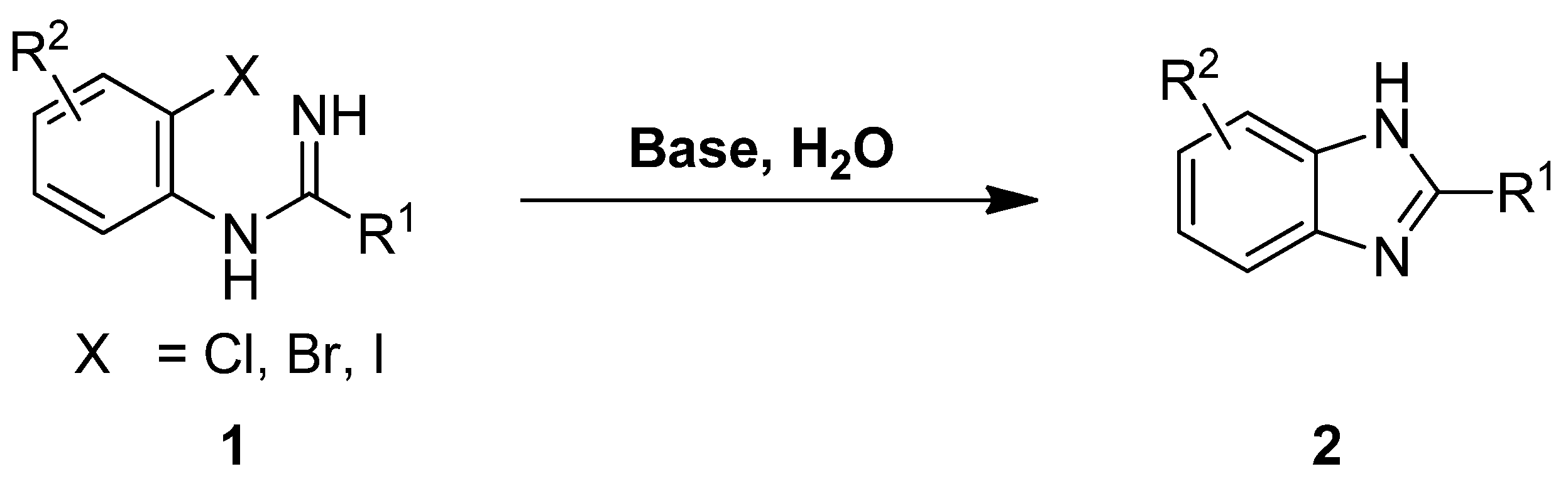
Aqueous Synthesis of 1-H-2-Substituted Benzimidazoles via Transition-Metal-Free Intramolecular Amination of Aryl Iodides

Abstract
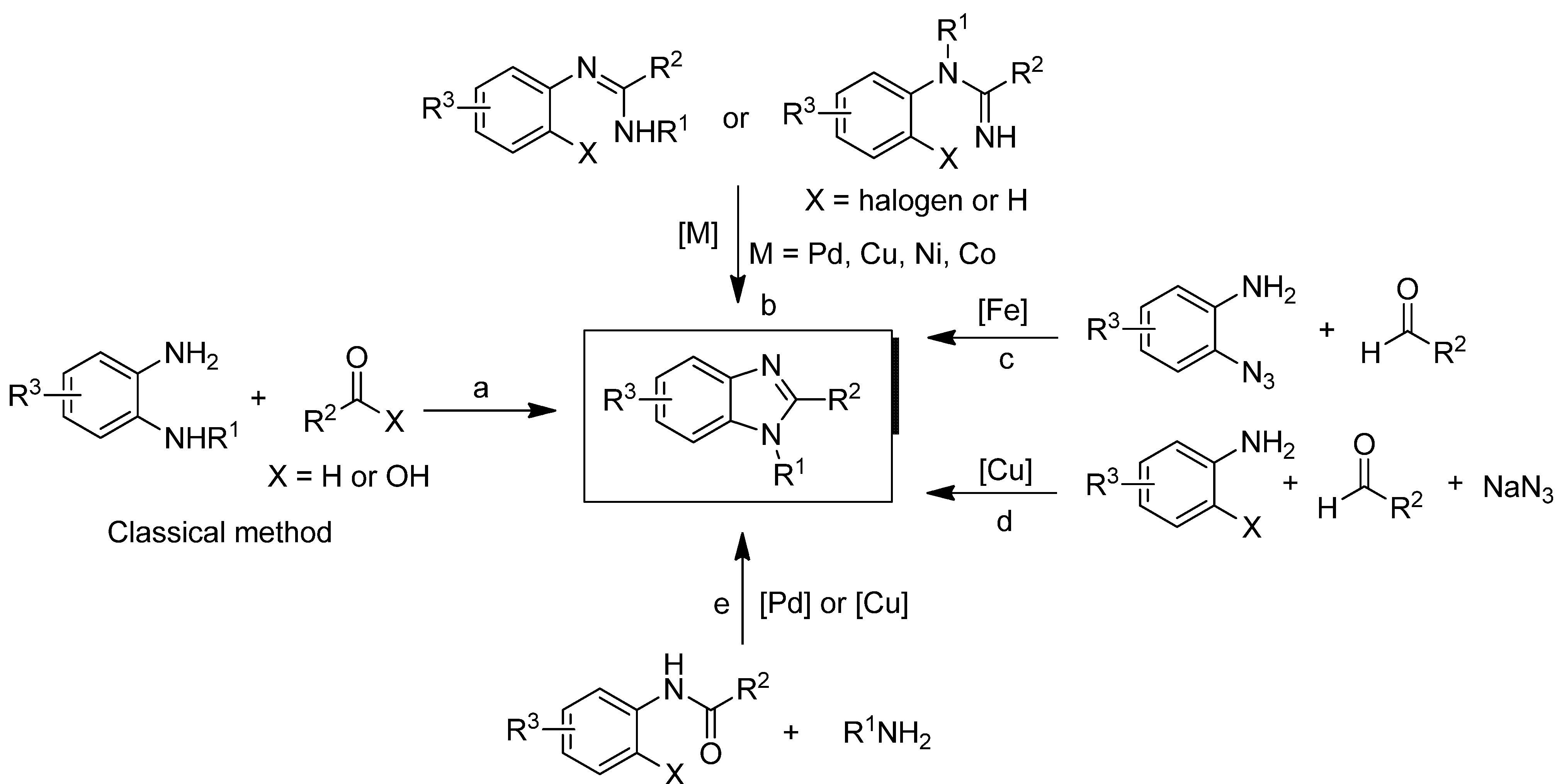
:1. Introduction
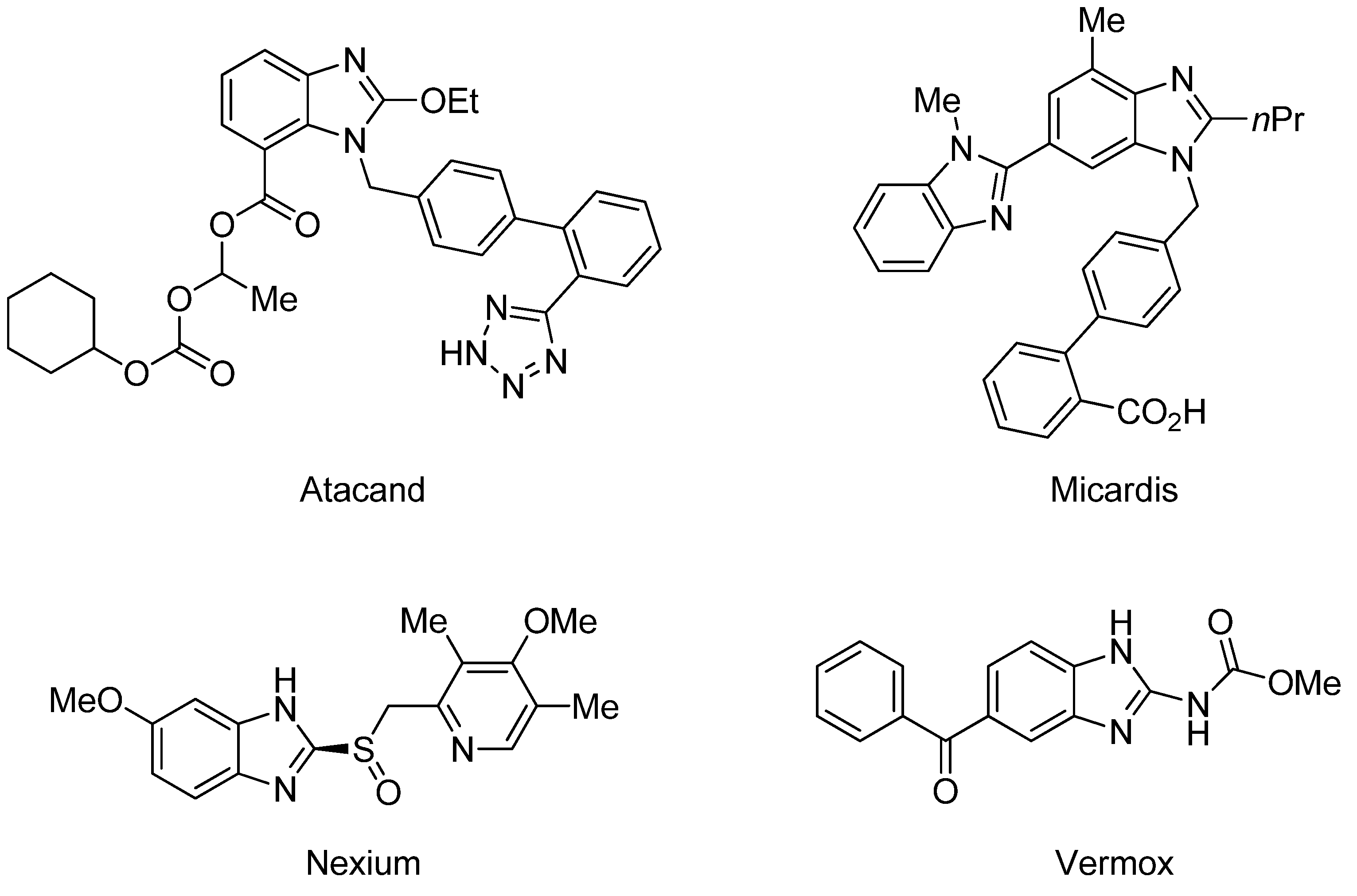
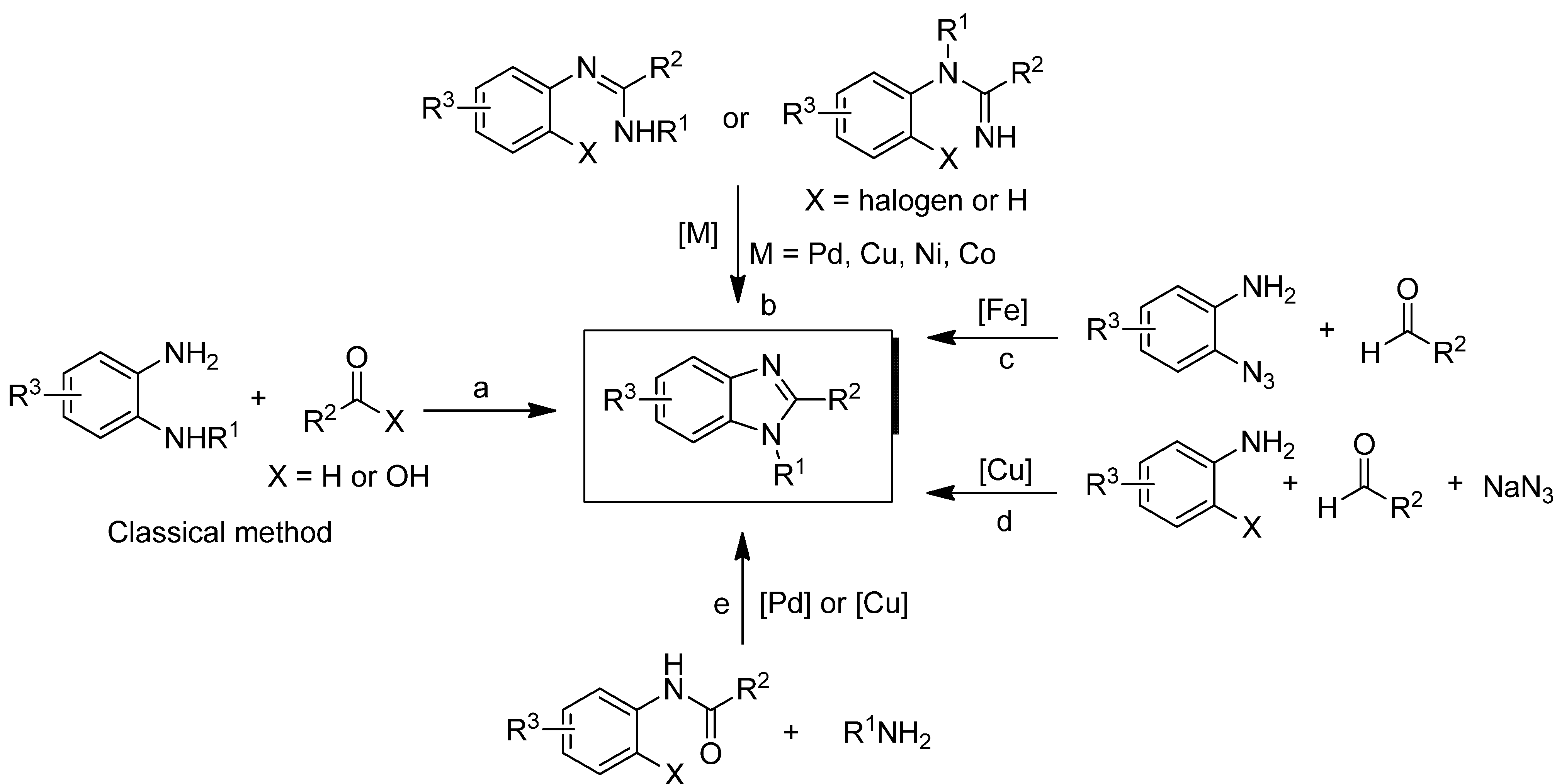
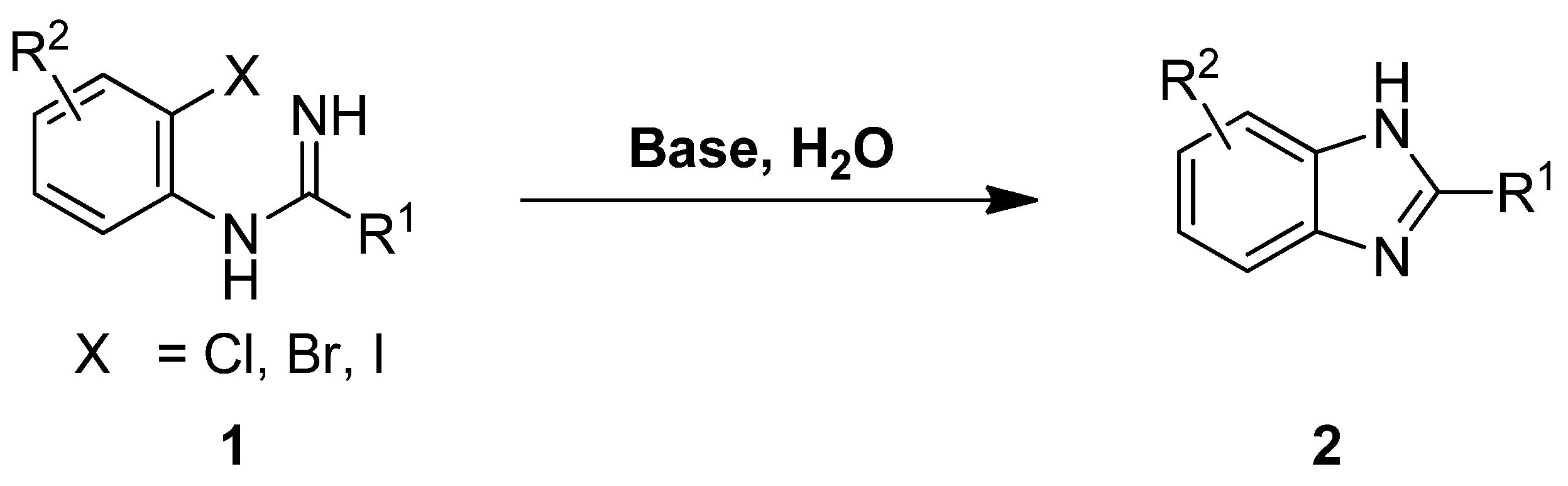
2. Results and Discussion


| Entry | Substrate | Base | Temperature (°C) | Time (h) | Yield (%) [b] |
|---|---|---|---|---|---|
| 1 | 1a | ― | 100 | 30 | 0 |
| 2 | 1a | K2CO3 | 100 | 30 | 80 |
| 3 | 1a | KOH | 100 | 30 | 63 |
| 4 | 1a | K3PO4 | 100 | 30 | trace |
| 5 | 1a | NaOH | 100 | 30 | 0 |
| 6 | 1a | NaHCO3 | 100 | 30 | 0 |
| 7 | 1a | Na2CO3 | 100 | 30 | 0 |
| 8 | 1a | Cs2CO3 | 100 | 30 | 84 |
| 9 | 1a | Et3N | 100 | 30 | 0 |
| 10 | 1a | Pyridine | 100 | 30 | 0 |
| 11 [c] | 1a | K2CO3 | 80 | 30 | trace |
| 12 | 1a | K2CO3 | 90 | 30 | 60 |
| 13 | 1a | K2CO3 | 100 | 20 | 50 |
| 14 | 1a | K2CO3 | 100 | 48 | 74 |
| 15 [d] | 1a | K2CO3 | 120 | 30 | 78 |
| 16 [d] | 1a | K2CO3 | 150 | 30 | 66 |
| 17 | 1a' | K2CO3 | 100 | 30 | 0 |
| 18 | 1a'' | K2CO3 | 100 | 30 | 0 |

| Entry | Substrate | Product | Yield (%) [b] |
|---|---|---|---|
| 1 |  1a 1a |  2a 2a | 80 |
| 2 |  1b 1b |  2b 2b | 77 |
| 3c |  1b' 1b' |  2b 2b | 0 |
| 4 |  1c 1c |  2c 2c | 66 |
| 5 |  1d 1d |  2d 2d | 54 |
| 6 |  1e 1e |  2e 2e | 67 |
| 7 |  1f 1f |  2f 2f | 67 |
| 8 |  1g 1g |  2g 2g | 44 |
| 9 [c] |  1g' 1g' |  2g 2g | 0 |
| 10 [c] |  1h 1h |  2h 2h | 0 |

{kind=link}
{kind=link}
{kind=link}
| Entry | Substrate | Product | Yield (%) [b] |
|---|---|---|---|
| 1 |  1i 1i |  2i 2i | 60 |
| 2 |  1j 1j |  2j 2j | 63 |
| 3 |  1k 1k |  2k 2k | 58 |
| 4 |  1l 1l |  2l 2l | 64 |
| 5 |  1m 1m |  2m 2m | 70 |
| 6 |  1n 1n |  2n 2n | 50 |
| 7 [c] |  1o 1o |  2o 2o | 0 |
| 8 [c] |  1p 1p |  2p 2p | 0 |
| 9 |  1q 1q |  2q 2q | 48 |
| 10 |  1r 1r |  2r 2r | 60 |
| 11 |  1s 1s |  2s 2s | 48 |
