Crossed and Linked Histories of Tetrapyrrolic Macrocycles and Their Use for Engineering Pores within Sol-Gel Matrices

Abstract
:
Nomenclature
| Acac | acetylacetone (CH3COCH2COCH3) |
| APTES | aminopropyltri-ethoxysilane |
| AmTEOS | amyltriethoxysilane (C5H11)Si(OC2H5)3 |
| AlyTEOS | allyl-triethoxysilane (CH2=CH-CH2-Si(OC2H5)3) |
| APDT | antimicrobial photodynamic therapy |
| B | Soret band |
| BJH | Barrett, Joyner and Halenda pore-size distribution method |
| dMeDEOS | dimethyldiethoxysilane, (CH3)2Si(OC2H5)2 |
| DCB | dicyanobenzene |
| DMF | dimethylformamide |
| DoTEOS | dodecyltriethoxysilane (C12H25-Si(OC2H5)3) |
| EtTEOS | ethyltriethoxysilane (C2H5)Si(OC2H5)3 |
| FA | functionalized alkoxide |
| h | ηH2O/ηalkoxy (water-alkoxide molar ratio) |
| Hexa | 1,6-hexanodiamine |
| HpD | hematoporphyrin derivative |
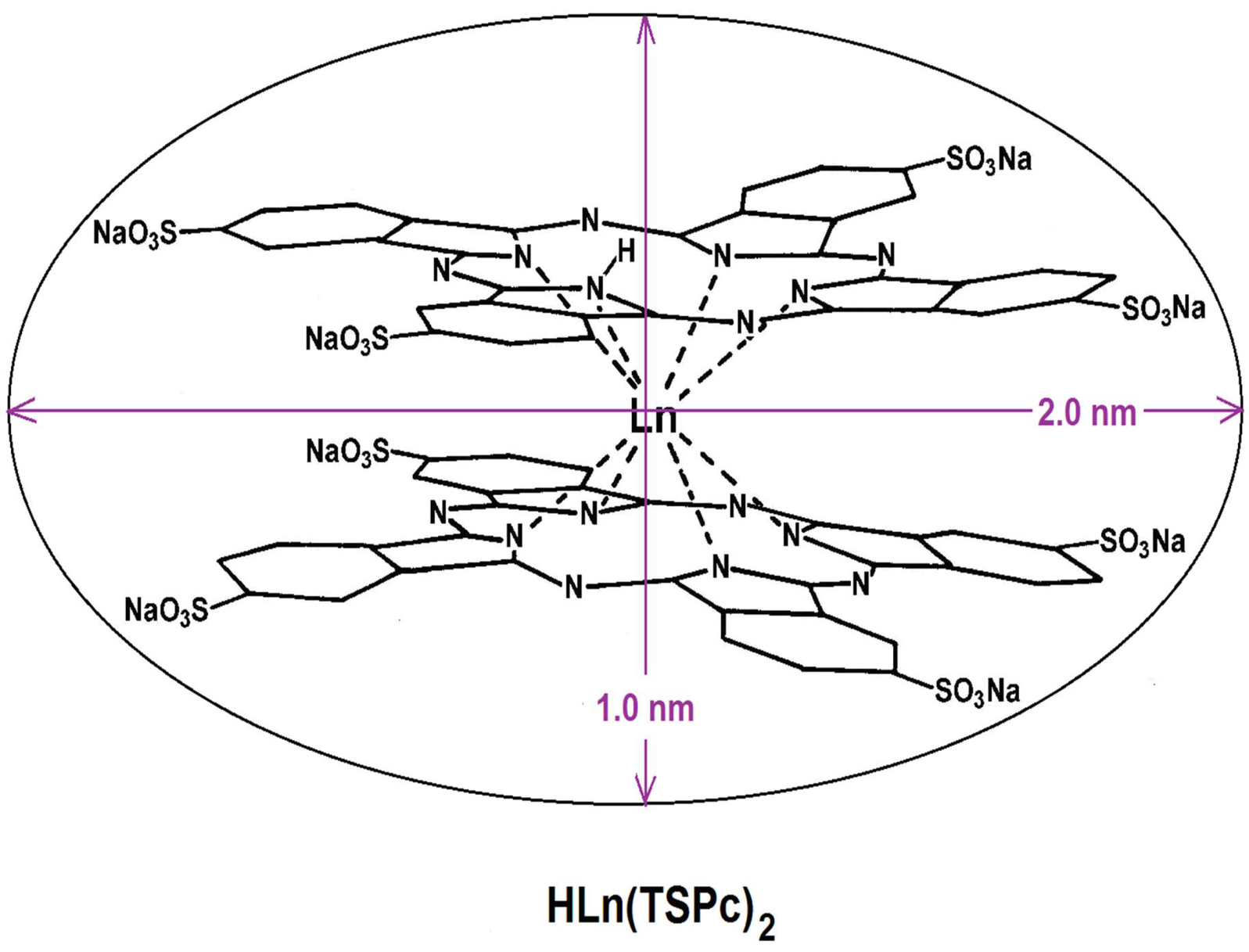
| HLn(Pc)2 | lanthanide bisphthalocyanine |
| H2P | free base of porphyrin |
| H2Pc | free phthalocyanine |
| HTES | hydride-triethoxysilane (HSi(OC2H5)3) |
| H2TPP | tetra-phenylporphyrin free base |
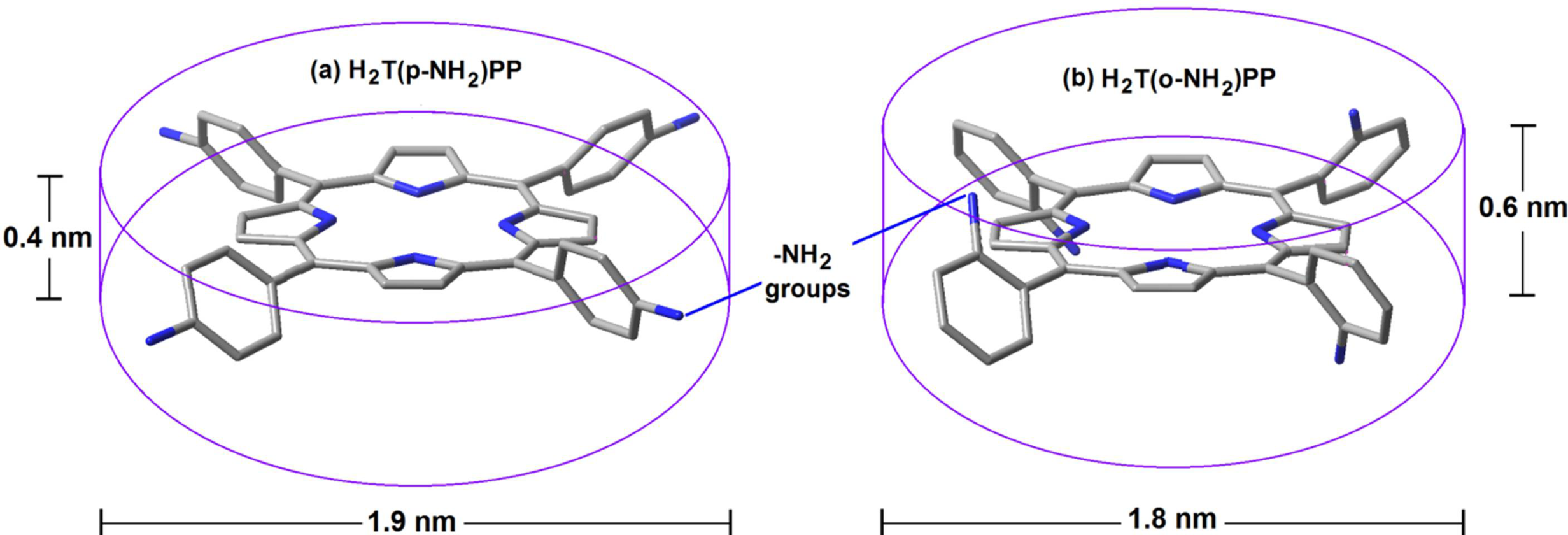
| H2T(o or p-S)PP | ortho (X) or para (Y) substituted tetraphenyl-porphyrins |
| IPTES | isocyanatopropyltriethoxysilane |
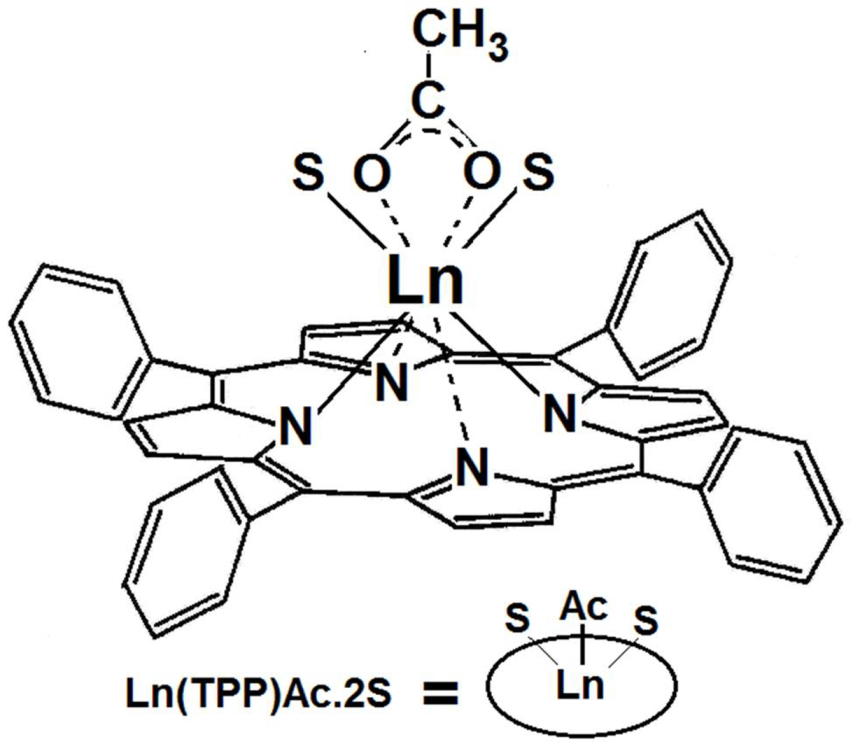
| Ln(TPP)Ac·2S | lanthanide acetate-tetraphenylporphyrin |
| Ln(Ac)•nH2O | lanthanide acetate |
| HLn(Pc)2 | lanthanide bisphthalocyanine |
| MCM-41, 48, 50, etc. | Mobil Corporation Material 41, 48, 50, etc. |
| MeTES | methyl-triethoxysilane (CH3Si(OC2H5)3) |
| MSNP | mesoporous silica nanoparticle |
| MTSPc | metal tetrasulfophthalocyanine |
| NAEPTES | N-(2-aminoethyl-amino)propyltrimethoxysilane |
| NHSG | non-hydrolytic sol gel |
| NIR | Near Infrared Spectroscopy |
| NMR | Nuclear Magnetic Resonance Spectroscopy |
| NLDFT | Non-Local Density Functional Theory |
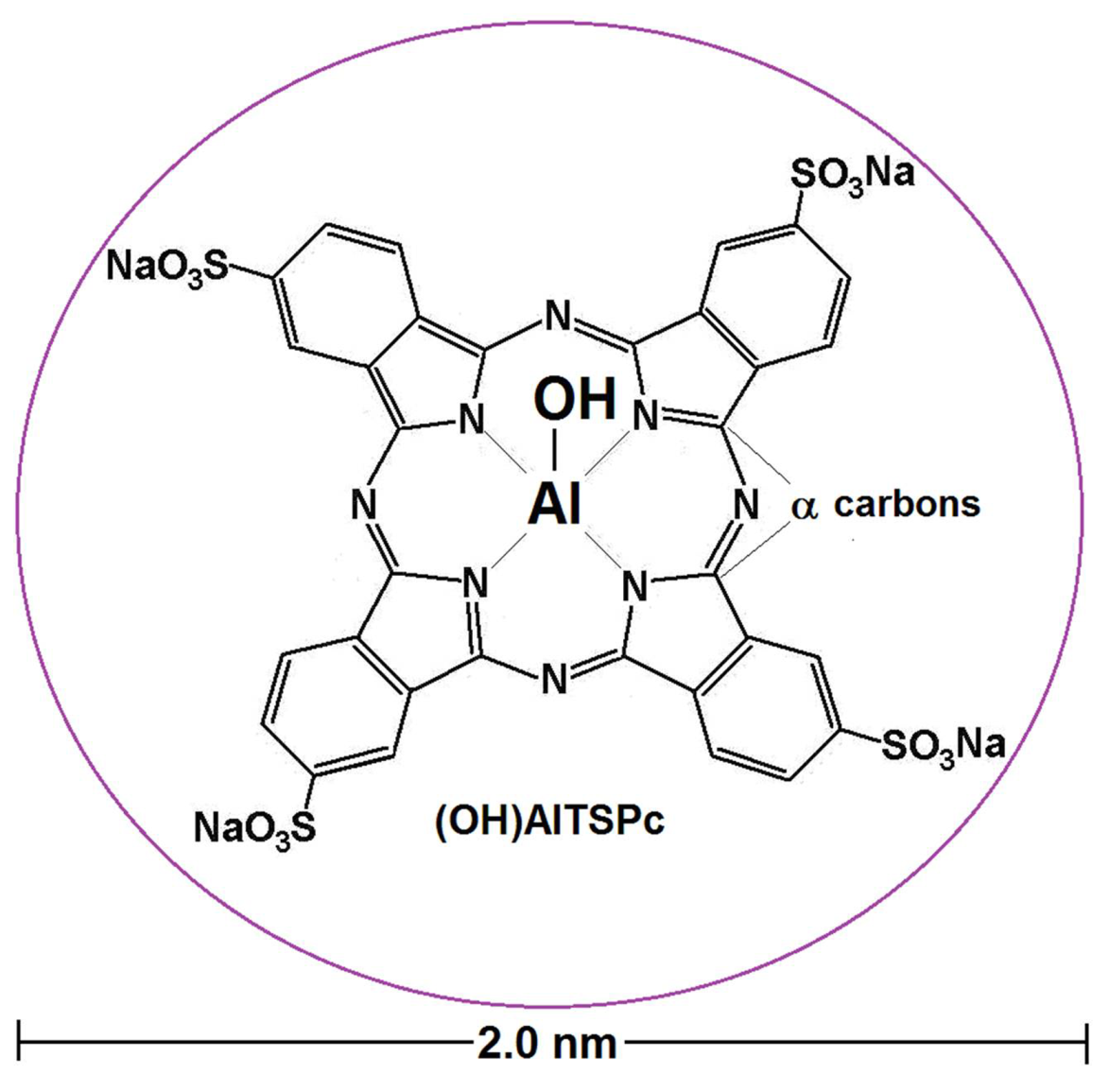
| (OH)AlTSPc | aluminum hydroxyl-tetrasulfophathalocyanine |
| OSA | organo-substituted alkoxide |
| PBG | porphobilinogen |
| PDT | photodynamic therapy |
| PMMA | polymethylmetacrylate |
| PMO’s | Periodic MesoporousOrgano-Silicas |
| Py | pyridine |
| QI, II, III or IV | Q bands of tetrapyrrolic macrocycles |
| R | alkyl or aryl group |
| rLn | lanthanide ionic radious |
| S | solvent molecule |
| SBA-n | Santa Barbara “n” Material |
| SBA-15 | Santa Barbara Material “15” |
| SEM | Scanning Electron Microscopy |
| TEOS | tetraethoxysilane (Si(OC2H5)4) |
| TMOS | tetramethoxysilane (Si(OCH3)4) |
| UV-Vis | Ultrviolet and Visible Spectroscopy |
| Vf | total volume of gelling mixture |
| VTEOS | tetraethoxysilane volume |
| VyTEOS | vynil-triethoxysilane (CH2=CH-)Si(OC2H5)3 |
| Zr(OnPr)4 | zirconium n-propoxide (Zr(OCH2CH2CH3)4 |
| [H2P or MP]n | oligomeric free- or metallo-porphyrin |
| Ф | average pore diameter |
| λ | wavelength |
| λex | fluorescence excitation wavelength |
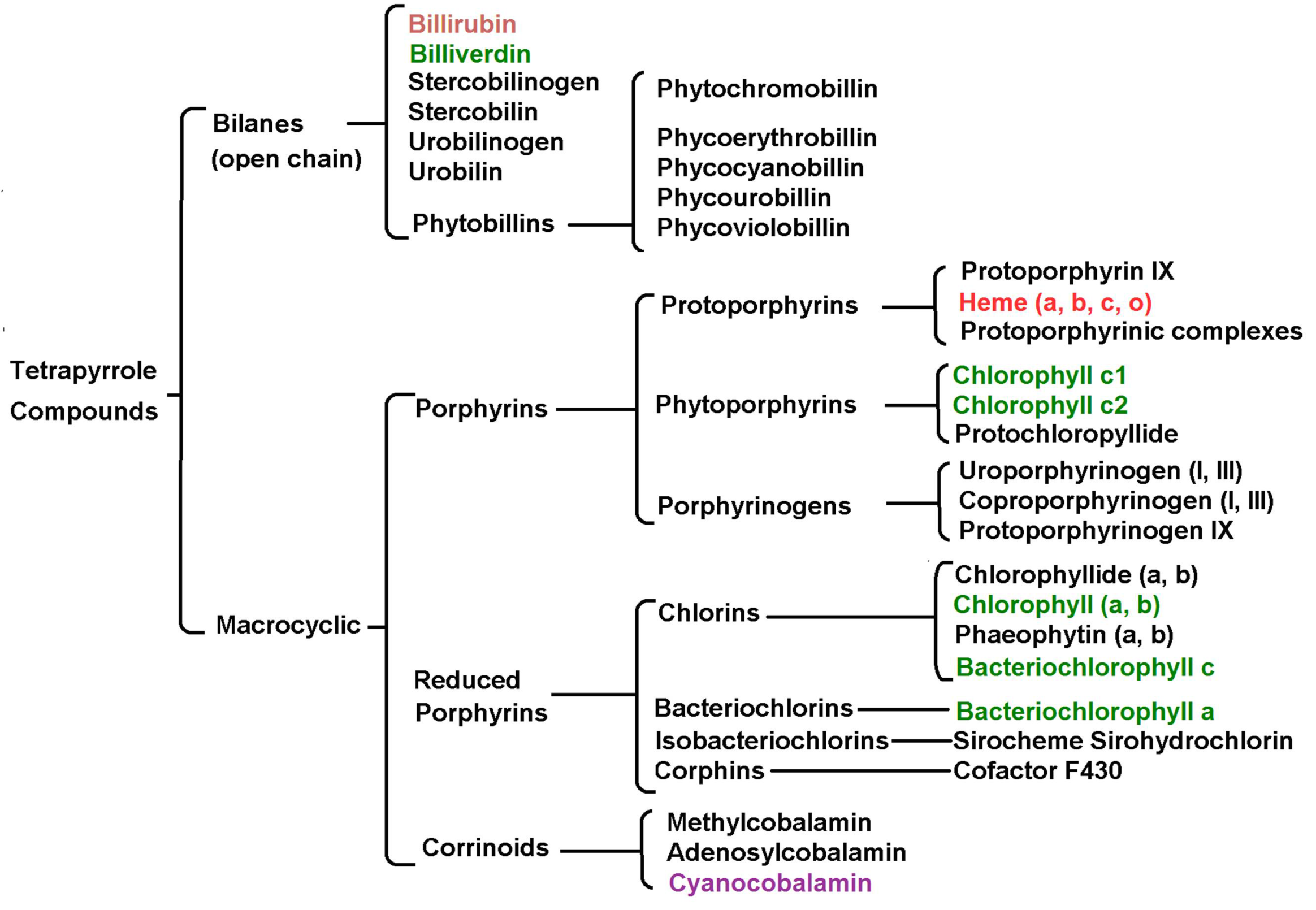
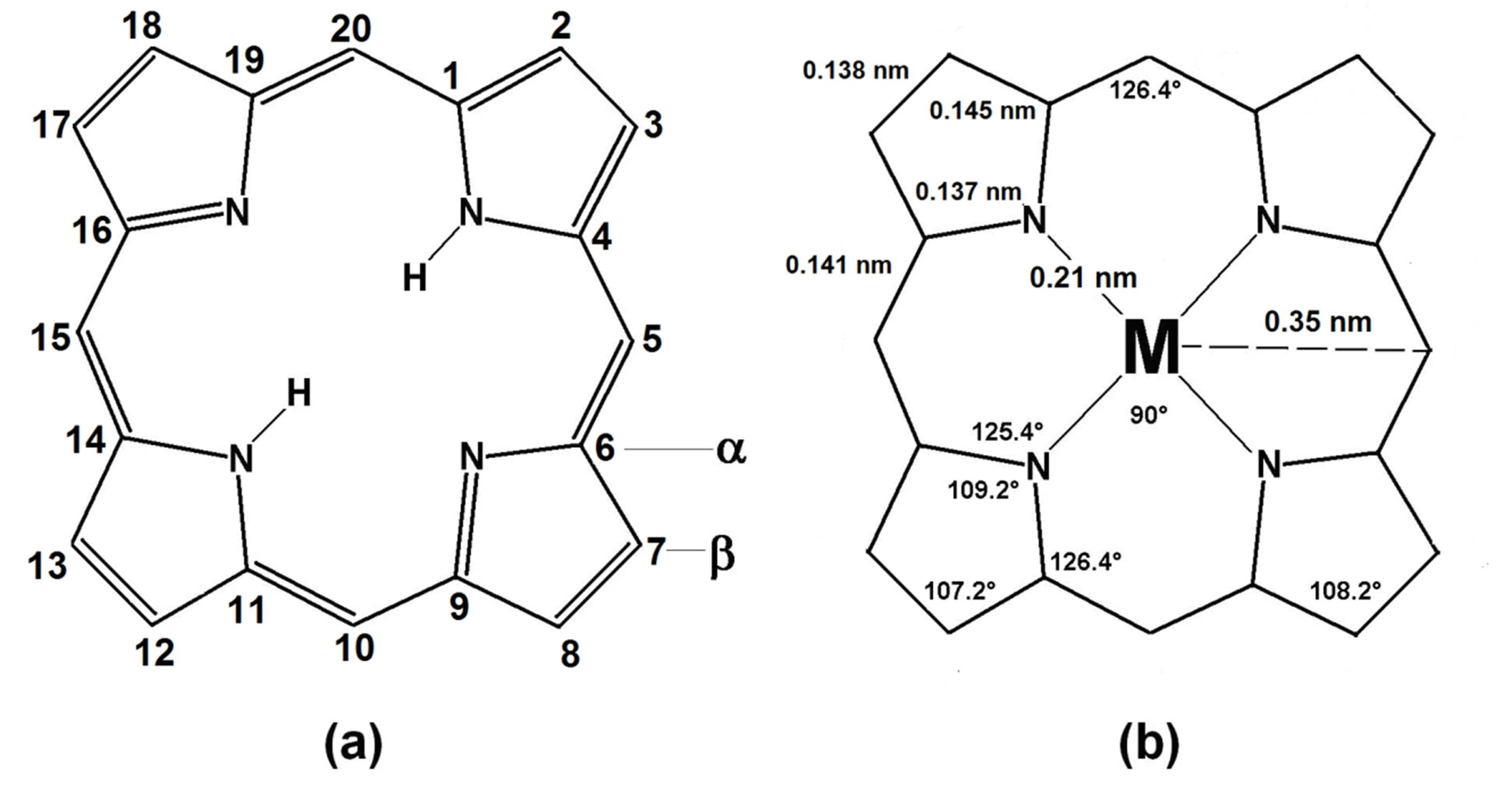
1. Crossed and Linked Histories of Tetrapyrrolic or How Nature Allows for No Mistakes
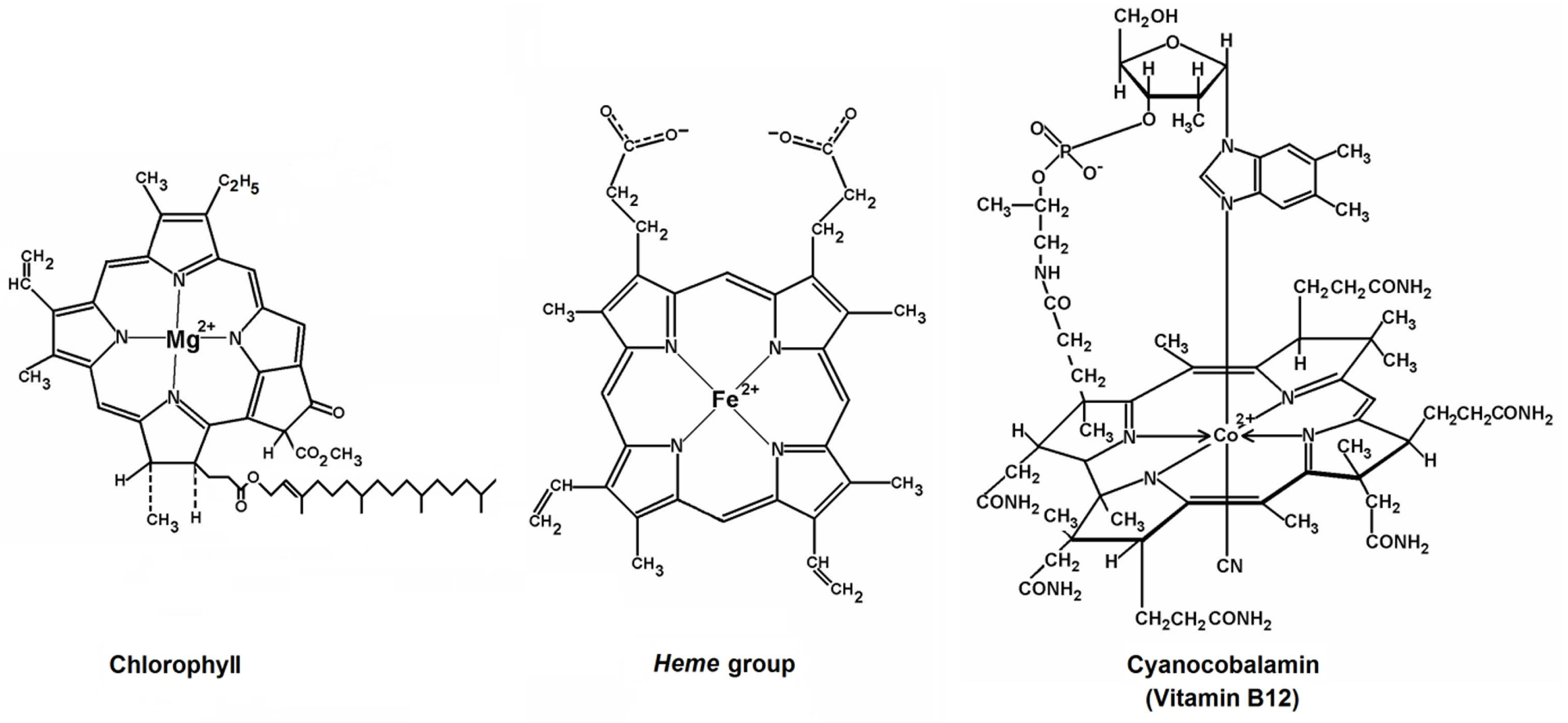
1.1. Hemoglobin and Chlorophyll

1.2. The Cytochromes
1.3. Cyanocobalamin
1.4. Tetrapyrrole Macrocycles and Photodynamic Therapy (PDT)
- To be readily synthesizable, homogeneous, and well characterized,
- To have low toxicity at therapeutic doses subjected under light or darkness conditions,
- To combat high tumor accumulation and facilitate its fast elimination
- To show absorption bands in the wavelength region at which radiation penetration of biological tissues is deeper (i.e., far red and near-infrared wavelengths)
