | 421 | 518 | 555 | 593 | 650 |
| 10 | o-NO2TPPFeCl | 422 | 510 | 579 | ||
| 11 | o-ClTPPFeCl | 418 | 505 | 576 | ||
| 12 | o-CH3TPPFeCl | 416 | 511 | 585 | ||
| 13 | o-OCH3TPPFeCl | 418 | 513 | |||
| 14 | TPPFeCl | 418 | 507 | 572 | ||
| 15 | p-NO2TPPFeCl | 421 | 514 | 584 | ||
| 16 | p-ClTPPFeCl | 417 | 509 | 573 | ||
| 17 | p-CH3TPPFeCl | 418 | 452 | 511 | ||
| 18 | p-OCH3TPPFeCl | 420 | 509 | 572 | ||
Table 3.
IR/FIR data of free base porphyrins and iron porphyrin compounds.
| No. | Compounds | IR (cm−1) | ||||||
|---|---|---|---|---|---|---|---|---|
| νN-H (δN-H) | ν=C-H | νC=C | νC=N | γ=C-H | νFe-N | νFe-Cl | ||
| 1 | o-NO2TPP | 3321 (968) | 3060 | 1606 | 1349 | 722 | — | — |
| 2 | o-ClTPP | 3325 (967) | 3056 | 1626 | 1346 | 750 | — | — |
| 3 | o-CH3TPP | 3315 (965) | 3016 | 1599 | 1348 | 739 | — | — |
| 4 | o-OCH3TPP | 3322 (966) | 3070 | 1580 | 1349 | 753 | — | — |
| 5 | TPP | 3309 (966) | 3051 | 1594 | 1352 | 732 | — | — |
| 6 | p-NO2TPP | 3322 (967) | 3055 | 1595 | 1347 | 800 | — | — |
| 7 | p-ClTPP | 3315 (965) | 3024 | 1627 | 1349 | 796 | — | — |
| 8 | p-CH3TPP | 3317 (967) | 3024 | 1561 | 1349 | 798 | — | — |
| 9 | p-OCH3TPP | 3320 (967) | 2925 | 1596 | 1346 | 805 | — | — |
| 10 | o-NO2TPPFeCl | — | 2925 | 1607 | 1345 | 740 | 999 | 367 |
| 11 | o-ClTPPFeCl | — | 2923 | 1673 | 1334 | 754 | 999 | 370 |
| 12 | o-CH3TPPFeCl | — | 3014 | 1598 | 1332 | 753 | 998 | 361 |
| 13 | o-OCH3TPPFeCl | — | 2934 | 1596 | 1334 | 756 | 998 | 360 |
| 14 | TPPFeCl | — | 2923 | 1597 | 1340 | 750 | 991 | 379 |
| 15 | p-NO2TPPFeCl | — | 2925 | 1595 | 1346 | 802 | 999 | 368 |
| 16 | p-ClTPPFeCl | — | 3133 | 1682 | 1338 | 805 | 999 | 359 |
| 17 | p-CH3TPPFeCl | — | 3022 | 1494 | 1338 | 799 | 999 | 360 |
| 18 | p-OCH3TPPFeCl | — | 2923 | 1605 | 1334 | 810 | 998 | 359 |
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
MDPI and ACS Style
Sun, Z.-C.; She, Y.-B.; Zhou, Y.; Song, X.-F.; Li, K. Synthesis, Characterization and Spectral Properties of Substituted Tetraphenylporphyrin Iron Chloride Complexes. Molecules 2011, 16, 2960-2970. https://doi.org/10.3390/molecules16042960
AMA Style
Sun Z-C, She Y-B, Zhou Y, Song X-F, Li K. Synthesis, Characterization and Spectral Properties of Substituted Tetraphenylporphyrin Iron Chloride Complexes. Molecules. 2011; 16(4):2960-2970. https://doi.org/10.3390/molecules16042960
Chicago/Turabian StyleSun, Zhi-Cheng, Yuan-Bin She, Yang Zhou, Xu-Feng Song, and Kai Li. 2011. "Synthesis, Characterization and Spectral Properties of Substituted Tetraphenylporphyrin Iron Chloride Complexes" Molecules 16, no. 4: 2960-2970. https://doi.org/10.3390/molecules16042960