| 12 [c] |  1t 1t |  2t 2t | 0 |
| 13 |  1u 1u |  2u 2u | 33 |
3. Experimental
3.1. General
3.2. Chemistry
3.2.1. General Procedure for the Preparation of Benzimidazoles 2a–u
4. Conclusions
Acknowledgments
References and Notes
- Desai, K.G.; Desai, K.R. Green route for the heterocyclization of 2-mercaptobenzimidazole into β-lactum segment derivatives containing –CONH– bridge with benzimidazole: Screening in vitro antimicrobial activity with various microorganisms. Bioorg. Med. Chem. 2006, 14, 8271–8279. [Google Scholar] [CrossRef]
- Erdoğan, T.; Göker, H.; Ylidiz, S. Synthesis and antimicrobial activity of some novel phenyl and benzimidazole substituted benzyl ethers. Bioorg. Med. Chem. Lett. 2007, 17, 2233–2236. [Google Scholar]
- Igual-Adell, R.; Oltra-Alcaraz, C.; Soler-Company, E.; Sánchez-Sánchez, P.; Matogo-Oyany, J.; Rodríguez-Dalabuig, D. Efficacy and safety of ivermectin and thiabendazole in the treatment of strongyloidiasis. Expert Opin. Pharmacother. 2004, 5, 2615–2619. [Google Scholar] [CrossRef]
- Mavrova, A.T.; Anichina, K.K.; Vuchev, D.I.; Tsenov, J.A.; Denkova, P.S.; Kondeva, M.S.; Micheva, M.K. Antihelminthic activity of some newly synthesized 5(6)-(un)substituted-1H-benzimidazol-2-ylthioacetylpiperazine derivatives. Eur. J. Med. Chem. 2006, 41, 1412–1420. [Google Scholar] [CrossRef]
- Barreca, M.L.; Chimirri, A.; de Clercq, E.; de Luca, L.; Monforte, A.-M.; Monforte, P.; Rao, A.; Zappala, M. Anti-HIV agents: Design and discovery of new potent RT inhibitors. Il Farmaco 2003, 58, 259–263. [Google Scholar] [CrossRef]
- Leonard, J.T.; Rajesh, O.S.; Jeyaseeli, L.; Murugesh, K.; Sivakumar, R.; Gunasekaran, V. Synthesis, Antiinflammatory and Antibacterial Activities of Substituted Phenyl Benzimidazoles. Asian J. Chem. 2007, 19, 116–120. [Google Scholar]