- To generate a high quantum yield of singlet oxygen in vivo
2. Synthetic Porphyrins and Phthalocyanines


2.1. The Porphyrins

2.2. The Phthalocyanines

2.3. Porphyrins and Phthalocyanines in Catalysis and Sensoring
3. The Sol-Gel Process and the Trapping of Chemical Species within Xerogels
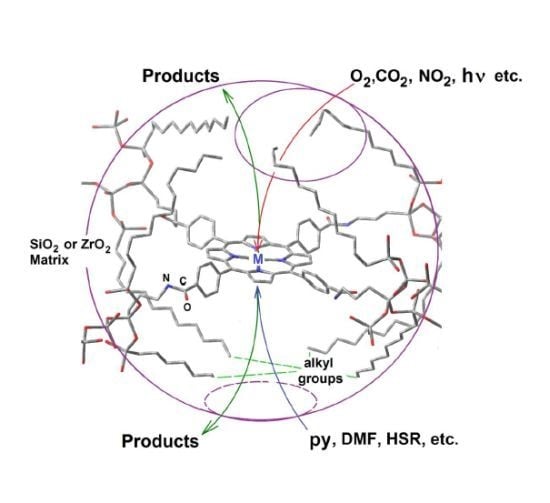
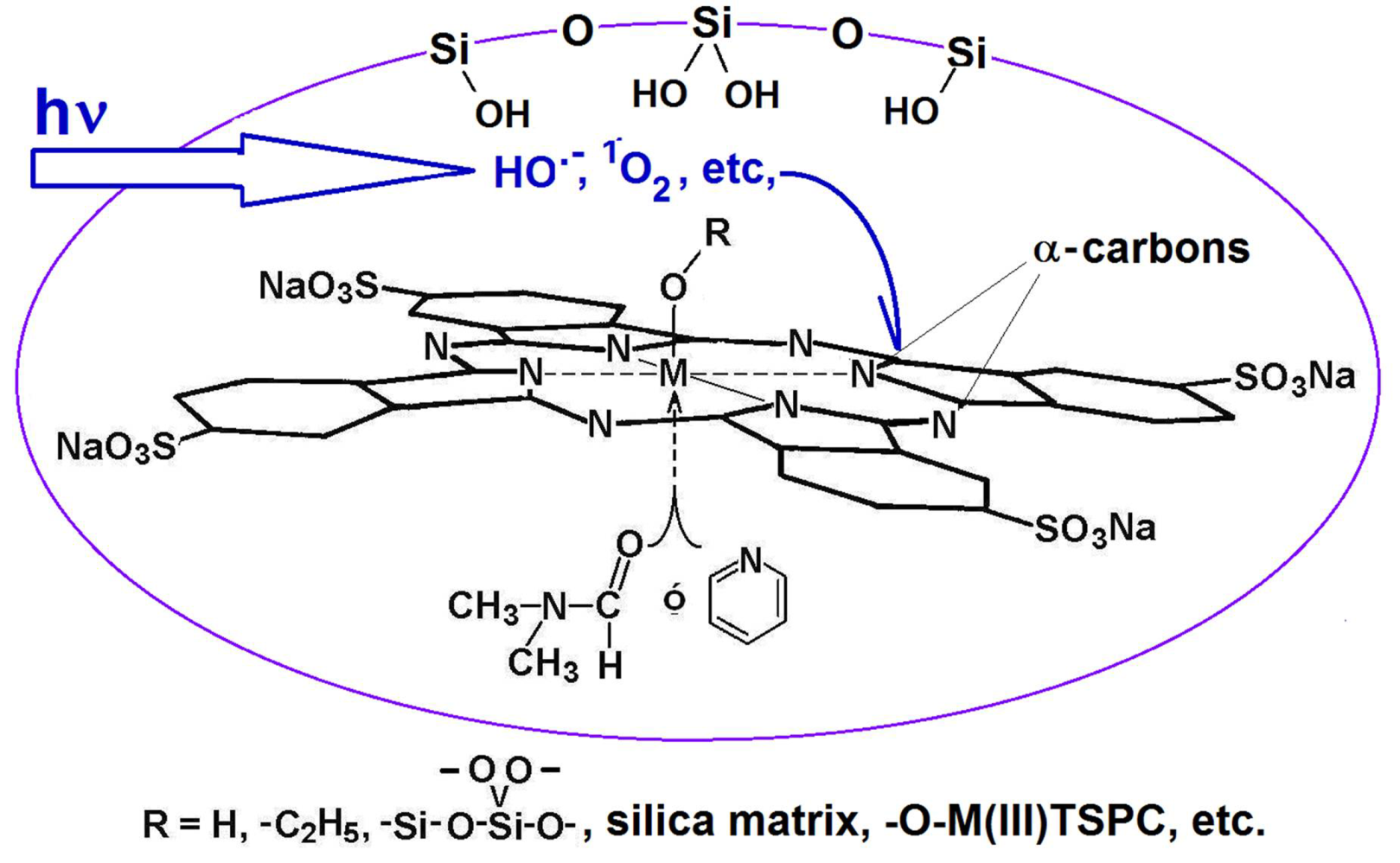
4. Tetrapyrrole Macrocycles and Cavity Pore Engineering Through the Sol-Gel Method
4.1. Aluminum μ-Hydroxytetrasulfo Phthalocyanine,(OH)AlTSPc as Molecular Probe


4.2. Physical Trapping or Covalent Bonding of Porphyrins





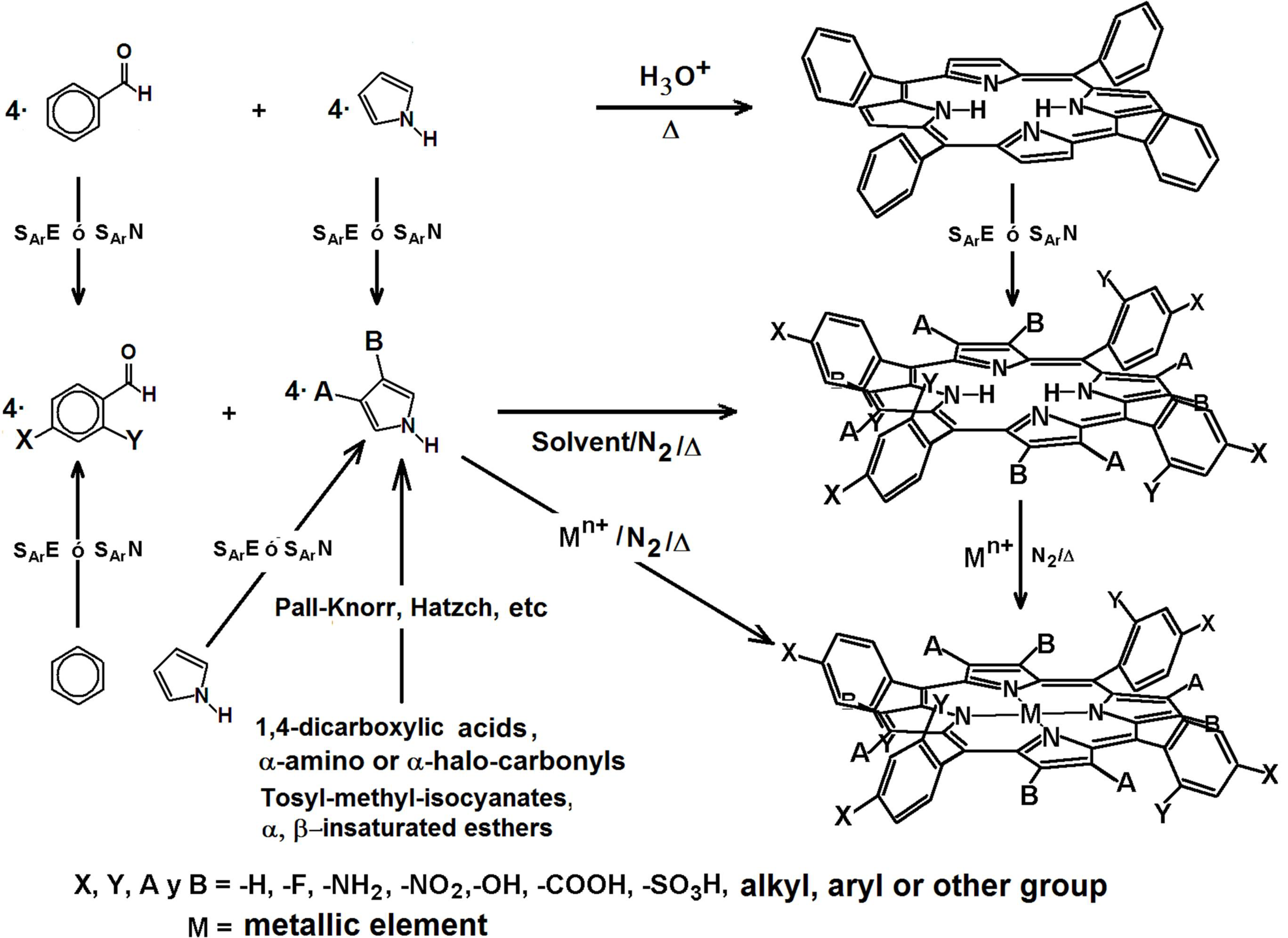
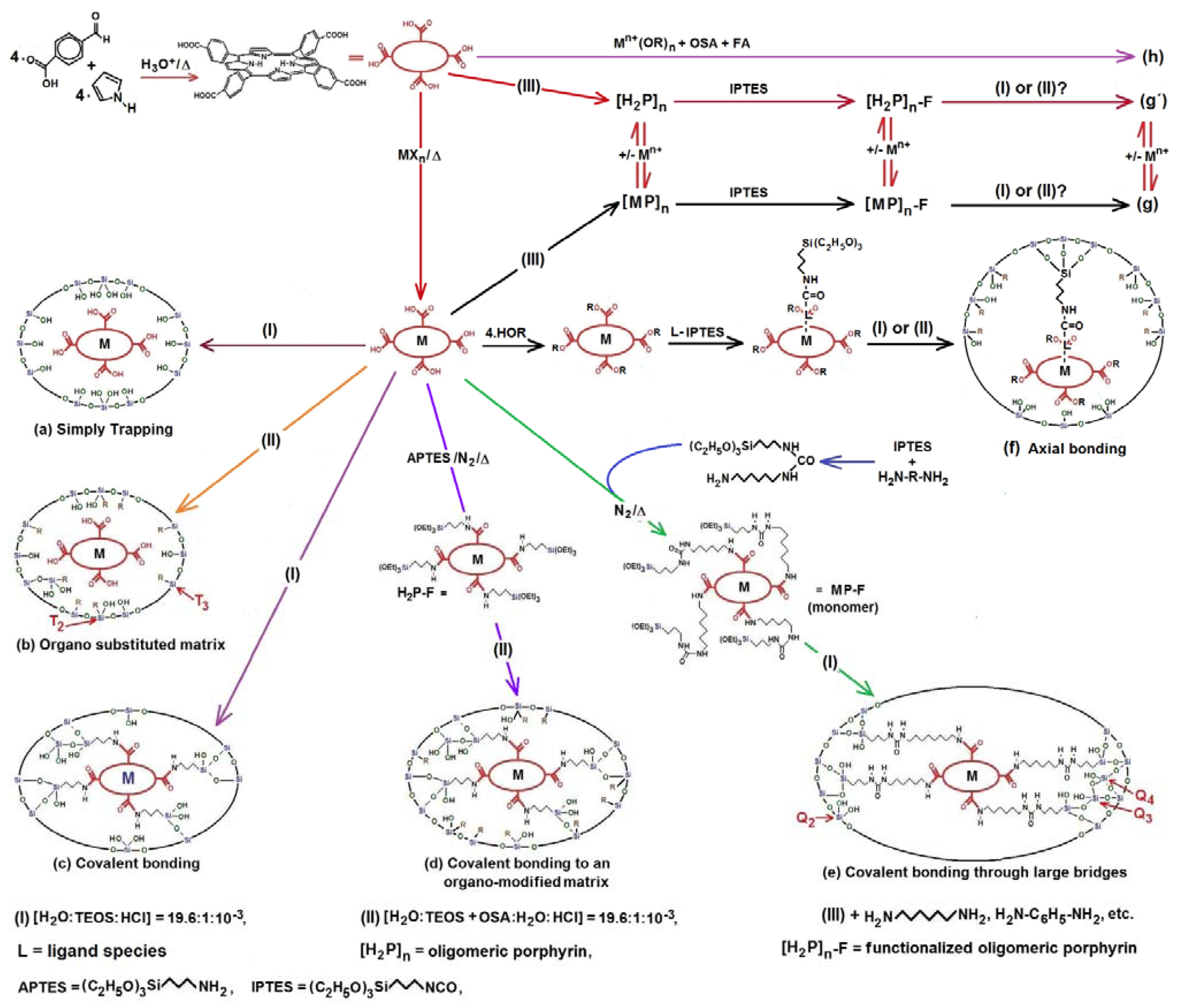
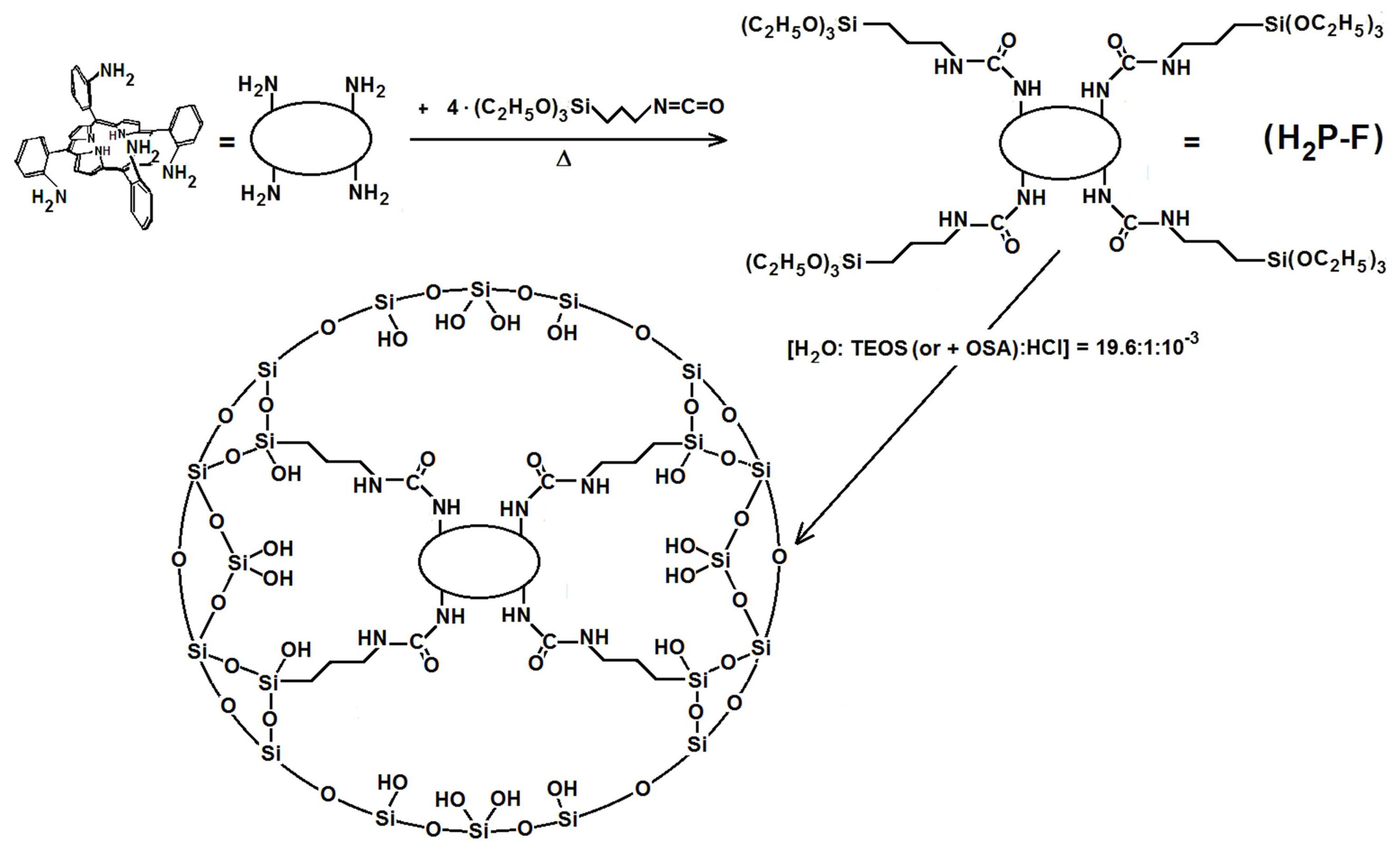
4.3. Development and Optimization of Methodologies for Trapping or Bonding Porphyrins and Phthalocyanines within Pore Networks
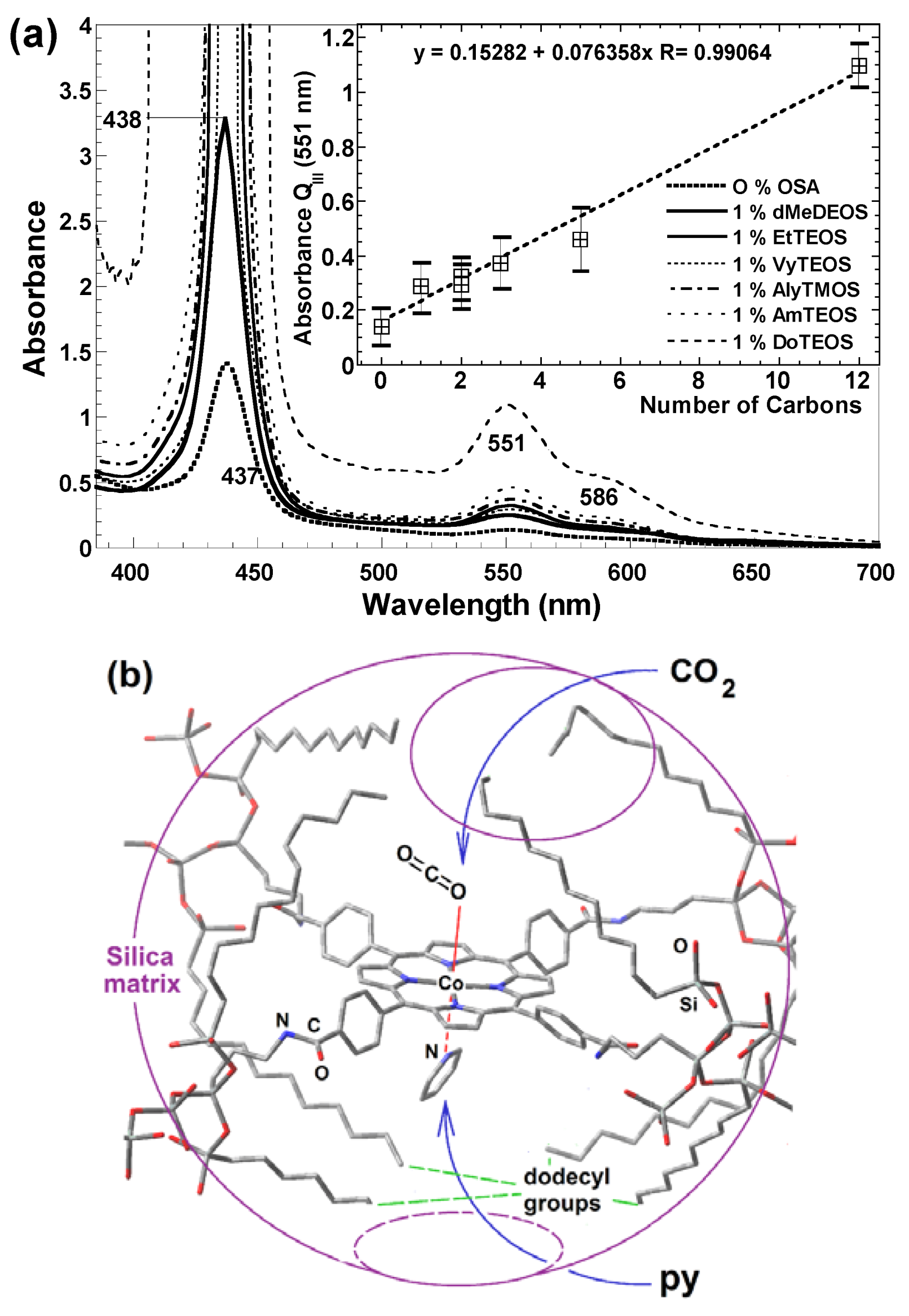
- To place the macrocyclic species far from the SiOH groups by establishing bridges between macrocyclic molecules endowed with FA species and the pore walls (Route c in Figure 13).
- To trap the macrocycle inside a network in which SiOH groups have been exchanged with alkyl or aryl groups proceeding from OSA compounds (Route b in Figure 13).
- To create large unions between the macrocycle and the pore walls by combining FA species with polymer precursors such as lactams, diamines or diacids, etc. (Route e in Figure 13) or to fix oligomeric species synthesized from tetrapyrrole macrocycles and polymer precursors, [H2P or MP]n, inside the cavities of inorganic networks (route g in Figure 13).
- To fix the macrocycle species to the pore walls via an organo-modified network in which the surface SiOH groups have been exchanged by alkyl or aryl groups (Route d in Figure 13).
- To trap or fix monogenic or oligomeric macrocyclic complexes, through axial ligands, inside inorganic or hybrid networks (Route f in Figure 13).
- To combine the use of both FA and OSA compounds for trapping or bonding tetrapyrrolic species inside pore networks of different metal oxides such as ZrO2, TiO2, Al2O3, etc. (Routes g and h in Figure 13).





4.4. Research in Progress

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Trapped species | Entrapment | Network | Average pore size φ/nm | Surface m2/g |
|---|---|---|---|---|
| Blank | physically trapped | pristine SiO2 | 3.4 | 729 |
| (OH)AlTSPc | physically trapped | SiO2 | 2.7 | 585 |
| MTSPc (M = Fe, Co, Ni, Cu, Al) | physically trapped | SiO2 | 2.7–3.5 | 540–631 |
| HLn(TSPc)2 (Ln = Eu and Sm) | physically trapped or covalently bonded | SiO2 | 2.2–2.4 | |
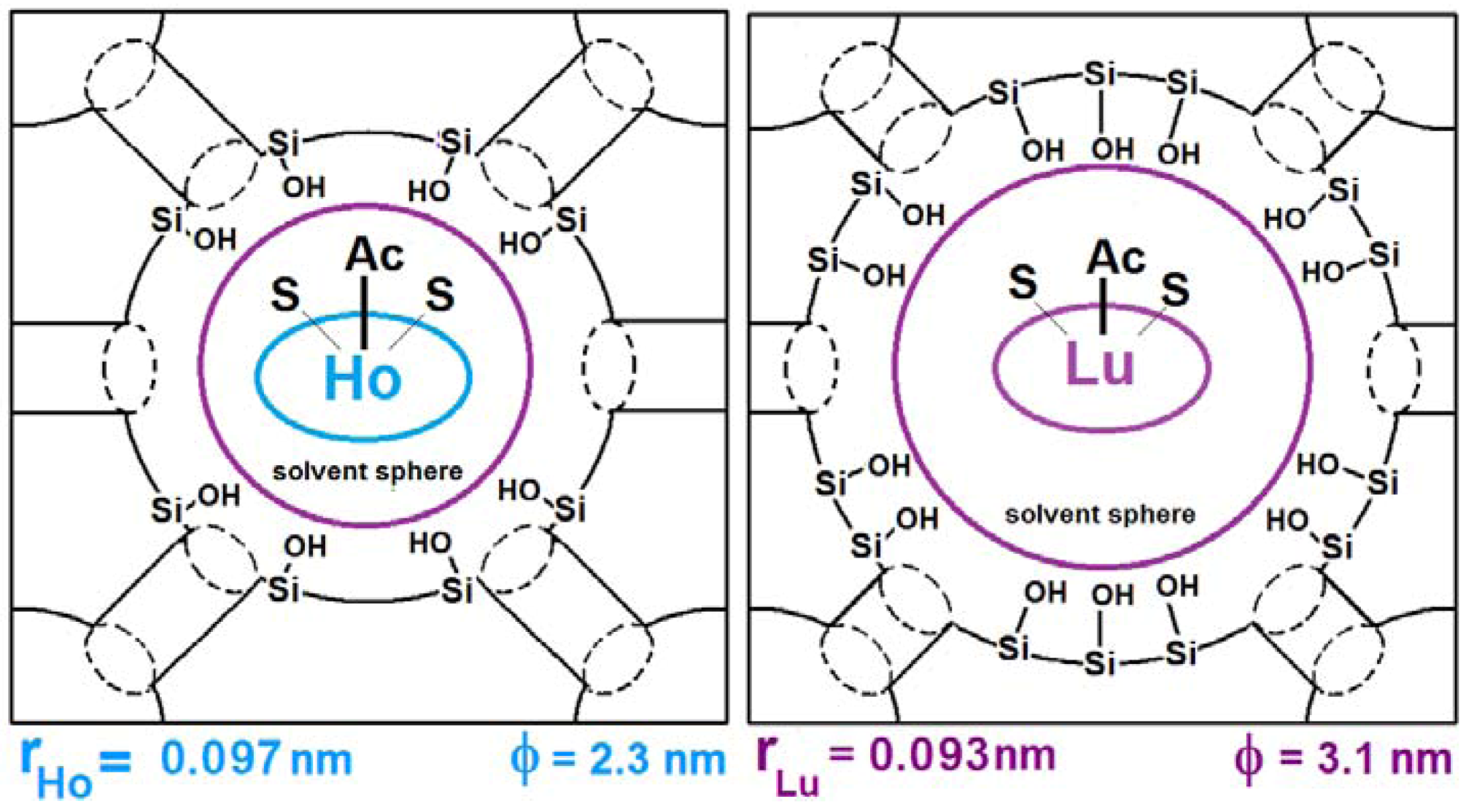
| Ln(TPP)Ac·2S (Ln = Ho to Lu) | physically trapped | SiO2 | 2.6–3.1 | |
| H2T(X or Y)PP (X = OH or Y = OH, NH2) | physically trapped | SiO2 | 2.0–3.5 | |
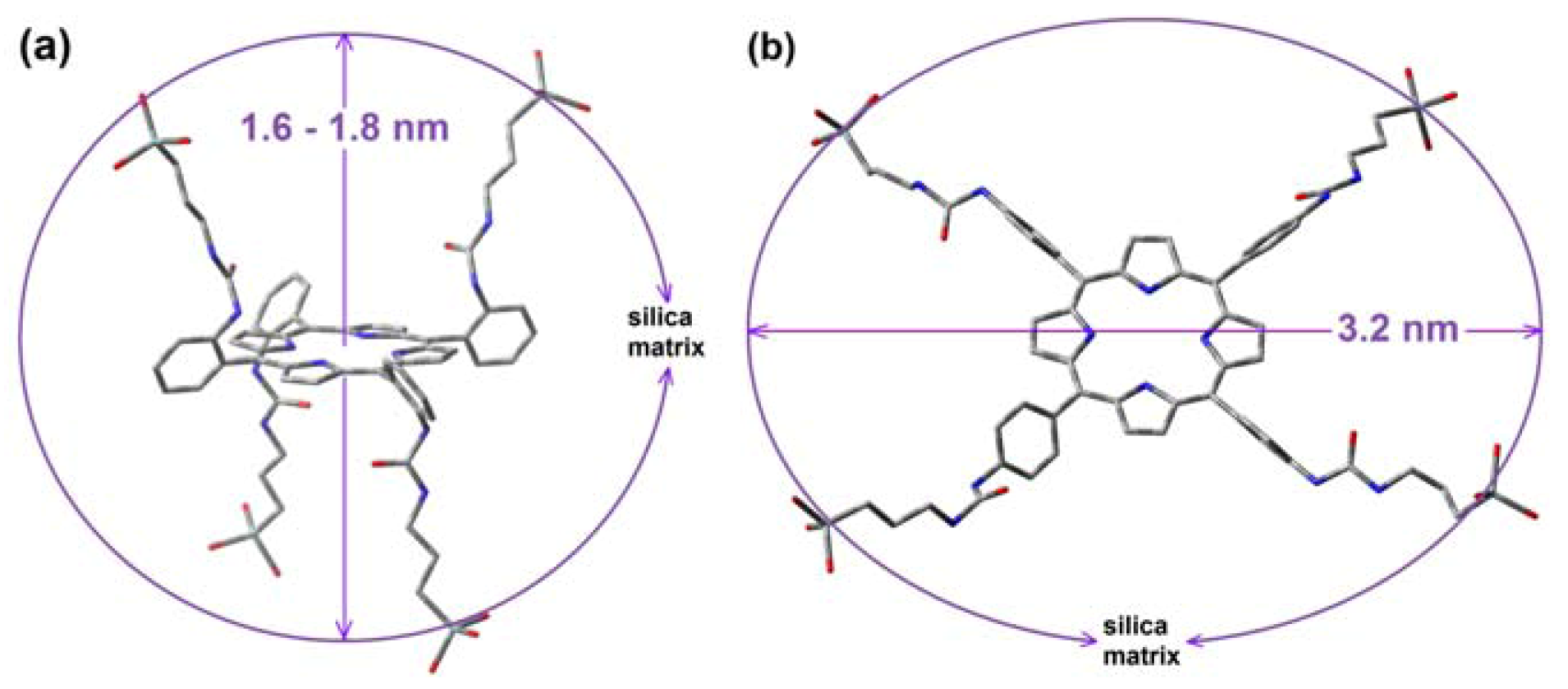
| H2T(o-X)PP | covalently bonded | SiO2 | 1.6–1.8 | 459–632 |
| H2T(p-Y)PP | covalently bonded | SiO2 | 3.2–4.0 | 459–632 |
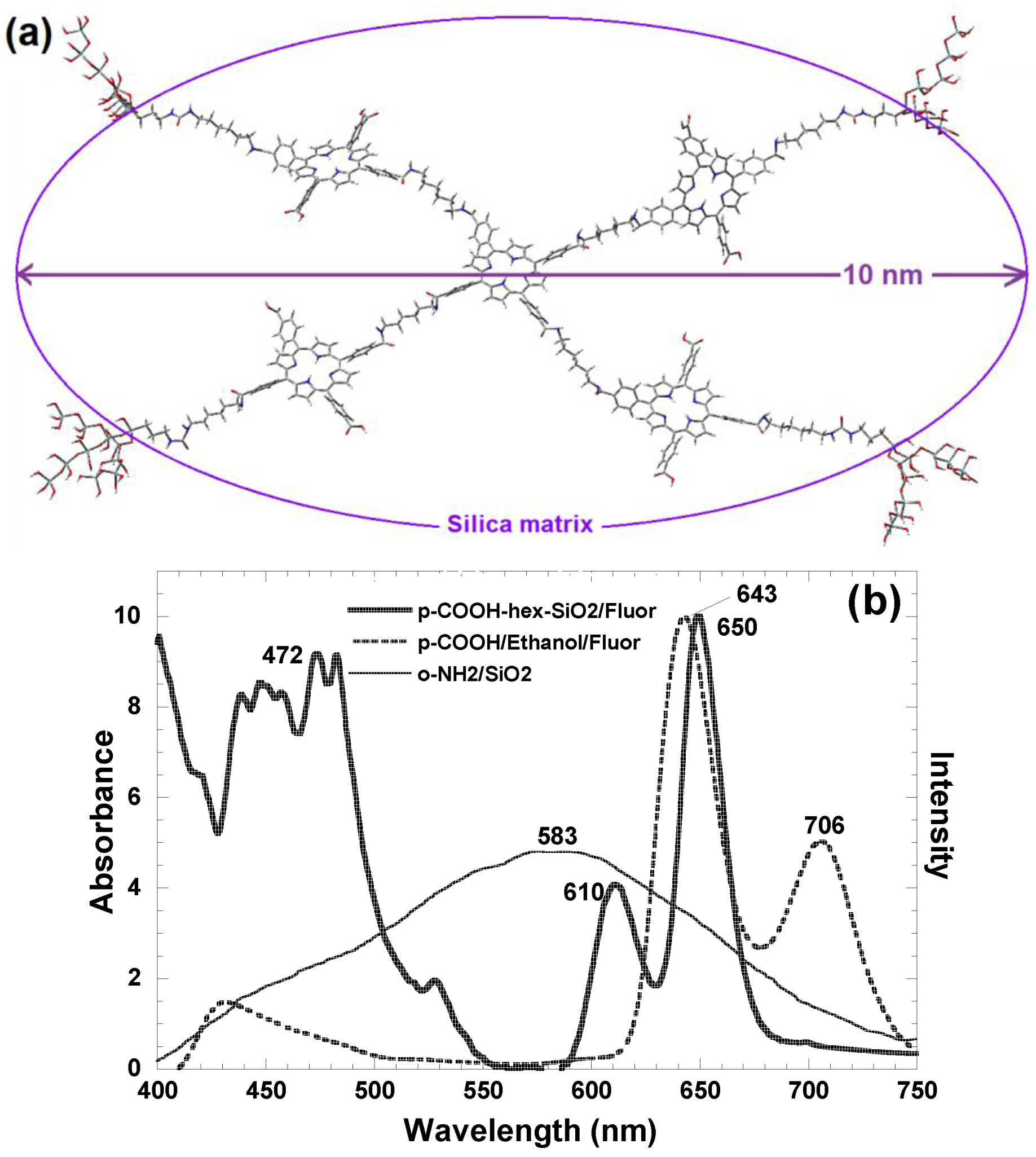
| [H2T(X or Y)PP]n (oligomers) | covalent bonded | SiO2 | 4.4–9.4 | 463–610 |
| (OH)AlTSPc | physically trapped | organo modified-SiO2 | 1.6–3.8 | 688–841 |
| CoT(p-COOH)PP | covalently bonded | organo modified-SiO2 | 2.9–3.5 | 410–621 |
| MTSPc (M = Fe, Co, Ni, Cu, Al) | physically trapped | ZrO2 | 1.9–2.0 |
5. Conclusions
Acknowledgments
References
- Moore, M.R. An Historical Introduction to Porphyrin and Chlorophyll Synthesis, Chapter I. In Tetrapyrroles: Birth, Life and Death; Warren, M.J., Smith, A.G., Eds.; Landes Bioscience and Springer Science+Business Media: Austin, TX, USA, 2009. [Google Scholar]