- Baraldi, P.G.; Bovero, A.; Fruttarolo, F.; Preti, D.; Tabrizi, M.A.; Pavani, M.G.; Romagnoli, R. DNA minor groove binders as potential antitumor and antimicrobial agents. Med. Res. Rev. 2004, 24, 475–528. [Google Scholar] [CrossRef]
- Singh, A.K.; Lown, J.W. Design, synthesis and antitumor cytotoxicity of novel bis-benzimidazoles. Anticancer Drug Des. 2000, 15, 265–275. [Google Scholar]
- Chaudhuri, P.; Ganguly, B.; Bhattacharya, S. An Experimental and Computational Analysis on the Differential Role of the Positional Isomers of Symmetric Bis-2-(pyridyl)-1H-benzimidazoles as DNA Binding Agents. J. Org. Chem. 2007, 72, 1912–1923. [Google Scholar]
- Alamgir, M.; Black, D.St.C.; Kumar, N. Synthesis, Reactivity and Biological Activity of Benzimidazoles. In Topics in Heterocyclic Chemistry; Khan, M.T.H., Ed.; Springer: Berlin, Germany, 2007; Volume 9, pp. 87–118. [Google Scholar]
- Pal, S.; Hwang, W.-S.; Lin, I.J.B.; Lee, C.-S. Benzene benzimidazole containing Pd(II) metallacycle: Synthesis, X-ray crystallographic characterization and its use as an efficient Suzuki coupling catalyst. J. Mol. Catal. A: Chem. 2007, 269, 197–203. [Google Scholar] [CrossRef]
- Hao, P.; Zhang, S.; Sun, W.-H.; Shi, Q.; Adewuyi, S.; Lu, X.; Li, P. Synthesis, Characterization and Ethylene Oligomerization Studies of Nickel Complexes Bearing 2-Benzimidazolylpyridine Derivatives. Organometallics 2007, 26, 2439–2446. [Google Scholar]
- Rajadhyaksha, D.D.; Rangnekar, D.W. Synthesis of pyrazolo[4′,3′:5,6]pyrido[1,2-a]benzimidazole derivatives and study of their fluorescence properties. J. Chem. Technol. Biotechnol. 1986, 36, 300–304. [Google Scholar] [CrossRef]
- Asensio, J.A.; Gomez-Romero, P. Recent Developments on Proton Conduc-ting Poly(2,5-benzimidazole) (ABPBI) Membranes for High Temperature Poly-mer Electrolyte Membrane Fuel Cells. Fuel Cells 2005, 5, 336–343. [Google Scholar] [CrossRef]
- Schwartz, G.; Fehse, K.; Pfeiffer, M.; Walzer, K.; Leo, K. Highly efficient white organic light emitting diodes comprising an interlayer to separate fluorescent and phosphorescent regions. Appl. Phys. Lett. 2006. [Google Scholar] [CrossRef]