- Pelletier, J.; Caventou, J.B. Sur la Matiere Verte des Feuilles. Ann. Chim. Phys. Ser. 1818, 2, 194–196. [Google Scholar]
- Verdeil, F. Recherches sur la Matiére Coloranre Verte des Plantes et sur la Matiére Rouge du Sang. Compt. Rend. Acad. Sci. (París) 1851, 33, 689. [Google Scholar]
- Hunfeld, F.L. Der Chemismus in der Thierischen Organisation; Brockhaus: Leipzig, Germany, 1840; pp. 158–163. [Google Scholar]
- Scherer, J. Chemische-Physiologische Untersuchungen. Ann. Chern. Phar. 1841, 40, 1–64. [Google Scholar]
- Mulder, G.H. Uber Eisenfreises Hamatin. J. Prakr. Chern. 1844, 32, 186–197. [Google Scholar]
- Thudichum, J.L.W. Report on Researches Intended to Promote an Improved Chemical Identification of Disease; 10th Report of The Medical Officer; Privy Council, H.M.S.O.: London, UK, 1867; pp. 152–233. [Google Scholar]
- Hoppe-Seyler, F. Ueber das Verhalten des Blutfarbstoffes im Spectrum des Sonnenlichtes. Virchows Arch. 1862, 23, 446–449. [Google Scholar]
- Hoppe-Seyer, F. Über die Chemischen und Optischen Eigenschaftern des Blutsfarbstoffs. Arch. F. Path. Anat. U. Physiol. (Virchow’s Archiv) 1864, 29, 233–235. [Google Scholar]
- Hoppe-Seyler, F. Über die Oxydation in Lebendem Blute. Med-chem Untersuch Lab. 1866, 1, 133–140. [Google Scholar]
- Hoppe-Seyler, F. Beitrage zur Kenntniss des Blutes des Menscheu und der Wirbelthiere Das hamatin. Tubinger Med. Chem. Untersuchungen 1871, 4, 523–533. [Google Scholar]
- Hoppe-Seyler, F. ber das Chlorophyll der Hamen. Physiol. Chem. 1879, 3, 339–341. [Google Scholar]
- Teichmann, L. Üeber die Krystallisation des Organischen Bestandtheile des Blutes. Zeitschr. F Rat. Med. 1853, 3, 375–388. [Google Scholar]
- Schalfejef, M. Preparation of Haemin. J. Soc. Phys. Chim. Russe 1885, 30. [Google Scholar]
- Nencki, M.; Zaleski, J. Untersuchungen uber den Bluffarbstoff. Z. Physiol. Chem. 1900, 30, 384–435. [Google Scholar] [CrossRef]
- Mac Munn, C.A. Researches on Myohaematin and the Histohaematins. Phil. Trans. R. Soc. London 1886, 177, 267–298. [Google Scholar] [CrossRef]
- Keilin, D. A Comparative Study of Turacin and Haematin and its Bearing on Cytochrome. Proc. R. Soc. Lond. Ser. B 1926, 100, 129–151. [Google Scholar] [CrossRef]
- Piloty, O. The Constitution of Coloured Components of the Blood Pigments. Justus Liebigs Ann. Chem. 1910, 377, 314–369. [Google Scholar] [CrossRef]
- Küster, W. Beitrage zur Kenntnis des Bilirubins und Hämins. Hoppe-Seyler’s Z. Physiol. Chern. 1912, 82, 463–483. [Google Scholar] [CrossRef]
- Küster, W. Über den Chemismus der Porphyrinbildung und die Konstitution des Hämin. Hoppe-Seyler’s Z. Physiol. Chem. 1927, 163, 267–283. [Google Scholar] [CrossRef]
- Hoppe-Seyler, F. Weitere Mittheilungen Über die Eigenschaftern des Blutsfarbstoffs. Physiol. Chem. 1880, 4, 193–203. [Google Scholar]
- Soret, J.L. Analyse spectrale: Sur le Spectre d’Absorption du Sang dans la Partie Violette et Ultra-Violette. Compt. Rend. 1883, 97, 1269–1270. [Google Scholar]
- Gamgee, A. Hemoglobin; its Compounds and the Principal Products of its Decompositions. In Textbook of Physiology; Schäfer: Edinburgh and London, UK, 1898; Volume 1, pp. 185–260. [Google Scholar]
- Willstätter, R.; Hocheder, F. Über die Einwirkung von Säiuren und Alkalien auf Chlorophyll. Ann Chem. 1907, 354, 205–258. [Google Scholar]
- Willstätter, R.; Utzinger, M. Untersuchungen über Chlorophyll; über die ersten Umwandlungen des Chlorophylls. Ann. Chem. 1911, 382, 129–194. [Google Scholar] [CrossRef]
- Willstätter, R.; Stoll, A. Untersuchungen uber Chlorophyll; Julius Springer: Berlin, Germany, 1913. [Google Scholar]
- Fischer, H.; Putzer, B. Natural porphyrin. XIX. Announcement. The Transfer of Hemanine in Protoporphyrin and it’s New Illustration of Mesoporphyrin. Z. Physiol. Chem. 1926, 154, 39–63. [Google Scholar] [CrossRef]
- Fischer, H.; Zeile, K. Synthesis of the Hamatoporphyrine Protoporphyrin and Hamin. Ann. Chem. 1929, 468, 98–116. [Google Scholar] [CrossRef]
- Fischer, H.; Treibs, A.; Zeile, K. Information on Hamins, Hamatins and Protoporphyrins. Z. Physiol. Chem. 1930, 193, 138–166. [Google Scholar] [CrossRef]
- Fischer, H.; Goldschmidt, M.; Nüssler, W. The Synthesis of Tetramethyl-Tetraporpyl-Porphine I-IV and Octapropyl-Porphin. Ann. Chem. 1931, 486, 1–54. [Google Scholar] [CrossRef]
- Fischer, H.; Hierneis, J. The New Synthesis of Coproporphyrin III and Coprorhodin II. Z. Physiol. Chem. 1931, 195, 155–168. [Google Scholar]
- Fischer, H.; Kürzinger, A. Bilirrubin pigment and coproporphyrin IV. Z. Physiol. Chem. 1931, 195, 213–240. [Google Scholar] [CrossRef]
- Fischer, H.; Orth, H. Die Chemie des Pyrrols, I; Akad. Verlagsges: Leipzig, Germany, 1937. [Google Scholar]
- Fischer, H.; Stern, A. Die Chemie des Pyrrols, II; Band, Pyrrolfarbstoffe, 2; Akademische Verlagsgesellschaft: Leipzig, Germany, 1940. [Google Scholar]
- Perutz, M.F.; Rossmann, M.G.; Cullis, A.F.; Muirhead, H. Structure of Haemoglobin: A Three-dimensional Fourier Synthesis at 5.5 Å. Resolution by X-Ray Analysis. Nature 1960, 185, 416–422. [Google Scholar] [CrossRef]
- Perutz, M.F.; Dickerson, R.E.; Strandberg, B.E. Structure of Myoglobin: A Three-Dimensional Fourier Synthesis at 2 Å. Resolution. Nature 1960, 185, 422–427. [Google Scholar] [CrossRef]
- Perutz, M.F.; Fermi, G.; Luisi, B. Stereochemistry of Cooperative Mechanism in Hemoglobin. Acc. Chem. Res. 1987, 20, 309–321. [Google Scholar] [CrossRef]
- Kendrew, J.C.; Bodo, G.; Dintzis, H.M. A Three-Dimensional Model of the Myoglobin Molecule Obtained by X-Ray Analysis. Nature 1958, 81, 662–666. [Google Scholar]
- Kendrew, J.C.; Dickerson, R.E.; Strandberg, B.E. Structure of Myoglobin 3-Dimensional Fourier Synthesis at 2 A Resolution. Nature 1960, 185, 422–427. [Google Scholar] [CrossRef]
- Woodward, R.B.; Ayer, W.A.; Beaton, J.M.; Bickelhaupt, F.; Bonnet, R.; Buchschacher, P.; Closs, G.L.; Dutler, H.; Hannah, J.; Hauck, F.P.; et al. The Total Synthesis of Chlorophyll. J. Am. Chem. Soc. 1960, 82, 3800–3802. [Google Scholar]
- Woodward, R.B. The Total Synthesis of Chlorophyll. Pure Appl. Chem. 1961, 2, 383–404. [Google Scholar] [CrossRef]
- Smith, K.M. Porphyrins and Metalloporphyrins; Elsevier Scientific Publishing Co: Amsterdam, The Netherlands, 1976. [Google Scholar]
- Dolphin, D. The Porphyrins, Physical Chemistry, Part A and B; Academic Press: New York, NY, USA, 1979. [Google Scholar]
- Nicholas, R.E.H.; Rimington, C. Isolation of Unequivocal Uroporphyrin-III a Further Study of Turacin. Biochem. J. 1951, 50, 194–201. [Google Scholar]
- Keilin, J.; McCosker, P.J. Reaction Between Uroporphyrin and Copper and their Biological Significance. Biochim. Biophys. Acta 1961, 52, 424–435. [Google Scholar] [CrossRef]
- Hodgson, G.W. Geochemistry of Porphyrins-Reactions During Diagenesis. Ann. NY Acad. Sci. 1973, 206, 670–684. [Google Scholar] [CrossRef]
- Humphrey, A.M. Chlorophyll. Food Chem. 1980, 5, 57–67. [Google Scholar] [CrossRef]
- Chlorophyll and Bacteriochloropylls: Biochemistry, Biophysics, Functions and Applications; Grimm, B.; Porra, R.J.; Rüdiger, W.; Scheer, H. (Eds.) Springer: Dordrecht, The Netherlands, 2006.