- Carvalho, L.C.R.; Fernandes, E.; Marques, M.M.B. Developments Towards Regioselective Synthesis of 1,2-Disubstituted Benzimidazoles. Chem. Eur. J. 2011, 17, 12544–12555. [Google Scholar]
- Grimmett, M.R. Ring Syntheses Involving Formation of Two Bonds: [4+1] Fragments. In Imidazole and Benzimidazole Synthesis; Meth-Cohn, O., Ed.; Academic: London, UK, 1997; pp. 63–102. [Google Scholar]
- Yang, D.; Fokas, D.; Li, J.; Yu, L.; Baldino, C.M. A Versatile Method for the Synthesis of Benzimidazoles from o-Nitroanilines and Aldehydes in One Step via a Reductive Cyclization. Synthesis 2005, 47–56. [Google Scholar]
- Bahrami, K.; Khodaei, M.M.; Naali, F. Mild and Highly Efficient Method for the Synthesis of 2-Arylbenzimidazoles and 2-Arylbenzothiazoles. J. Org. Chem. 2008, 73, 6835–6837. [Google Scholar] [CrossRef]
- Goossen, L.J.; Knauber, T. Concise Synthesis of Telmisartan via Decarboxylative Cross-Coupling. J. Org. Chem. 2008, 73, 8631–8634. [Google Scholar] [CrossRef]
- Brain, C.T.; Brunton, S.A. An intramolecular palladium-catalysed aryl amination reaction to produce benzimidazoles. Tetrahedron Lett. 2002, 43, 1893–1895. [Google Scholar] [CrossRef]
- Brain, C.T.; Brunton, S.A. An Improved Procedure for the Synthesis of Benzimidazoles, Using Palladium-Catalyzed Aryl-Amination Chemistry. J. Org. Chem. 2003, 68, 6814–6816. [Google Scholar] [CrossRef]
- Zhao, D.; Hu, J.; Wu, N.; Huang, X.; Qin, X.; Lan, J.; You, J. Regiospecific Synthesis of 1,2-Disubstituted (Hetero)aryl Fused Imidazoles with Tunable Fluorescent Emission. Org. Lett. 2011, 13, 6516–6519. [Google Scholar]
- Xiao, Q.; Wang, W.-H.; Liu, G.; Meng, F.-K.; Chen, J.-H.; Yang, Z.; Shi, Z.-J. Direct Imidation to Construct 1H-Benzo[d]imidazole through PdII-Catalyzed CH Activation Promoted by Thiourea. Chem. Eur. J. 2009, 15, 7292–7296. [Google Scholar] [CrossRef]
- Zheng, N.; Anderson, K.W.; Huang, X.H.; Nguyen, H.N.; Buchwald, S.L. A Palladium-Catalyzed Regiospecific Synthesis of N-Aryl Benzimidazoles. Angew. Chem. 2007, 119, 7653–7656. Angew. Chem. Int. Ed. Engl. 2007, 46, 7509–7512. [Google Scholar]
- Evindar, G.; Batey, R.A. Copper- and Palladium-Catalyzed Intramolecular Aryl Guanidinylation: An Efficient Method for the Synthesis of 2-Aminobenzimidazoles. Org. Lett. 2003, 5, 133–136. [Google Scholar] [CrossRef]
- Szczepankiewicz, B.G.; Rohde, J.J.; Kurukulasuriyz, R. Synthesis of Purines and Other Fused Imidazoles from Acyclic Amidines and Guanidines. Org. Lett. 2005, 7, 1833–1835. [Google Scholar] [CrossRef]
- Peng, J.; Ye, M.; Zong, C.; Hu, F.; Feng, L.; Wang, X.; Wang, Y.; Chen, C. Copper-Catalyzed Intramolecular C−N Bond Formation: A Straightforward Synthesis of Benzimidazole Derivatives in Water. J. Org. Chem. 2011, 76, 716–719. [Google Scholar] [CrossRef]
- Brasche, G.; Buchwald, S.L. C–H Functionalization/C–N Bond Formation: Copper-Catalyzed Synthesis of Benzimidazoles from Amidines. Angew. Chem. 2008, 120, 1958–1960. Angew. Chem. Int. Ed. Engl. 2008, 47, 1932–1934. [Google Scholar] [CrossRef]
- Zou, B.L.; Yuan, Q.L.; Ma, D.-W. Synthesis of 1,2-Disubstituted Benzimidazoles by a Cu-Catalyzed Cascade Aryl Amination/Condensation Process. Angew. Chem. 2007, 119, 2652–2655. Angew. Chem. Int. Ed. Engl. 2007, 46, 2598–2601. [Google Scholar] [CrossRef]
- Kim, Y.; Kumar, M.R.; Park, N.; Heo, Y.; Lee, S. Copper-Catalyzed, One-Pot, Three-Component Synthesis of Benzimidazoles by Condensation and C–N Bond Formation. J. Org. Chem. 2011, 76, 9577–9583. [Google Scholar]
- Deng, X.; Mani, N.S. Reactivity-Controlled Regioselectivity: A Regiospecific Synthesis of 1,2-Disubstituted Benzimidazoles. Eur. J. Org. Chem. 2010, 680–686. [Google Scholar] [CrossRef]