- Blankenship, R.E. Molecular Mechanisms of Photosynthesis; Blackwell Science: Oxford, UK, 2002. [Google Scholar]
- Gregory, R.L. Biochemistry of Photosynthesis; Wiley-Interscience: New York, NY, USA, 1971. [Google Scholar]
- Munekaga, Y.; Hashimoto, M.; Miyake, C.; Tomizawa, K.; Endo, T.; Tasaka, M.; Shikanai, T. Cyclic Electron Flow Around Photosystem I is Essential for Photosynthesis. Nature 2004, 429, 579–582. [Google Scholar] [CrossRef]
- Asimov, I. Photosynthesis; Basic Books,Inc: New York, NY, USA, 1968. [Google Scholar]
- Springer, B.A.; Sligar, S.G.; Olson, J.S.; Phillips, G.N., Jr. Mechanisms of Ligand Recognition in Myoglobin. Chem. Rev. 1994, 94, 699–714. [Google Scholar] [CrossRef]
- Shikama, K. The Molecular Mechanism of Autoxidation for Myoglobin and Hemoglobin: A Venerable Puzzles. Chem. Rev. 1998, 98, 1357–1373. [Google Scholar] [CrossRef]
- Sugawara, Y.; Shikama, K. Autoxidation of Native Oxymyoglobin Thermodynamic Analysis of pH Profile. Eur. J. Biochem. 1980, 110, 241–246. [Google Scholar] [CrossRef]
- Mansouri, A.; Winterhalter, K.H. Nonequivalence of Chains in Hemoglobin Oxidation. Biochemistry 1973, 12, 4946–4949. [Google Scholar] [CrossRef]
- Pauling, L.; Weiss, J.J. Nature of Iron-oxygen Bond in Oxyhaemoglobin. Nature 1964, 203, 182–183. [Google Scholar] [CrossRef]
- Phillips, S.E.V.; Schoenborn, B.P. Neutron-Diffraction Reveals Oxygen-Histidine Hydrogen-Bond in Oxymyoglobin. Nature 1981, 292, 81–82. [Google Scholar] [CrossRef]
- Shaanan, B. The Iron Oxygen Bond in Human Oxyhemoblobin. Nature 1982, 296, 683–684. [Google Scholar] [CrossRef]
- Shaanan, B. Structure of Human Oxyhemoglobin at 2.1 A Resolution. J. Mol. Biol. 1983, 171, 31–59. [Google Scholar] [CrossRef]
- Kitagawa, T.; Ondrias, M.R.; Rousseau, D.L.; IkedaSaito, M.; Yonetani, T. Evidence for Hydrogen-bonding of Bound Dioxygen to the Distal Histidine of Oxycobalt Myoglobin and Hemoglobin. Nature 1982, 298, 869–871. [Google Scholar] [CrossRef]
- Tani, M.; Matsu-ura, F.; Nakayama, S.; Ichimura, M.; Nakamura, N.; Naruta, Y. Synthesis and Characterization of Alkanethiolate-Coordinated Iron Porphyrins and their Dioxygen Adducts as Models for the Active Center of Cytochrome P450: Direct Evidence for Hydrogen Bonding to Bound Dioxygen. J. Am. Chem. Soc. 2001, 123, 1133–1142. [Google Scholar]
- Lemberg, R.; Barret, J. Cytochromes; Academic Press: London, UK, 1973. [Google Scholar]
- Crofts, A.R. The Cytochrome bc1 Complex: Function in the Context of Structure. Annu. Rev. Physiol. 2004, 66, 689–733. [Google Scholar] [CrossRef]
- Kurisu, G.; Zhang, H.; Smith, J.L.; Cramer, W.A. Structure of the Cytochrome b(6)f Complex of Oxygenic Photosynthesis: Tuning the Cavity. Science 2003, 302, 1009–1014. [Google Scholar] [CrossRef]
- Rieske, J.S. Composition, Structure, and Function of Complex III of Respiratory-chain. Biochim. Biophys. Acta 1976, 456, 195–243. [Google Scholar] [CrossRef]
- Tsukihara, T.; Aoyama, H.; Yamashita, E.; Tomizaki, T.; Yamaguchi, H.; Shinzawa-Itoh, K.; Nakashima, R.; Yaono, R.; Yoshikawa, S. Structures of Metal Sites of Oxidized Bovine Heart Cytochrome C Oxidase at 2.8 A. Science 1995, 269, 1069–1074. [Google Scholar]
- Shimokata, K.; Katayama, Y.; Murayama, H.; Suematsu, M.; Tsukihara, T.; Muramoto, K.; Aoyama, H.; Yoshikawa, S.; Shimada, H. The Proton Pumping Pathway of Bovine Heart Cytochrome C Oxidase. Proc. Natl. Acad. Sci. USA 2007, 104, 4200–4205. [Google Scholar]
- Ortiz de Montellano, P.R. Cytochrome P450: Structure, Mechanism, and Biochemistry, 3rd ed; Kluwer Academic/Plenum Publishers: New York, NY, USA, 2005. [Google Scholar]
- Meunier, B.; de Visser, S.P.; Shaik, S. Mechanism of Oxidation Reactions Catalyzed by Cytochrome p450 Enzymes. Chem. Rev. 2004, 104, 3947–3980. [Google Scholar] [CrossRef]
- Bendall, D.S.; Manasse, R.S. Cyclic Photophosphorylation and Electron Transport. Biochim. Biophys. Acta 1995, 1229, 23–38. [Google Scholar] [CrossRef]
- Wikstrom, M.; Krab, K.; Saraste, M. Proton-translocating Cytochrome Complexes. Annu. Rev. Biochem. 1981, 50, 623–655. [Google Scholar] [CrossRef]
- Kramer, D.M.; Roberts, A.G.; Muller, F.; Cape, J.; Bowman, M.K. Q-cycle Bypass Reactions at the Q(o) Site of the Cytochrome bc(1) (and Related) Complexes. Methods Enzymol. 2004, 382, 21–45. [Google Scholar] [CrossRef]
- Combs, G.F. The Vitamins: Fundamental Aspects in Nutrition and Health, 3rd ed; Elsevier Scientific Publishing Co: Amsterdam, The Netherlands, 2008. [Google Scholar]
- Martens, J.H.; Barg, H.; Warren, M.J.; Jahn, D. Microbial Production of Vitamin B-12. Appl. Biochem. Biotechnol. 2002, 58, 275–285. [Google Scholar]
- Combe, J.S. History of a Case of Anaemia. Transcripts of the Medical-Chirurgical Society Edinburgh 1824, 1, 193–198. [Google Scholar]
- Addison, T. On anaemia: Disease of the supra-renal capsules. Lond. Med. Gaz. 1849, 43, 517–518. [Google Scholar]
- Biermer, M.A. Eine Eigenthunliche Form von Progressiver Pernicioser Anaemie. Correspondenz Blatt für Schweizer Aerzte Basel 1872, 2, 15–18. [Google Scholar]
- Whipple, G.H.; Hooper, C.W.; Robscheit-Robbins, F.S. Robscheit-Robbins, F.S. Blood Regeneration Following Simple Anemia I. Mixed Diet Reaction. Am. J. Physiol. 1920, 53, 151–166. [Google Scholar]
- Whipple, G.H.; Robscheit-Robbins, F.S. Blood Regeneration in Severe Anemia II. Favorable Influence of Liver, Heart and Skeletal Muscle in Diet. Am. J. Physiol. 1925, 72, 408–418. [Google Scholar]
- Whipple, G.H.; Robscheit-Robbins, F.S. Blood Regeneration in Severe Anemia III. Iron Reaction Favorable—Arsenic and Germanium Dioxide Almost Inert. Am. J. Physiol. 1925, 72, 419–430. [Google Scholar]
- Hooper, C.W.; Robscheit-Robbins, F.S.; Whipple, G.H. Blood Regeneration Following Simple Anemia V. The Influence of Blaud’s Pills. Am. J. Physiol. 1920, 53, 263–282. [Google Scholar]
- Minot, G.R.; Murphy, L.P. Treatment of Pernicious Anemia by a Special Diet. JAMA 1926, 87, 470–476. [Google Scholar] [CrossRef]
- Minot, G.R.; Murphy, L.P. Response of reticulocytes to liver theraphy: Particularly in pernicious anemis. Am. J. Med. Sci. 1928, 175, 581–599. [Google Scholar] [CrossRef]
- Castle, W.B.; Townsend, W.C.; Heath, C.W. Observations on the Etiologic Relationship of Achylia Gastrica to Pernicious Anemia—The Nature of the Reaction Between Normal Human Gastric Juice and Beef Muscle Leading to Clinical Improvement and Increased Blood Formation Similar to the Effect of Liver Feeding. Am. J. Med. Sci. 1930, 180, 305–355. [Google Scholar] [CrossRef]
- Doniach, D.; Roitt, I.M.; Taylor, A.B. Autoimmune Phenomena in Pernicious Anaemia-serological Overlap with Thyroiditis, Thyrotoxicosis, and Systemic Lupus Erythematosus. BMJ 1963, 2, 1374–1380. [Google Scholar]
- Smith, E.L. Purification of Anti-pernicious Anaemia Factors from Liver. Nature 1948, 161, 638–639. [Google Scholar] [CrossRef]
- Rickes, E.L.; Brink, N.G.; Koniuszy, F.R.; Wood, T.R.; Folkers, K. Crystalline Vitamin-B12. Science 1948, 107, 396–397. [Google Scholar]
- Hodgkin, D.C.; Camper, J.; Mackay, M.; Pickworth, J.; Trueblood, T.K.M.; White, J.G. Structure of Vitamin-B12. Nature 1956, 178, 64–66. [Google Scholar] [CrossRef]
- Friedrich, W.; Gross, G.; Bernhauer, K.; Zeller, P. Synthesen auf dem vitamin-B12-gebiet. 4. Partialsynthese von Vitamin-B12. Helv. Chim. Acta 1960, 43, 704–712. [Google Scholar] [CrossRef]
- Woodward, R.B. Recent Advances in the Chemistry of Natural Products. Pure Appl. Chem. 1971, 25, 283–304. [Google Scholar] [CrossRef]
- Woodward, R.B. The total synthesis of Vitamin B12. Pure Appl. Chem. 1973, 33, 145–178. [Google Scholar] [CrossRef]
- Eschenmoser, A.; Wintner, C.E. Natural Product Synthesis and Vitamin B12. Science 1977, 196, 1410–1420. [Google Scholar]
- Eschenmoser, A. Vitamin-B12. Experiments Concerning the Origin of its Molecular-Structure. Angew. Chem. Int. Ed. Engl. 1988, 27, 5–39. [Google Scholar] [CrossRef]
- Daugherty, T.J. Photodynamic Therapy: Principles and Chemical Applications; Henderson, B., Dougherty, T.J., Eds.; Marcel Dekker Inc.: New York, NY, USA, 1992. [Google Scholar]
- Henderson, B.; Dougherty, T.J. How does Photodynamic Pherapy Work? Photochem. Photobiol. 1992, 55, 145–157. [Google Scholar] [CrossRef]
- Daugherty, T.J.; Gomer, C.J.; Henderson, B.W.; Jori, G.; Kessel, D.; Korbelik, M.; Moan, J.; Peng, Q. Photodynamic Therapy. J. Natl. Cancer Inst. 1998, 90, 889–905. [Google Scholar] [CrossRef]
- Darwent, J.R.; Mccubbin, I.; Phillips, D. Excited Singlet and Triplet-State Electron-Transfer Teactions of Aluminum (III) Sulfonated Phthalocyanine. J. Chem. Soc. Faraday Trans. 1982, 78, 347–357. [Google Scholar]
- Rosenthal, I.; Ben-Hur, E. Role of Oxygen in the Phototoxicity of Phthalocyanines. Int. J. Radiat. Biol. 1995, 67, 85–91. [Google Scholar] [CrossRef]
- Ben-Hur, E.; Carmichael, A.; Riesz, P.; Rosenthal, I. Photochemical Generation of Superoxide Radical and the Cytotoxicity of Phthalocyanines. Int. J. Radiat. Biol. 1985, 48, 837–846. [Google Scholar] [CrossRef]
- Sternberg, E.D.; Dolphin, D.; Brickner, C. Porphyrin-based Photosensitizers for Use in Photodynamic Therapy. Tetrahedron 1998, 54, 4151–4202. [Google Scholar] [CrossRef]
- Wan, S.; Parrish, J.A.; Anderson, R.R.; Madden, M. Transmittance of Nonionizing Radaiation in Human Tissues. Photochem. Photobiol. 1981, 34, 679–681. [Google Scholar]
- Wilson, B.C.; Patterson, M.S. The Physics of Photodynamic Therapy. Phys. Med. Biol. 1986, 31, 327–360. [Google Scholar] [CrossRef]
- Marijnissen, J.P.A.; Star, W.M. Quantitative Light Dosimetry In Vitro and In Vivo. Lasers Med. Sci. 1987, 2, 235–242. [Google Scholar] [CrossRef]
- Welch, A.J.; Yoon, G.; van Gemert, M.J.C. Practical Models for Light Distribution in Laser-Irradiated Tissue. Lasers Surg. Med. 1987, 6, 488–493. [Google Scholar] [CrossRef]
- Harriman, A. Luminescence of Porphyrins and Metalloporphyrins Part I. J. Chem. Soc. Faraday Trans. 1 1980, 76, 1979–1985. [Google Scholar]
- Harriman, A. Luminescence of Porphyrins and Metalloporphyrins Part II. J. Chem. Soc. Faraday Trans. 1 1981, 77, 369. [Google Scholar] [CrossRef]
- Harriman, A. Luminescence of Porphyrins and Metalloporphyrins Part III. J. Chem. Soc. Faraday Trans. 1 1981, 77, 1281–1291. [Google Scholar]
- Bethea, D.; Fullmer, B.; Syed, S.; Seltzer, G.; Tiano, J.; Rischko, C.; Gillespie, L.; Brown, D.; Gasparro, F.P. Psoralen Photobiology and Potochemotherapy: 50 Years of Science and Medicine. J. Dermatol. Sci. 1999, 19, 78–88. [Google Scholar] [CrossRef]
- Tamesis, M.E.; Morelli, J.G. Vitiligo Treatment in Childhood: A State of the Art Review. Pediatr. Dermatol. 2010, 27, 437–445. [Google Scholar] [CrossRef]
- Witkop, B. Paul Ehrlich and His Magic Bullets Revisited. Proc. Am. Philos. Soc. 1999, 143, 540–557. [Google Scholar]
- De Kruif, P. Microbe Hunters (1926); Harvest Book,Harcourt Inc: New York, NY, USA, 1996. [Google Scholar]
- Raab, O. Ueber die Wirkung Fluorescierenden Stoffe auf Infusorien. Z. Biol. 1904, 39, 524–546. [Google Scholar]
- von Tappeiner, H.; Jodlbauer, A. Uber die Wirking der Photodynamischen (Fluorescierenden) Stoffe auf Photozen und Enzyme. Dtsch. Arch. Klin. Med. 1904, 80, 427–487. [Google Scholar]
- Blum, H.F. Photodynamic Action and Diseases Caused by Light, Reinhold; Reprinted by Hafner: New York, NY, USA, 1964; Reprinted by Hafner: New York, NY, USA, 1964. [Google Scholar]
- von Tappeiner, H.; Jesionek, H. Therapeutische Versuche mit Fluoreszierenden Stoffen. Munch. Med. Wschr. 1903, 50, 2042–2044. [Google Scholar]
- Jesionek, H.; von Tappeiner, H. Zur Behandlung der Hautcarcinome mit fluoreszierenden Stoffen. Dtsch. Arch. Klin. Med. 1905, 82, 223–226. [Google Scholar]
- Hausmann, W.H. Die Sensibilisierende Wirkung des Hamatoporphyrins. Biochem. Z. 1910, 30, 276–316. [Google Scholar]
- Meyer-Betz, F. Untersuchung Uber die Biologische (Photodynamische) Wirkung des Hamatoporphyrins und Anderer Derivate des Blut- und Gallenfarbstoffs. Dtsch. Arch. Klin. Med. 1913, 112, 476–503. [Google Scholar]
- Policard, A. Etudes sur les Aspects Offerts par des Tumeurs Experimentales Examines a la Lumiere de Wood. C. R. Soc. Biol. 1924, 91, 1423–1424. [Google Scholar]
- Figge, F.H.; Weiland, G.S.; Manganiello, O.J. Cancer Detection and Therapy. Affinity of Neoplastic, Embryonic, and Traumatized Tissues for Porphyrins and Metalloporphyrins. Proc. Soc. Exp. Biol. Med. 1948, 68, 640–641. [Google Scholar]
- Rasmussen, D.S.; Ward, G.E.; Figge, F.H.J. Fluorescence of Human Lymphatic and Cancer Tissues Following High Doses of Intravenous Hematoporphyrin. Cancer 1955, 1, 78–81. [Google Scholar]
- Schwartz, S.; Absolon, K.; Vermund, H. Some Relationships of Porphyrins, X-rays, and Tumor. Bull. Minn. Univ. School Med. 1955, 27, 7–13. [Google Scholar]
- Schwartz, S.; Winkelman, J.W.; Lipson, R.L. Historical Perspective. In Photodynamic Therapy: Principles and Chemical Applications; Henderson, B., Dougherty, T.J., Eds.; Marcel Dekker Inc: New York, NY, USA, 1992; pp. 1–15. [Google Scholar]
- Lipson, R.L.; Baldes, E.J. The Photodynamic Properties of a Particular Hematoporphyrin Derivative. Arch. Dermatol. 1960, 82, 508–516. [Google Scholar] [CrossRef]
- Lipson, R.L.; Baldes, E.J.; Olsen, A.M. The Use of a Derivative of Hematoporphyrin in Tumor Detection. J. Natl. Cancer Inst. 1961, 26, 1–8. [Google Scholar]
- Lipson, R.L.; Grey, M.J.; Baldes, E.J. Hematoporphyrin Derivative for Detection and Management of Cancer. In Proceedings of 9th International Cancer Congress, Tokyo, Japan, 26 October 1966; p. 393.