- Deng, X.; McAllister, H.; Mani, N.S. CuI-Catalyzed Amination of Arylhalides with Guanidines or Amidines: A Facile Synthesis of 1-H-2-Substituted Benzimidazoles. J. Org. Chem. 2009, 74, 5742–5745. [Google Scholar] [CrossRef]
- Amrani, R.O.; Thomas, A.; Brenner, E.; Schneider, R.; Fort, Y. Efficient Nickel-Mediated Intramolecular Amination of Aryl Chlorides. Org. Lett. 2003, 5, 2311–2314. [Google Scholar]
- Shen, M.; Driver, T.G. Iron(II) Bromide-Catalyzed Synthesis of Benzimidazoles from Aryl Azides. Org. Lett. 2008, 10, 3367–3370. [Google Scholar] [CrossRef]
- Saha, P.; Ali, M.A.; Ghosh, P.; Punniyamurthy, T. Cobalt-catalyzed intramolecular C–N and C–O cross-coupling reactions: Synthesis of benzimidazoles and benzoxazoles. Org. Biomol. Chem. 2010, 8, 5692–5699. [Google Scholar] [CrossRef]
- Roberts, J.D.; Semenow, D.A.; Simmons, H.E., Jr.; Carlsmith, L.A. The Mechanism of Aminations of Halobenzenes. J. Am. Chem. Soc. 1956, 78, 601–611. [Google Scholar]
- Ross, S.D. Nucleophilic Displacement Reactions in Aromatic Systems. IV. Rates of Reaction of 1-Halo-2,4-dinitrobenzene with n-Butylamine in Chloroform and with n-Butylamine and t-Butylamine in Dimethylformamide. J. Am. Chem. Soc. 1959, 81, 2113–2115. [Google Scholar] [CrossRef]
- Beller, M.; Breindl, C.; Riermeier, R.H.; Tillack, A. Synthesis of 2,3-Dihydroindoles, Indoles, and Anilines by Transition Metal-Free Amination of Aryl Chlorides. J. Org. Chem. 2001, 66, 1403–1412. [Google Scholar] [CrossRef]
- Shi, L.; Wang, M.; Fan, C.-A.; Zhang, F.-M.; Tu, Y.-Q. Rapid and Efficient Microwave-Assisted Amination of Electron-Rich Aryl Halides without a Transition-Metal Catalyst. Org. Lett. 2003, 5, 3515–3517. [Google Scholar] [CrossRef]
- Varala, R.; Ramu, E.; Alam, M.M.; Adapa, S.R. Scope and Utility of CsOH·H2O in Amination Reactions via Direct Coupling of Aryl Halides and sec-Alicyclic Amines. Synlett 2004, 1747–1750. [Google Scholar]