- Lipson, R.L.; Baldes, E.J.; Gray, M.J. Hematoporphyrin Derivative for Detection and Management of Cancer. Cancer 1967, 20, 2255–2257. [Google Scholar] [CrossRef]
- Diamond, I.; McDonagh, A.F.; Wilson, C.B. Photodynamic Therapy of Malignant Tumors. Lancet 1972, 300, 1175–1177. [Google Scholar] [CrossRef]
- Kelly, J.F.; Snell, M.E. Hematoporphyrin Derivative: A Possible Aid in the Diagnosis and Therapy of Carcinoma of the Bladder. J. Urol. 1976, 115, 150–151. [Google Scholar]
- Dougherty, T.J.; Kaufman, J.E.; Goldfarb, A.; Weishaupt, K.R.; Boyle, D.; Mittleman, A. Photoradiation Therapy for the Treatment of Malignant Tumors. Cancer Res. 1978, 38, 2628–2635. [Google Scholar]
- Bugelski, P.J.; Porter, C.W.; Dougherty, T.J. Autoradiographic Distribution of Hematoporphyrin Derivative in Normal and Tumor Tissue of Mouse. Cancer Res. 1981, 41, 4606–4612. [Google Scholar]
- Dougherty, T.J. Studies on the Structure of Porphyrins Contained in Photofrin II. Photochem. Photobiol. 1987, 46, 569–573. [Google Scholar] [CrossRef]
- Dougherty, T.J.; Thoma, R.E.; Boyle, D.; Weishaupt, K.R. Photoradiation Therapy for the Treatment of Malignant Tumors: Role of the Laser. In Laser in Photomedicine and Photobiology; Pratesi, R., Sacchi, C.A., Eds.; Springer: New York, NY, USA, 1980; pp. 67–75. [Google Scholar]
- Mironov, A.F.; Nizhnik, A.N.; Nockel, A.Y. On the Nature of Chemical Bonds in Haematoporphyrin Derivative. J. Photochem. Photobiol. B 1990, 6, 337–341. [Google Scholar] [CrossRef]
- Mironov, A.F.; Nizhnik, A.N.; Nockel, A.Y. Hematoporphyrin Derivatives: An Oligomeric Composition Study. J. Photochem. Photobiol. B 1990, 4, 297–306. [Google Scholar] [CrossRef]
- Mironov, A.F.; Seylanov, A.S.; Seylanov, J.A.; Pizhik, V.M.; Deruzhenko, I.V.; Nockel, A.J. Hematoporphyrin Derivative: Distribution in a Living Organism. J. Photochem. Photobiol. B 1992, 16, 341–346. [Google Scholar] [CrossRef]
- Stranadko, E.F.; Skobelkin, O.K.; Mironov, A.F. Photodynamic Therapy of Cancer by Photochem. In Photodynamic Therapy of Cancer I; Jori, G., Moan, J., Star, W.M., Eds.; Proceedings of the Photo-Optical Instrumentation Engineers (SPIE): Bellingham, WA, USA, 1994; Volume 2078, pp. 499–501. [Google Scholar]
- Stranadko, E.F.; Skobelkin, O.K.; Vorozhtsov, G.N.; Mironov, A.F. Photodynamic Therapy of Cancer, Five-Year Clinical Experience. In Photochemotherapy: Photodynamic Therapy and other Modalities III; Berg, K., Ehrenberg, B., Malik, Z., Moan, J., Eds.; Proceedings of the photo-optical instrumentation engineers (SPIE): Bellingham, WA, USA, 1997; Volume 3191, pp. 253–362. [Google Scholar]
- Huang, Z. Photodynamic Therapy in China: Over 25 Years of Unique Clinical Experience: Part One, History and Domestic Photosensitizers. Photodiagnosis Photodyn. Ther. 2006, 3, 3–10. [Google Scholar] [CrossRef]
- Xu, D.Y. Research and Development of Photodynamic Therapy Photosensitizers in China. Photodiagnosis Photodyn. Ther. 2007, 4, 13–25. [Google Scholar] [CrossRef]
- Stranadko, E.F.; Skobelldn, O.K.; Litwin, G.D.; Astrakhankina, T.A. Photodynamic Therapy of Human Malignant Tumours: A Comparative Study between Photoheme and Tetrasulfonated Aluminium Phthalocyanine. In Photochemotherapy: Photodynamic Therapy and Other Modalities; Ehrenberg, B., Jori, G., Moan, J., Eds.; Proc SPIE: Barcelona, Spain, 1996; Volume 2625, pp. 440–448. [Google Scholar]
- Stranadko, E.F.; Skobelkin, O.K.; Vorozhtsov, G.N. Clinical Trials of a Second Generation Phorosensitizers-Photosense. In Abstracts of Biennal 7th Congress of International Photodynamic Association, Nantes, France, 7-9 July 1998; Patrice, T., Ed.;
- Stranadko, E.F.; Ponomarev, G.V.; Mechkov, V.M.; Riabo, M.V.; Ivanov, A.V.; Reshetnikov, A.V.; Koraboyev, U.M. The First Experience of Photodithazine Clinical Application for Photodynamic Therapy of Malignant Tumors. In Optical Methods for Tumor Treatment and Detection: Mechanisms and Techniques in Photodynamic Therapy IX; Proc. SPIE: San Jose, CA, USA, 22 January 2000; Volume 3909, pp. 138–144. [Google Scholar]
- Gomer, C.J. Preclinical Examination of First and Second Generation Photosensitizers Used in Photodynamic Theraphy. Photochem. Photobiol. 1991, 54, 1093–1107. [Google Scholar] [CrossRef]
- Snyder, E.G. Preparation of Chlorine. U.S. Patent 2,274,101, February 1942. [Google Scholar]
- Sakata, I.; Nakajima, S. Pheophorbid Derivatives and Alkaline Salts Thereof. U.S. Patent 4,709,022, 24 November 1987. [Google Scholar]
- Bommer, J.C.; Burnham, B.F. Tetrapyrrol Therapeutic Agents. U.S. Patent 4,656,186, 7 April 1987. [Google Scholar]
- Morgan, A.R.; Selman, S.H.; Kreimer-Birnbaum, M. Production and Use of Purpurins, Chlorins and Purpurins- and Chlorin-Containing Compositions. U.S. Patent 5,216,012, June 1993. [Google Scholar]
- Smith, K.M.; Lee, S-J.H. Long Wavelength Water Soluble Chlorine Photosensitizers Useful for Photodynamic Theraphy and Diagnosis of Tumors. U.S. Patent 5,330,741, 19 July 1994. [Google Scholar]
- Reshetnikov, A.V.; Zalevsky, I.D.; Kemov, J.V.; Ivanov, A.V.; Karmenyan, A.V.; Gradjushko, A.T.; Laptev, V.P.; Neugodova, N.P.; Abakumova, O.Y.; Privalov, V.A.; et al. Photosensitizers and Method for Production Thereof. U.S. Patent 6, 969,765 B2, 29 November 2005. [Google Scholar]
- Bae, S.M.; Kim, Y.W.; Lee, J.M.; Namkoong, S.E.; Han, S.J.; Kim, J.K.; Lee, C.H.; Chun, H.J.; Jin, H.S.; Ahn, W.S. Photodynamic Effects of Radachlorin® on Cervical Cancer Cells. Cancer Res. Treat. 2004, 36, 389–394. [Google Scholar] [CrossRef]
- Wainwright, M. Photodynamic Antimicrobial Chemotherapy (PACT). J. Antimicrob. Chemother. 1998, 42, 13–28. [Google Scholar] [CrossRef]
- Zeina, B.; Greeman, J.; Purcell, W.; Das, B. Killing Cutaneous Microbial Species by Photodynamic Therapy. Br.J. Deratol. 2001, 144, 274–278. [Google Scholar] [CrossRef]
- Nitzan, Y.; Gozhansky, S.; Malik, Z. Effect of Photoactivated Hematoporphyrin Derivative on the Viability of Staphylococcoes aureus. Curr. Microbiol. 1983, 8, 279–284. [Google Scholar]
- Malik, Z.; Hanania, J.; Nitzan, Y. New Trends in Photobiology Bactericidal Effects of Photoactivated Porphyrins, an Alternative Approach to Antimicrobial Drugs. J. Photochem. Photobiol. B 1990, 5, 281–293. [Google Scholar] [CrossRef]
- Malik, Z.; Landan, H.; Erenberg, B.; Nitzan, Y. Bacterial and Viral Photodinamic Inactivation. In Photodynamic Theraphy: Medical Applications; Henderson, B.W., Dougherty, T.J., Eds.; Marcel Dekker Inc.: Buffalo, NY, USA, 1992; pp. 97–113. [Google Scholar]
- Minnock, A.; Vernon, D.I. Photoinactivation of Bacteria. Use of a Cationic Water-Soluble Zinc Phthalocyanine to Photoinactivate both Gram-Negative and Gram-Positive Bacteria. J. Photochem. Photobiol. B 1996, 32, 159–164. [Google Scholar] [CrossRef]
- Minnock, A.; Vernon, D.I.; Schofield, J. Mechanism of Uptake of a Cationic Water-Soluble Pyridinium Zinc Phthalocyanine across the Outer Membrane of Escherichia coli. Antimicrob. Agents Chemother. 2000, 44, 522–527. [Google Scholar] [CrossRef]
- Bertoloni, G.; Salvato, B.; DallÁcqua, M.; Vazzoler, M.; Jori, G. Hemoporphyrin-Sensitized Photoinactivation of Straptococcus fecalis. Photochem. Photobiol. 1984, 39, 811–816. [Google Scholar] [CrossRef]
- Reddi, E.; Ceccon, M.; Valduga, G.; Jori, G.; Bommer, J.C.; Elisei, F.; Latterini, L.; Mazzucato, U. Photophysical Properties and Antibacterial Activity of Meso-Substituted Cationic Porphyrins. Photochem. Photobiol. 2002, 75, 462–470. [Google Scholar] [CrossRef]
- Horowitz, B.; Williams, B.; Rywkin, S.; Prince, A.M.; Pascual, D.; Geacintov, N.; Valinsky, J. Inactivation of Viruses in Blood with Aluminum Phthalocyanine Derivatives. Transfusion 1991, 31, 102–108. [Google Scholar]
- Jackson, Z.; Meghji, S.; McRobert, A.M.; Henderson, B.; Wilson, M. Killing of the Yeast and Hyphal Forms of Candida albicans Using a Light-Activated Antimicrobial Agent. Laser Med. Sci. 1999, 14, 150–177. [Google Scholar] [CrossRef]
- Bachmann, B.; Knuver-Hopf, J.; Lambrecht, B. Target Structures for HIV-1 Inactivation by Methylene Blue and Light. J. Med. Virology. 1995, 47, 172–178. [Google Scholar] [CrossRef]
- Bedvell, J.; Holton, J.; Vaira, D.; Macrobert, A.J.; Bown, S.G. In Vitro Killing of Helicobacter Pylori with Photodynamic Therapy. Lancet 1990, 335, 1287. [Google Scholar] [CrossRef]
- Millson, C.E.; Wilson, M.; MacRobert, A.J.; Bown, S.G. Ex-vivo Treatment of Gastric Helicobacter Infection by Photodynamic Therapy. J. Photochem. Photobiol. B 1996, 32, 59–65. [Google Scholar] [CrossRef]
- Millson, C.E.; Wilson, M.; MacRobert, A.J.; Bown, S.G. The Killing of Helicobacter pylori by Low-Power Laser Light in the Presence of a Photosensitiser. J. Med. Microbiol. 1996, 44, 245–252. [Google Scholar] [CrossRef]
- Måkvy, P.; Messmann, H.; Regula, J.; Conio, M.; Pauer, M.; Millson, C.E.; MacRobert, A.J.; Bown, S.G. Photodynamic Therapy for Gastrointestinal Tumors Using Three Photosensitizers-ALA Induced PPIX, Photofri® and MTHPC. A Pilot Study. Neoplasma 1998, 45, 1257–161. [Google Scholar]
- Capella, M.; Menezes, S. Synergism Between Electrolysis and Methylene Blue Photodynamic Action in Escherichia coli. Int. J. Radiat. Biol. 1992, 62, 321–326. [Google Scholar] [CrossRef]
- Soukos, N.; Ximenez-Fyvie, L.A.; Hamblin, M.R.; Socransky, S.S.; Hasanl, T. Mechanisms of Action: Physiological Effects, Targeted Antimicrobial Photochemotherapy. Antimicrob. Agents Chemother. 1998, 42, 2595–2601. [Google Scholar]
- Devanathan, S.; Dahl, T.; Midden, W. Readily Available Fluorescein Isothiocyanate-Conjugated Antibodies can be Easily Converted into Targeted Phototoxic Agents for Antibacterial, Antiviral, and Anticancer Therapy. Proc. Natl. Acad. Sci. USA 1990, 87, 2980–2984. [Google Scholar] [CrossRef]
- Milgrom, R.L. The Colour of Life; An Introduction to the Chemistry of Porphyrins and Related Compounds; Oxford University Press: Oxford, UK, 1997. [Google Scholar]
- Braun, A.; Tcherniac, J. Über die Produkte der Einwirkung von Acetanhydrid auf Phthalamid. Berichte der Deutschen Chemischen Gesellschaft 1907, 40, 2709–2714. [Google Scholar]
- Diesbach, H.; von der Weid, E. Quelques sels Complexes des o-dinitriles avec le Cuivre et la Pyridine. Helev. Chim. Acta 1927, 10, 886–888. [Google Scholar] [CrossRef]
- Lever, A.B.P. The Phthalocyanines. Adv. Inorg. Chem. Radiochem. 1965, 7, 27–114. [Google Scholar] [CrossRef]
- Moser, F.H.; Thomas, A.L. The Phthalocyanines; CRC, Press Inc: Boca Raton, FL, USA, 1983; Volume 1, p. 127. [Google Scholar]
- McKeown, N.B. Phthalocyanine Materials. In Synthesis, Structure and Function; Cambridge University Press: Cambridge, UK, 1998. [Google Scholar]
- Leznoff, C.C.; Lever, A.B.P. Phtalocyanines Properties and Applications; VCH Publishers Inc: New York, NY, USA, 1989; Volume 1, 1993, Volume II-III; 1996, Volume IV. [Google Scholar]
- Kadish, K.; Smith, K.M.; Guilard, R. The Porphyrin Handbook (20 Volumes); Academic Press: San Diego, CA, USA, 2003. [Google Scholar]
- Stuzhin, P.A.; Khelevina, O.G. Azaporphyrins: Structure of the Reaction Centre and Reactions of Complex Formation. Coord. Chem. Rev. 1996, 147, 41–86. [Google Scholar]
- Meller, A.; Ossko, A. Phthalocyaninartige Bor-Komplexe. Monatsh. Chem. 1972, 103, 150–155. [Google Scholar] [CrossRef]
- Lux, F.; Dempf, D.; Graw, D. Bis(phthalocyaninato)thorium(IV) und -uran(IV). Angew. Chem. 1968, 80, 792–793. [Google Scholar] [CrossRef]
- Lux, F.; Demf, D. Diphthalocyaninato-thorium(IV) and -uranium(IV). Angew. Chem. Int. Ed. 1968, 7, 819–820. [Google Scholar] [CrossRef]
- Marks, T.J.; Stojakovic, D.R. Large Metal Ion-centered Templated Reactions. Chemical and Spectral Studies of the “Superphthalocyanine” Dioxocyclopentakis(1 iminoisoindolinato) uranium(IV) and its Derivatives. J. Am. Chem. Soc. 1978, 100, 1695–1705. [Google Scholar] [CrossRef]
- Chmielewski, P.J.; Latos-Grażyński, L.; Rachlewicz, K. 5,10,15,20-Tetraphenylsapphyrin-Identification of a Pentapyrrolic Expanded Porphyrin in the Rothemund Synthesis. Chem. Eur. J. 1995, 1, 68–73. [Google Scholar]
- Sessler, J.L.; Weghorn, S.J. Expanded Contracted and Isomeric Porphyrins; Elsevier: Oxford, UK, 1997. [Google Scholar]
- Sessler, J.L.; Seidel, D. Synthetic Expanded Porphyrin Chemistry. Angew. Chem., Int. Ed. Engl. 2003, 42, 5134–5175. [Google Scholar] [CrossRef]
- Sessler, J.L.; Vivian, A.E.; Seidel, D.; Burrell, A.K.; Hoehner, M.; Mody, T.D.; Gebauer, A.; Weghorn, S.J.; Lynch, V. Actinide Expanded Porphyrin Complexes. Coord. Chem. Rev. 2001, 216-217, 411–434. [Google Scholar]
- Jasat, A.; Dolphin, D. Expanded Porphyrins and their Heterologs. Chem. Rev. 1997, 97, 2267–2340. [Google Scholar] [CrossRef]
- Toganoh, M.; Gokulnath, S.; Kawabe, Y.; Furuta, H. Unique Interaction between Directly-linked Laminated π-Planes in Benzonorrole Dimer. Chem. Eur. J. 2012, 18, 4380–4391. [Google Scholar] [CrossRef]
- Pandey, R.K.; Gerzevske, K.R.; Zhoue, H.; Smith, K.M. New Syntheses of Biliverdins, Corroles and Araporphyrins from 1,19-Dibromo-ac-biladiene Salts. J. Chem. Soc. Perkin Trans. 1994, 1, 971–977. [Google Scholar]
- Rambo, B.M.; Sessler, J.L. Oligopyrrole Macrocycles: Receptors and Chemosensors for Potentially Hazardous Materials. Chem. Eur. J. 2011, 17, 4946–4959. [Google Scholar] [CrossRef]
- Chmielewski, P.J.; Latos-Grazynski, L.; Rachlewicz, K.; Glowiak, T. Tetra-p-Tolylporphyrin with an Inverted Pyrrole Ring: A Novel Isomer of Porphyrin. Angew. Chem. Int. Ed. Engl. 1994, 33, 779–781. [Google Scholar] [CrossRef]
- Latos-Grażyński, L.; Pacholska, E.; Chmielewski, P.J.; Olmstead, M.M.; Balch, A.L. Alteration of the Reactivity of Tellurophene within a Core-Modified Porphyrin Environment. Synthesis and Oxidation of 21-Tellurapophyrin. Angew.Chem. Int. Ed. Engl. 1995, 34, 2252–2254. [Google Scholar] [CrossRef]
- Sprutta, N.; Świderska, M.; Latos-Grażyński, L. Dithiadiazuliporphyrin: Facile Generation of Carbaporphyrinoid Cation Radical and Dication. J. Am. Chem. Soc. 2005, 127, 13108–13109. [Google Scholar]
- Furuta, H.; Asano, T.; Ogawa, T. N-Confused Porphyrin: A New Isomer of Tetraphenylporphyrin. J. Am. Chem. Soc. 1994, 116, 767–768. [Google Scholar] [CrossRef]
- Furuta, H.; Maeda, H.; Furuta, T.; Osuka, A. First Synthesis of Tetrapyrrolylporphyrin. Org. Lett. 2000, 2, 187–189. [Google Scholar] [CrossRef]
- Maeda, H.; Furuta, H. A dozen years of N-confusion: From Synthesis to Supramolecular Chemistry. Pure Appl. Chem. 2006, 78, 29–44. [Google Scholar] [CrossRef]
- Toganoh, M.; Yamamoto, T.; Hihara, T.; Akimaru, H.; Furuta, H. Regulation of NH-Tautomerism in N-Confused Porphyrin by N-Alkylation. Org. Biomol. Chem. 2012, 10, 4367–4374. [Google Scholar] [CrossRef]