- Bolliger, J.L.; Frech, C.M. Transition metal-free amination of aryl halides—A simple and reliable method for the efficient and high-yielding synthesis of N-arylated amines. Tetrahedron 2009, 65, 1180–1187. [Google Scholar] [CrossRef]
- Poirier, M.; Goudreau, S.; Poulin, J.; Savole, J.; Beaulieu, P.L. Metal-Free Coupling of Azoles with 2- and 3-Haloindoles Providing Access to Novel 2- or 3-(Azol-1-yl)indole Derivatives. Org. Lett. 2010, 12, 2334–2337. [Google Scholar] [CrossRef]
- Yuan, Y.; Thomé, I.; Kim, S.H.; Chen, D.; Beyer, A.; Bonnamour, J.; Zuidema, E.; Chang, S.; Bolm, C. Dimethyl Sulfoxide/Potassium Hydroxide: A Superbase for the Transition Metal-Free Preparation of Cross-Coupling Products. Adv. Synth. Catal. 2010, 352, 2892–2898. [Google Scholar] [CrossRef]
- March, J. Aromatic Nucleophilic Substitution. In Advanced Organic Chemistry, Reactions, Mechanisms and Structure, 4th ed; John Wiley & Sons: New York, NY, USA, 1992; p. 641. [Google Scholar]
- Huisgen, R.; König, H. Erweitertes Makromolekulares Kolloquium, Freiburg. Angew. Chem. 1957, 69, 268–272. [Google Scholar] [CrossRef]
- Huisgen, R.; Sauer, J. Nucleophile aromatische Substitutionen über Arine. Angew. Chem. 1960, 72, 91–108. [Google Scholar] [CrossRef]
- Hrutford, B.F.; Bunnett, J.F. A General Principle for the Synthesis of Heterocyclic and Homocyclic Compounds. J. Am. Chem. Soc. 1958, 80, 2021–2022. [Google Scholar] [CrossRef]
- Bunnett, J.F.; Hrutford, B.F. Ring Closure via Aryne Intermediates: A General Principle of Synthesis. J. Am. Chem. Soc. 1961, 83, 1691–1697. [Google Scholar]
- Sielecki, T.M.; Meyers, A.I. Formamidines in synthesis. An efficient one-pot synthesis of 7-substituted indolines via intramolecular cyclization of (2-phenethyl)formamidines. An asymmetric route to benzopyrrocoline alkaloids. J. Org. Chem. 1992, 57, 3673–3676. [Google Scholar] [CrossRef]
- Organic Synthesis in Water; Grieca, P.A. (Ed.) Blackie A & P: London, UK, 1998.
- Chanda, A.; Fokin, V.V. Organic Synthesis “On Water”. Chem. Rev. 2009, 109, 725–748. [Google Scholar]
- Butler, R.N.; Coyne, A.G. Water: Nature’s Reaction Enforcer—Comparative Effects for Organic Synthesis “In-Water” and “On-Water”. Chem. Rev. 2010, 110, 6302–6337. [Google Scholar] [CrossRef]
- Simon, M.-O.; Li, C.-J. Green chemistry oriented organic synthesis in water. Chem. Soc. Rev. 2012, 41, 1415–1427. [Google Scholar]
- Peng, J.; Zong, C.; Ye, M.; Chen, T.; Gao, D.; Wang, Y.; Chen, C. Direct transition-metal-free intramolecular C–O bond formation: Synthesis of benzoxazole derivatives. Org. Biomol. Chem. 2011, 9, 1225–1230. [Google Scholar]
- Wang, Y.; Song, Z.; Chen, C.; Peng, J. Pictet-Spengler condensation reactions catalyzed by a recyclable H+-montmorillonite as a heterogeneous Brønsted acid. Sci. Chin. Chem. 2010, 53, 562–568. [Google Scholar] [CrossRef]
- Pirrung, M.C. Acceleration of Organic Reactions through Aqueous Solvent Effects. Chem. Eur. J. 2006, 12, 1312–1317. [Google Scholar] [CrossRef]
- Jung, Y.; Marcus, R.A. On the Theory of Organic Catalysis “On Water”. J. Am. Chem. Soc. 2007, 129, 5492–5502. [Google Scholar]
- Buchwald, S.L.; Bolm, C. On the Role of Metal Contaminants in Catalyses with FeCl3. Angew. Chem. 2009, 121, 5694–5695. Angew. Chem. Int. Ed. Engl. 2009, 5586–5587. [Google Scholar] [CrossRef] [Green Version]
- Larsson, P.-F.; Correa, A.; Carril, M.; Norrby, P.-O.; Bolm, C. Copper-Catalyzed Cross-Couplings with Part-per-Million Catalyst Loadings. Angew. Chem. 2009, 121, 5801–5803. Angew. Chem. Int. Ed. Engl. 2009, 48, 5691–5693. [Google Scholar]
- Thomé, I.; Nijs, A.; Bolm, C. Trace metal impurities in catalysis. Chem. Soc. Rev. 2012, 41, 979–987. [Google Scholar]
- Leadbeater, N.E. When is free really free? Nat. Chem. 2010, 2, 1007–1009. [Google Scholar] [CrossRef]
- Trace metal amounts in the commercially available bases were analyzed by inductively coupled plasma atomic emission spectroscopy (ICP-AES): for K2CO3 2 ppm of Pd, <1 ppm of Cu, 2 ppm of Ni, 3 ppm of Fe; for KOH 5 ppm of Pd, <1 ppm of Cu, 2 ppm of Ni, 3 ppm of Fe; for NaOH 4 ppm of Pd, <1 ppm of Cu, 4 ppm of Ni, 3 ppm of Fe; for K3PO4 6 ppm of Pd, <1 ppm of Cu, <1 ppm of Ni, 3 ppm of Fe; for NaHCO3 8 ppm of Pd, <1 ppm of Cu, 1 ppm of Ni, 5 ppm of Fe; for Na2CO3 10 ppm of Pd, <1 ppm of Cu, 3 ppm of Ni, 4 ppm of Fe; for Cs2CO3 6 ppm of Pd, <1 ppm of Cu, 2 ppm of Ni, 5 ppm of Fe.
- Briner, G.P.; Miller, J.; Liveris, M.; Lutz, P.G. The SN mechanism in aromatic compounds. Part VII. J. Chem. Soc. 1954, 1265–1266. [Google Scholar]
- Bartoli, G.; Todesco, P.E. Nucleophilic substitution. Linear free energy relations between reactivity and physical properties of leaving groups and substrates. Acc. Chem. Res. 1977, 10, 125–132. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds are available from the authors.
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Chen, C.; Chen, C.; Li, B.; Tao, J.; Peng, J. Aqueous Synthesis of 1-H-2-Substituted Benzimidazoles via Transition-Metal-Free Intramolecular Amination of Aryl Iodides. Molecules 2012, 17, 12506-12520. https://doi.org/10.3390/molecules171112506
Chen C, Chen C, Li B, Tao J, Peng J. Aqueous Synthesis of 1-H-2-Substituted Benzimidazoles via Transition-Metal-Free Intramolecular Amination of Aryl Iodides. Molecules. 2012; 17(11):12506-12520. https://doi.org/10.3390/molecules171112506
Chicago/Turabian StyleChen, Chunxia, Chen Chen, Bin Li, Jingwei Tao, and Jinsong Peng. 2012. "Aqueous Synthesis of 1-H-2-Substituted Benzimidazoles via Transition-Metal-Free Intramolecular Amination of Aryl Iodides" Molecules 17, no. 11: 12506-12520. https://doi.org/10.3390/molecules171112506