- Nikels, D.E.; Jones, R.S.; Kuder, J.E. Use of Anthracyanine and Phenanthracyanine Chromophores in Optical Information Media. U.S. Patent 4,783,386, November 1988. [Google Scholar]
- Kobayashi, N.; Nakajima, S.; Ogata, H.; Fukuda, T. Synthesis, Spectroscopy, and Electrochemistry of Tetra-tert-butylated Tetraazaporphyrins, Phthalocyanines, Naphthalocyanines, and Anthracocyanines, together with Molecular Orbital Calculations. Chem. Eur. J. 2004, 10, 6294–6312. [Google Scholar] [CrossRef]
- Muranaka, A.; Yonehara, M.; Uchiyama, M. Azulenocyanine: A New Family of Phthalocyanines with Intense Near-IR Absorption. J. Am. Che. Soc. 2010, 132, 7845–7845. [Google Scholar]
- Lash, T.D.; Chandrasekar, P. Synthesis of Tetraphenyltetraacenaphthoporphyrin: A New Highly Conjugated Porphyrin System with Remarkably Red Shifted Electronic Absorption Spectra. J. Am. Chem. Soc. 1996, 118, 8767–8768. [Google Scholar] [CrossRef]
- Davis, N.K.S.; Thompson, A.L.; Anderson, H.L. A Porphyrin Fused to Four Anthracenes. J. Am. Chem Soc. 2011, 133, 30–31. [Google Scholar]
- Kobayashi, N.; Ashida, T.; Osa, T.; Konami, H. Phthalocyanines of a Novel Structure: Dinaphthotetraazaporphyrins with D2h Symmetry. J. Am. Chem. Soc. 1994, 33, 1737–1740. [Google Scholar]
- Islyaikin, M.K.; Danilova, E.A. Structural Analogs of Tetrapyrrole Macrocycles and their Biological Properties. Russ. Chem. Bull. Int. Ed. 2007, 56, 689–706. [Google Scholar] [CrossRef]
- Hoard, J.L. Stereochemistry of Hemes and Other Metalloporphyrins. Science 1971, 174, 1295–1302. [Google Scholar]
- Milroy, J.A. A Stable Derivative of Hæmochromogen. J. Physiol. 1909, 38, 384–391. [Google Scholar]
- Treibs, A. Metal Complexes of Porphyrins. Justus Liebing Ann. Chem. 1969, 728, 115–148. [Google Scholar] [CrossRef]
- Tsutsui, M.; Ichakawa, M.; Vohwinkel, F.; Suzuki, K. Unusual Metalloporphyrins. I. Preparation of Chromium Mesoporphyrin IX Dimethyl Ester1. J. Am. Chem. Soc. 1966, 88, 854–855. [Google Scholar] [CrossRef]
- Ostfeld, D.; Tsutsui, M.; Hrung, C.P.; Conway, D.C. Unusual Metalloporphyrins. VII. Porphyrin Bridging Two Metal Atoms: μ[mesoporphyrin IX dimethyl esterato]bis[tricarbonylrhenium(I)]. J. Am. Chem. Soc. 1971, 93, 2548–2549. [Google Scholar] [CrossRef]
- Tsutsui, M.; Hrung, C.P. Unusual Metalloporphyrins. XVIII. First Heterodinuclear Metalloporphyrin. [Tricarbonyltechnetium(I)]-μ-[mesoporphyrin IX dimethyl esterato]-[tricarbonylrhenium(I)]. J. Am. Chem. Soc. 1973, 95, 5777–5778. [Google Scholar] [CrossRef]
- Fleischer, E.B.; Lavalle, D. Phenylrhodium Tetraphenylporphine. Novel Synthesis of a Rhodium-carbon σ-bond. J. Am. Chem. Soc. 1967, 89, 7132–7233. [Google Scholar] [CrossRef]
- Srivastava, T.S.; Fleischer, E.B. New Oxomolybdenum(V) Compounds of Tetraphenylporphine. J. Am. Chem. Soc. 1970, 92, 5518–5519. [Google Scholar] [CrossRef]
- Horrocks, W.D., Jr.; Sipe, J.P., III. Lanthanide Complexes as Nuclear Magnetic Resonance Structural Probes: paramagnetic Anisotropy of Shift Reagent Adducts. Science 1972, 177, 994–996. [Google Scholar]
- Horrocks, W.D., Jr.; Wong, C.P.; Venteicher, R. Lanthanide Porphyrin Complexes. Potential New Class of Nuclear Magnetic Resonance Dipolar Probe. J. Am. Chem. Soc. 1974, 96, 7149–7150. [Google Scholar] [CrossRef]
- Wong, C.P.; Horrocks, W.D., Jr. New Metalloporphyrins. Thorium and Yttrium Complexes of Tetraphenylporphin. Tetrahedron Lett. 1975, 31, 2637–2640. [Google Scholar]
- Horrocks, W.D., Jr.; Venteicher, R.F.; Spilburg, C.A.; Vallee, B.L. Lanthanide Porphyrin Probes of Heme Proteins. Insertion of Itterbium(III)mesoporphyrin IX into Apomyoglobin. Biochem. Biophys. Res. Commun. 1975, 64, 317–322. [Google Scholar] [CrossRef]
- Martarano, L.A.; Wong, C.P.; Horrocks, W.D., Jr. Lanthanide Porphyrin Complexes. Evaluation of Nuclear Magnetic Resonance Dipolar Probe and Shift Reagent Capabilities. J. Phys. Chem. 1976, 98, 7157–7162. [Google Scholar]
- Buchler, J.W.; Rohbock, K. Metallkomplexe mit Tetrapyrrol-liganden: IX. Octaäthylporphinato-osmium(II)-carbonyl-komplexe mit Trans-ständigen Donator-liganden. J. Organometal. Chem. 1974, 65, 223–234. [Google Scholar]
- Buchler, J.W.; Scharbert, B. Metal Complexes with Tetrapyrrole Ligands. 40. Cerium(IV) bis(octaethylporphyrinate) and Dicerium(III) Tris(octaethylporphyrinate): Parents of a New Family of Lanthanoid Double-decker and Triple-decker Molecules. J. Am. Chem. Soc. 1986, 108, 3652–3659. [Google Scholar] [CrossRef]
- Buchler, J.W.; Scharbert, B. Metal Complexes with Tetrapyrrole Ligands. 50. Redox Potentials of Sandwichlike Metal Bis(octaethylporphyrinates) and their Correlation with Ring-ring Distances. J. Am. Chem. Soc. 1988, 110, 4272–4276. [Google Scholar] [CrossRef]
- Buchler, J.W.; Hammerschmitt, P.; Kaufeld, I.; Loffer, J. Redoxpotentiale, Nah-Infrarot-Absorption, Ionenradien sowie Ring-Ring-Abstand in Metall(II)-bis(tetraphenylporphyrinat)-Systemen mit Porphyrinringen in Unterschiedlichen Oxidationsstufen. Chem. Ber. 1991, 124, 2151–2159. [Google Scholar] [CrossRef]
- Kasuga, K.; Tsutsui, M. Some New Developments in the Chemistry of Metallophthalocyanines. Coord. Chem. Rev. 1980, 32, 67–95. [Google Scholar]
- De Cian, A.; Moussavi, M.; Fisher, J.; Weiss, R. Synthesis, Structure, and Spectroscopic and Magnetic Properties of Lutetium(III) Phthalocyanine Derivatives: LuPc2•CH2Cl2 and [LuPc(OAc)(H2O)2]•H2O•2CH3OH. Inorg. Chem. 1985, 24, 3162–3167. [Google Scholar] [CrossRef]
- Gurevich, M.G.; Sevchenko, G.P.; Solov’ev, K.N. The Spectroscopy of Porphyrins. Sov. Phy. Uspekhi. 1963, 6, 67–105. [Google Scholar] [CrossRef]
- Combe, S.; Bunzli, J.C.G. Lanthanide Near-infrared Luminescence in Molecular Probes and Devices. In Handbook of the Physics and Chemistry of Rare Earths; Gschneider, K.A., Jr., Bunzli, J.-C.G., Pecharsky, V.K., Eds.; Elsevier Science: Oxford, MS, USA, 2007; Volume 37, pp. 244–253. [Google Scholar]
- He, H.H.; Zhu, X.X.; Cheah, K.W. Reactivity of Aqua Coordinated Monoporphyrinate Lanthanide Complexes: Synthetic, Structure and Photoluminiscence Studies of Lanthanide Porphyrinate Dimer. Dalton Trans. 2004, 23, 4064–5003. [Google Scholar]
- Cook, C.E.; Twine, M.E.; Myers, M.; Amerson, E.; Kepler, J.A.; Taylor, G.F. Theophylline Radioimmunoassay: Synthesis of Antigen and Characterization of Antiserum. Res. Comm. Chem. Pathol. Pharmacol. 1976, 13, 497–505. [Google Scholar]
- Huo, A.X.; Xue, Z.; Qu, S.S.; Wong, W.K. Microcalorimetric and Spectroscopic Investigation of the Antibacterial Properties of Cationic Ytterbium(III)-Porphyrin Complexes Lacking Charged Peripheral Groups. Chem. Biodivers. 2007, 4, 2889–2899. [Google Scholar]
- Huo, A.X.; Xue, Z.; Wong, W.K. Antibacterial Effects of a Monoporphyrinato Ytterbium(III) Complex and its Free Components on Staphylococcus Aureus as Determined by Stop-flow Microcalorimetry. Chem. Biodivers. 2007, 4, 1492–1500. [Google Scholar] [CrossRef]
- Friedrich, J.; Wolfrum, H.; Haarer, D. Photochemical Holes: A Spectral Probe of the Amorphous State in the Optical Domain. J. Chem. Phys. 1982, 77, 2309–2316. [Google Scholar] [CrossRef]
- Wei, T.H.; Hagan, D.J.; Sence, M.J.; Stryland, E.W.; Perry, J.W.; Coulter, D.R. Direct Measurements of Nonlinear Absorption and Refraction in Solutions of Phthalocyanines. Appl. Phys. B 1992, 54, 46–51. [Google Scholar] [CrossRef]
- Inoue, H.; Iwamoto, T.; Makishima, A.; Ikemoto, M.; Horie, K. Preperation and Properties of Sol-gel Thin Films with Porphins. J. Opt. Soc. Am.B 1992, 9, 816–818. [Google Scholar] [CrossRef]
- Liu, Y.; Shigehara, K.; Yamada, A. Preparation of Bis(phthalocyaninato)lutetium with Various Substituents and Their Electrochemical Properties. Bull. Chem. Soc. Jpn. 1992, 65, 250–257. [Google Scholar] [CrossRef]
- Krivokopic, M.; Anderson, H.L.; Bourhill, G.; Ives, R.; Clark, S.; McEwan, K.J. Meso-tetra-alkynyl Porrphyrins for Optical Limiting—A Survey of Group III and IV Metal Complexes. Adv. Mater. 2001, 13, 652–656. [Google Scholar] [CrossRef]
- Mori, T.; Santa, T.; Hirobe, M. Synthesis and Cytochrome P-450-like Reactivity of Polypeptide-bound Porphyrinatoiron (III). Tetrahedron Lett. 1985, 26, 5555–5558. [Google Scholar] [CrossRef]
- Gunter, M.J.; Turner, P. Metalloporphyrins as Models for Cytochrome P-450. Coord. Chem. Rev. 1991, 108, 115–161. [Google Scholar]
- Katsuki, T. Catalytic Asymmetric Oxidation Using Optically Active (Salen) Manganese(III) Complexes as Catalysts. Coord. Cheem. Rev. 1995, 140, 189–214. [Google Scholar]
- Collman, J.P.; Gagne, R.R.; Reed, C.A. Paramagnetic Dioxygen Complex of Iron(II) Derived from a Picket Fence porphyrin. Further Models for Hemoproteins. J. Am Chem. Soc. 1974, 96, 2629–2631. [Google Scholar] [CrossRef]
- Collman, J.P.; Gagne, R.R.; Reed, C.; Halbert, T.R.; Lang, G.; Robinson, W.T. Picket Fence Porphyrins. Synthetic Models for Oxygen Binding Hemoproteins. J. Am. Chem. Soc. 1975, 97, 1427–1439. [Google Scholar] [CrossRef]
- Collman, J.P.; Sorrell, T.N. Model for the Carbonyl Adduct of Ferrous Cytochrome P 450. J. Am. Chem. Soc. 1975, 97, 4133–4134. [Google Scholar] [CrossRef]
- Collman, J.P.; Fu, L.; Herrmann, P.C.; Zhang, X. A Functional Model Related to Cytochrome c Oxidase and its Electrocatalytic Four-electron Reduction of O2. Science 1997, 275, 949–951. [Google Scholar] [CrossRef]
- Zingg, A.; Felber, B.; Gramlich, V.; Fu, L.; Collman, J.P.; Diederich, F. Dendritic Iron(II) Porphyrins as Models for Hemoglobin and Myoglobin: Specific Stabilization of O2 Complexes in Dendrimers with H-Bond-Donor Centers. Helv. Chim. Acta 2002, 85, 333–351. [Google Scholar] [CrossRef]
- Collman, J.P.; Gosh, S.; Dey, A.; Decreau, R.A.; Yang, Y. Catalytic Reduction of O2 by Cytochrome c Using a Synthetic Model of Cytochrome c Oxidase. J. Am. Chem. Soc. 2009, 131, 5034–5035. [Google Scholar] [CrossRef]
- Linstead, R.P. Phthalocyanines. Part I. A new Type of Synthetic Colouring Matters. J. Chem. Soc. 1934, 1016–1017. [Google Scholar]
- Robertson, M.; Woodward, I. An X-ray Study of the Phthalocyanines. Part IV. Direct Quantitative Analysis of the Platinum Compound. J. Chem. Soc. 1940, 36–48. [Google Scholar] [CrossRef]
- Berezin, B.D. The Thermodynamic Characteristics of Phthalocyuanines in Aulphuric acid Solutions. Russ. J. Inorg. Chem. 1962, 7, 1300–1303. [Google Scholar]
- Berezin, B.D. Metal Phthalocyanines in Solution. VI. Effect of the Nature of the Central ion on the Stability of the Phthalocyanine Macroring in Sulphuric Acid Solutions. Russ. J. Physic Chem. 1962, 36, 258–261. [Google Scholar]
- Eley, D.D. Phthalocyanines as Semiconductors. Nature 1948, 162, 819–819. [Google Scholar] [CrossRef]
- Fielding, P.E.; MacKay, A.G. Electrical Conduction in the Phthalocyanines. I. Optical Properties. Aust. J. Chem. 1964, 17, 750–758. [Google Scholar] [CrossRef]
- Hoffman, C.J. Semiconductivity of the Phthalocyanines. Q. Rev. Chem. Soc. 1964, 18, 113–121. [Google Scholar] [CrossRef]
- Linsky, J.P.; Paul, T.R.; Nohr, R.S.; Kenney, M.E. Studies of a Series of Halo Aluminum, Gallium, and Indium Phthalocyanine. Inorg. Chem. 1980, 19, 3131–3135. [Google Scholar] [CrossRef]
- Ferraudi, G. Photochemical Properties of Metallophthalocyanines in Homogeneous Solution. In Phtalocyanines Properties and Applications; Leznoff, C.C., Lever, A.B.P., Eds.; VCH Publishers: Weinheim, Germany, 1989; pp. 295–338. [Google Scholar]
- Ashley, K.; Trent, I. Kinetic and Equilibrium Study of the Reaction of [Meso-tetrakis(psulfonatophenyl)porphyrinato]diaquacromate (III) with Pyridine in Aqueous Solution. Inorg. Chim. Acta 1989, 163, 159–166. [Google Scholar] [CrossRef]
- Ostler, R.B.; Scully, A.D.; Taylor, A.G.; Gould, I.R.; Smith, T.A.; Waite, A.; Phillips, D. The Effect of pH on the Photophysics and Photochemistry of Disulphonated Aluminum Phthalocyanine. Photochem. Photobiol. 2007, 71, 397–404. [Google Scholar] [CrossRef]
- Weber, J.H.; Buch, D.H. Complexes Derived from Strong Field Ligands. XIX. Magnetic Properties of Transition Metal Derivatives of 4,4',4",4'''-Tetrasulfophthalocyanine. Inorg. Chem. 1965, 4, 469–471. [Google Scholar] [CrossRef]
- Weber, J.H.; Buch, D.H. Complexes Derived from Strong Field Ligands. XX. The Effect of Extraplanar Ligands on the Properties of Transition Metal 4,4',4",4'"-Tetrasulfophthalocyanines. Inorg. Chem. 1965, 4, 472–475. [Google Scholar] [CrossRef]
- Zagal, J.; Sen, R.K.; Yeager, E. Oxygen Reduction by Co(III) Tetrasulfonatephthalocyanine Irreversibly Adsorbed on a Stress Anneled Pyrolytic Graphite Electrode Surface. J. Electroanal. Chem. 1977, 83, 207–213. [Google Scholar]
- Schneider, O.; Hanack, M. Phthalocyaninatoeisen mit Pyrazin als Zweizähnigen Brückenliganden. Angew. Chem.Int. Ed. Engl. 1980, 19, 392–393. [Google Scholar] [CrossRef]
- Hanack, M. Uberbruckte Makrocyclische Metallkomplexe als Organische Leiter. Org. Chem. 1987, 2, 75–78. [Google Scholar]
- Hanack, M.; Deger, U.; Keppeler, U.; Lang, M.; Leverenz, A.; Rein, M. Phthalocyanines. In Conducting Polymers; Alcacer, L., Ed.; D. Reidel. Publ. Comp.: Amsterdam, The Netherlands, 1987. [Google Scholar]
- Hanack, M.; Polley, R.; Deger, S. μ-cyano-2,3-naphthalocyaninatocobalt(III). Synth. Met. 1997, 89, 47–49. [Google Scholar] [CrossRef]
- Hanack, M.; Kamenzin, S.; Kamenzin, C.; Subramanian, L.R. Synthesis and Properties of Axially Disubstituted Monomeric and Oligomeric Phthalocyaninato Ruthenium (II) Compounds. Synth. Met. 2000, 110, 93–103. [Google Scholar] [CrossRef]
- Calvete, M.; Yang, G.Y.; Hanack, M. Porphyrins and Phthalocyanines as Materials for Optical Limiting. Synth. Met. 2004, 141, 231–243. [Google Scholar] [CrossRef]
- Schramm, C.J.; Scaringe, R.P; Stojakovic, D.R.; Hoffman, B.M.; Ibers, J.A.; Marks, T.J. Chemical, Spectral, Structural, and Charge Transport Properties of the Molecular Metals Produced by Iodination of Nickel Phthalocyanine. J. Am. Chem. Soc. 1980, 102, 6702–6713. [Google Scholar]
- Hoffman, B.M.; Ibers, J.A. Porphyrinic Molecular Metals. Acc. Chem. Res. 1983, 16, 15–21. [Google Scholar] [CrossRef]
- Barret, P.A.; Dent, C.E.; Linstead, R.P. Phthalocyanines. Part VII. Phthalocyanine as a Coordinating Group. A General Investigation of the Metallic Derivatives. J. Chem. Soc. 1936, 1719–1736. [Google Scholar]
- Kirin, I.S.; Moskalev, P.N.; Makashev, Y.A. Formation of Unusual Phthalocyanines of the Rare-earth Elements. Russ. J. Inorg. Chem. 1965, 10, 1065–1066. [Google Scholar]
- Kirin, I.S.; Moskalev, P.N.; Ivannikova, N.V. Preparation and Properties of Neodymium Phthalocyanine. Russ. J. Inorg. Chem. 1967, 12, 944–945. [Google Scholar]
- Kirin, I.S.; Moskalev, P.N. Preparation and Properties of Yttrium and Erbium Diphthalocyanines. Russ. J. Inorg. Chem. 1970, 15, 7–8. [Google Scholar]
- Kirin, I.S.; Kolyadin, A.B.; Moskalev, P.N. Preparation and Properties of Uranium Bis(phthalocyanine). Russ. J. Inorg. Chem. 1971, 16, 1455. [Google Scholar]
- Kirin, I.S.; Moskalev, P.N. The Electrochromism of Lanthanide-Di-Phthalocyanines. Russ. J. Phy. Chem. 1972, 46, 1019. [Google Scholar]
- Moskalev, P.N.; Kirin, I.S. A Spectrophotometric Study of the Sulphonated Diphthalocyanine Complexes of Yttrium, Gadolinium, and Luthetium in Aqueous Solution. Russ. J. Inorg. Chem. 1971, 16, 57–60. [Google Scholar]
- Moskalev, P.N.; Mishin, V.Y.; Rubtsov, E.M.; Kirin, I.S. Preparation and Thermal Stability of Bisphthalocyanine Complexes of Lanthanides: Hafnium, Thorium and Uranium. Russ. J. Inorg. Chem. 1976, 21, 1243–1245. [Google Scholar]
- Kasuga, K.; Tsutsuo, M. Some New Developments in the Chemistry of Metallophthalocyanines. Coord. Chem. Rev. 1980, 32, 67–95. [Google Scholar] [CrossRef]
- Kasuga, K.; Tsutsuo, M. Bis(tetrasulfophthalocyaninato)samarium (III). J. Coord. Chem. 1981, 11, 177–178. [Google Scholar] [CrossRef]
- De Cian, A.; Moussavi, M.; Fisher, J.; Weiss, R. Synthesis, Structure, and Spectroscopic and Magnetic Properties of Lutetium(III) Phthalocyanine Derivatives: LuPc2•CH2C2 and [LuPc(OAc)(H2O)2].H2O.2CH3OH. Inorg. Chem. 1985, 24, 3162–3167. [Google Scholar] [CrossRef]
- André, J.J.; Holczer, K.; Petit, P.; Riou, M.T.; Clarisse, C.; Even, R.; Fourmigue, M.; Simon, J. Electrical and Magnetic Properties of Thin Films and Single Crystals of Bis(phthalocyaninato)lutetium. Chem. Phys. Lett. 1985, 115, 463–466. [Google Scholar] [CrossRef]
- Nicholson, M.M.; Galiardi, R.V. U.S. NTIS, AD Rep. 1977, AD-A039596. Chem. Abstr. 1977, 87, 144073. [Google Scholar]
- Nicholson, M.M.; Pizzarello, F.A. Charge Transport in Oxidation Product of Lutetium Diphthalocyanine. J. Electrochem. Soc. 1979, 126, 1490–1495. [Google Scholar] [CrossRef]
- Nicholson, M.M.; Pizzarello, F.A. Galvanostatic Transients in Lutetium Diphthalocyanine Films. J. Electrochem. Soc. 1980, 127, 821–827. [Google Scholar] [CrossRef]
- Nicholson, M.M.; Pizzarello, F.A. Cathodic Electrochromic of Lutetium Diphthalocyanine Films. J. Electrochem. Soc. 1981, 128, 1740–1743. [Google Scholar]
- Nicholson, M.M. Electrochromic display using rare-earth diphthalocyanines and a low freezing-point electrolyte. U.S. Patent 4,371,236, 1 February 1983. [Google Scholar]
- Sammells, A. Multi-color Electrochromic Cells Having Solid Polymer Electrolytes and a Distinct Electrochromic Layer. U.S. Patent 4,807,977, 28 February 1989. [Google Scholar]
- Hara, T.; Toriyama, M.; Tsukagoshi, K. Immunoassay Using a Metal-complex Compound as a Chemiluminescent Catalyst. I. Iron(III) Phthalocyanine as a Labeling Reagent. Bull. Chem. Soc. Jpn. 1983, 56, 2267–2271. [Google Scholar] [CrossRef]
- Hara, T.; Toriyama, M.; Miyoshi, H.; Syogase, S. Immunoassay Using a Metal-complex Compound as a Chemiluminescent Catalyst. IV. The Investigation of a Metal porphine Complex as a Labeling Reagent. Bull. Chem. Soc. Jpn. 1984, 57, 3009–3010. [Google Scholar] [CrossRef]
- Hara, T.; Toriyama, M.; Imaki, M. Immunoassay Using a Metal-complex Compound as a Chemiluminescent Catalyst. V. Continuous Immunoassay by the Use of CLCCIA. Bull. Chem. Soc. Jpn. 1985, 58, 1299–1303. [Google Scholar] [CrossRef]
- Mew, D.; Wat, C.K.; Towers, G.H.; Levy, J.G. Photoimmunotherapy: Treatment of Animal Tumors with Tumor-specific Monoclonal Antibody-hematoporphyrin Conjugates. J. Immunol. 1983, 130, 1473–1477. [Google Scholar]
- Mew, D.; Lum, V.; Wat, C.K.; Towers, G.H.; Sun, C.H.; Walter, R.J.; Wright, W.; Berns, M.W.; Levy, J.G. Ability of Specific Monoclonal Antibodies and Conventional Antisera Conjugated to Hematoporphyrin to Label and Kill Selected Cell Lines Subsequent to Light Activation. Cancer Res. 1985, 45, 4380–4386. [Google Scholar]
- Ben-Hur, E.; Rosenthal, I. Photosensitized Inactivation of Chinese Hamster Cells by Phthalocyanines. Photochem. Photobiol. 1985, 42, 129–133. [Google Scholar] [CrossRef]
- Ben-Hur, E.; Rosenthal, I. Photosensitization of Chinese Hamster Cells by Water-soluble Phthalocyanines. Photochem. Photobiol. 1986, 43, 615–619. [Google Scholar] [CrossRef]
- Ben-Hur, E.; Rosenthal, I. Action Spectrum (600-700 nm) for Chloraluminum Phthalocyanine-induced Phototoxicity in Chinese Hamster Cells. Lasers Life Sci. 1986, 1, 79–86. [Google Scholar]
- Ben-Hur, E.; Green, M.; Prager, A.; Kol, R.; Rosenthal, I. Phthalocyanine Photosensitization of Mammalian Cells: Biochemical and Ultrastructural Effects. Photochem. Photobiol. 1987, 46, 651–656. [Google Scholar] [CrossRef]
- Spikes, J.D.; Bommer, J.C. Zinc Tetrasulphophthalocyanine as a Photodynamic Sensitizer for Biomolecules. Int. J. Radiat. Biol. 1986, 50, 41–45. [Google Scholar] [CrossRef]
- Spikes, J.D. Phthalocyanines as Photosensitizers in Biological Systems and for the Photodynamic Therapy of Tumors. Photochem. Photobiol. 1986, 43, 691–699. [Google Scholar] [CrossRef]
- Brasseur, N.; Ali, H.; Autenrieth, D.; Langlois, R.; van Lier, J.E. Biological Activities of Phthalocyanines III. Photoinactivation of V-79 Chinese Hamster Cells by Tetrasulfophthalocyanines. Photochem. Photobiol. 1985, 42, 515–521. [Google Scholar] [CrossRef]
- Brasseur, N.; Ali, H.; Langlois, R.; van Lier, J.E. Biological activities of phthalocyanines—VII. Photoinactivation of V-79 Chinese Hamster Cells by Selectively Sulfonated Gallium Phthalocyanines. Photochem. Photobiol. 1987, 46, 739–744. [Google Scholar] [CrossRef]
- Langlois, R.; Ali, H.; Brasseur, N.; Wagner, J.R.; van Lier, J.E. Biological Activities of Phthalocyanines-IV. Type II Sensitized Photooxidation of L-tryptophan and Cholesterol by Sulfonated Metallo Phthalocyanines. Photochem. Photobiol. 1986, 44, 117–123. [Google Scholar] [CrossRef]
- Chan, W.S.; Marshall, J.F.; Hart, I.R. Photodynamic Therapy of a Murine Tumor Following Sensitisation with Chloro Aluminum Sulfonated Phthalocyanine. Photochem. Photobiol. 1987, 46, 867–871. [Google Scholar] [CrossRef]
- Selman, S.H.; Kreimer-Birnbaum, M.; Chaudhuri, K.; Garbo, G.M.; Seaman, D.A.; Keck, R.W.; Ben-Hur, E.; Rosenthal, I. Photodynamic Treatment of Transplantable Bladder Tumors in Rodents After Pretreatment with Chloroaluminum Tetrasulfophthalocyanine. J. Urol. 1986, 136, 141–145. [Google Scholar]
- Tralau, C.J.; Barr, H.; Sandeman, D.R.; Barton, T.; Lewin, M.R.; Bown, S.G. Aluminum Sulfonated Phthalocyanine Distribution in Rodent Tumors of the Colon, Brain and Páncreas. Photochem. Photobiol. 1987, 46, 777–781. [Google Scholar] [CrossRef]
- Algermissen, B.; Jamil, B.; Osterloh, K.; Berlien, H.-P. Proceedings of SPIE Conference on Photochemotheraphy: Phothodynamic Theraphy and Other Modalities III; Berg, K., Ehrenberg, B., Mohan, J., Eds.; SPIE: Bellingham, WA, USA, 1997; 3191.
- Lavi, A.; Weitman, H.; Holmes, R.T.; Smith, K.M.; Ehrenberg, B. The Depth of Porphyrin in a Membrane and the Membrane’s Physical Properties Affect the Photosensitizing Efficiency. Biophys. J. 2002, 82, 2101–2110. [Google Scholar] [CrossRef]
- Weizman, E.; Rothman, C.H.; Greenbaum, L.; Shainberg, A.; Adamek, M.; Ehrenberg, B.; Malik, Z. Mitocondrial Localization and Photodamage During Photodynamic Therapy with Tetraphenylporphines. J. Photochem. Photobiol. B 2000, 59, 92–102. [Google Scholar] [CrossRef]
- Lane, N. New Light on Medicine. Sci. Am. 2003, 288, 26–33. [Google Scholar]
- Roginski, S.S.; Sakharov, M.M. Catalytic Properties of Organic Semiconductors. Russ. J. Phys. Chem. 1968, 42, 696. [Google Scholar]
- Kropf, H.; Gebert, W.; Franke, K. Zur autoxidation von 1.1—Diphenyläthan in Gegenwart von Kupfer-Phthalocyanin. Tetrahedron Lett. 1968, 53, 5527–5528. [Google Scholar]
- Cullis, C.F.; Trimm, D.L. Homogeneous Catalysis of the Oxidation of Thiols by Metal Ions. Discuss Faraday Soc. 1968, 46, 144–149. [Google Scholar] [CrossRef]
- Beck, F. Voltammetrische Untersuchungen an Elektrokatalytisch Wirksamen Phthalocyaninen und Tetraazaannulenen in Konzentrierter Schwefelsäure. Ber. Bunsenges. Phys. Chem. 1973, 77, 353–364. [Google Scholar]
- Beck, F.; Heiss, J.; Hiller, H.; Polster, R. Symposium Katalyse an Phthalocyaninen, Hamburg, 5 October 1972; Kropf, H., Steinbach, F., Eds.; Thieme Publisher: Stuttgart, Germany, 1973; p. 53. [Google Scholar]
- Steinbach, F.; Hiltner, K. Heterogene Katalyse an Komplex Gebundenen Metallatomen am Beispiel der Propanol(2)dampfoxydation an Phthalocyaninpulvern. Z. Phys. Chem. (N. F.) 1973, 83, 126–140. [Google Scholar] [CrossRef]
- Steinbach, F.; Schmidt, H. The Influence of Sulphur Compounds on β-Cu-Phthalocyanine Used as an Oxidation Catalyst. J. Catal. 1973, 29, 515–516. [Google Scholar] [CrossRef]
- Steinbach, F.; Zobel, M. Die katalytische Zersetzung von Hydrazindampf an Monomeren β-Cu-Phthalocyanin (Catalytic Decomposition of Hydrazine Vapor on Monomeric β-Copper Phthalocyanine). Z. Phys. Chem. (N. F.) 1973, 87, 142–150. [Google Scholar]
- Griffith, J.S. On the Magnetic Properties of Some Haemoglobin Complexes. Proc. Roy. Soc. Lond. A 1956, 235, 23–36. [Google Scholar] [CrossRef]
- Heilmeir, G.H.; Zanoni, L.A. Surface Studies of α-Copper Phthalocyanine Films. J. Phy. Chem. Solids 1964, 25, 603–611. [Google Scholar] [CrossRef]
- Alt, H.; Binder, T.; Lindner, W.; Sandstede, G. Metal chelates as electrocatalysts for oxygen reduction in acid electrolytes. J. Electroanal. Chem. 1971, 31, A19–A22. [Google Scholar]
- Contour, J.P.; Lenfant, P.; Vijh, A.K. Gas-Phase Chemisorption and Electroreduction of Oxygen on Phthalocyanines. J. Catal. 1973, 29, 8–14. [Google Scholar] [CrossRef]
- Maxted, E.B. The Poisoning of Metallic Catalysts. Adv. Catal. 1951, 3, 129–178. [Google Scholar] [CrossRef]
- Steinbach, F.; Schmidt, H.H. Metal Phthalocyanines Used as Catalysts in Gas Phase Reactions: IV. Oxidation of 2-Propanol Catalyzed by Monomeric β-Cu-Phthalocyanine in the Presence of Sulfur Compounds. J. Catal. 1975, 39, 190–197. [Google Scholar] [CrossRef]
- Manassen, J.; Bar-ilan, A. Metal Complexes of Phthalocyanine and aβγδ-Tetraphenylporphyrin as Heterogeneous Catalysts in Oxidative Dehydrogenation. Correlation Between Catalytic Activity and Redox Potential. J. Catal. 1970, 17, 86–92. [Google Scholar] [CrossRef]
- Manassen, J. A Comparison Between the Chemical and Electrochemical Catalysis by Tetraphenylporphyrin and Phthalocyanine Complexes. J. Catal. 1974, 33, 133–137. [Google Scholar] [CrossRef]
- Manassen, J. Metal Complexes of Porphyrinlike Compounds as Heterogeneous Catalysts. Cat. Rev. Sci. Eng. 1974, 9, 223–243. [Google Scholar] [CrossRef]
- Appleby, A.J.; Savy, M. Oxygen Reduction on Ultra Thin Carbon Supported Polymeric Phthalocyanine Electrodes in 6 N KOH. Electrochim. Acta 1975, 21, 567–574. [Google Scholar] [CrossRef]
- Cook, A.H. Catalytic properties of the phthalocyanines. Part I. Catalase properties. J. Chem. Soc. 1938, 1761–1768. [Google Scholar] [CrossRef]
- Hara, T.; Ohkatsu, Y.; Osa, T. The Oxidation of Cumene Catalyzed by Metal Polyphthalocyanines. Chem. Lett. 1973, 103, 103–106. [Google Scholar]
- Hara, T.; Ohkatsu, Y.; Osa, T. The Oxidation of Acrolein Catalyzed by a Metal Polyphthalocyanine. Chem. Lett. 1973, 103, 953–956. [Google Scholar]
- Hara, T.; Ohkatsu, Y.; Osa, T. Kinetics of the Oxidation of Acrolein Initiated by Di-t-Butyl Diperoxyoxalate. Bull. Chem. Soc. Jpn. 1974, 47, 156–159. [Google Scholar] [CrossRef]
- Hara, T.; Ohkatsu, Y.; Osa, T. Catalytic Activity of Metal Polyphthalocyanines in Autoxidation Reactions. Bull. Chem. Soc. Jpn. 1975, 48, 85. [Google Scholar] [CrossRef]
- Kothari, V.M.; Tazuma, J.J. Selective autoxidation of some phenols using salcomines and metal phthalocyanines. J. Catal. 1976, 41, 180–189. [Google Scholar] [CrossRef]
- Chatti, I.; Ghorbel, A.; Grange, P.; Colin, J.M. Oxidation of Mercaptans in Light Oil Sweetening by Cobalt(II) Phthalocyanine-Hydrotalcite Catalysts. Catal. Today 2002, 75, 113–117. [Google Scholar]
- Naito, S.; Tamaru, K. Oxidative Dehydrogenation of Alcohols over Metal Polyphthalocyanines. Z. Phys. Chem. (N. F.) 1975, 94, 150–152. [Google Scholar] [CrossRef]
- Darwent, J.R.; Douglas, P.; Harriman, A.; Porter, G.; Richoux, M.C. Metal Phthalocyanines and Porphyrins as Photosensitizers for Reduction of water to Hydrogen. Coord. Chem. Rev. 1982, 44, 83–126. [Google Scholar]
- Basu, B.; Satapathy, S.; Bhatnagar, A.K. Merox and Related Metal Phthalocyanine Catalyzed Oxidation Processes. Catal. Rev. Sci. Eng. 1993, 35, 571–609. [Google Scholar] [CrossRef]
- Balkus, K.J., Jr.; Gavrielov, A.G. Zeolite Encapsulated Metal Complexes. J. Incl. Phenom. Mol. Recog. Chem. 1995, 21, 159–184. [Google Scholar]
- Bedioui, F. Zeolite-Encapsulated and clay-intercalated Metal Porphyrin, Phthalocyanine and Schiff-base Complexes as Models for Biomimetic Oxidation Catalysts: An Overview. Coord. Chem. Rev. 1995, 144, 39–68. [Google Scholar] [CrossRef]
- Herron, N. Toward Si-Based Life: Zeolites as Enzyme Mimics. Chemtech 1989, 19, 542–548. [Google Scholar]
- Kannan, S.; Awate, S.V.; Agashe, M.S. Incorporation of Anionic Copper Phthalocyanine Complexes in to the Intergallery of Mg-Al Layered Double Hydroxides. Recent Adv. Basic Appl. Surf. Sci. Catal. 1998, 113, 927–935. [Google Scholar]
- Carrado, K.A.; Forman, J.E.; Botto, R.E.; Winans, R.E. Incorporation of Phthalocyanines by Cationic and Anionic Clays Via Ion Exchange and Direct Synthesis. Chem. Matter. 1993, 5, 472–478. [Google Scholar] [CrossRef]
- Drezdzon, M.A. Synthesis of Isopolymetalate-Pillared Hydrotalcite Via Organic-Anion-Pillared Precursors. Inorg. Chem. 1988, 27, 4628–4632. [Google Scholar] [CrossRef]
- Perez-Bernal, M.E.; Ruano-Casero, R.; Pinnavaia, T.J. Catalytic Autoxidation of 1-Decanethiol by Cobalt(II) Phthalocyaninetetrasulfonate Intercalated in a Layered Double Hydroxide. Catal. Lett. 1991, 11, 55–61. [Google Scholar] [CrossRef]
- Yeager, E. External link Electrocatalysts for O2 Reduction. Electrochim. Acta 1984, 29, 1527–1537. [Google Scholar] [CrossRef]
- Van der Putten, A.; Elzing, A.; Visscher, W.; Barendrecht, E. Redox Potential and Electrocatalysis of O2 Reduction on Transition Metal Chelates. J. Electroanal. Chem. 1987, 221, 95–104. [Google Scholar] [CrossRef]
- Van Veen, J.A.R.; Colijn, H.A.; van Baar, J.F. On the Effect of a Heat Treatment on the Structure of Carbon-supported Metalloporphyrins and Phthalocyanines. Electrochim. Acta 1988, 33, 801–804. [Google Scholar] [CrossRef]
- Handbook of Petroleum Refining Processes; Mayers, R.A. (Ed.) McGraw-Hill: New York, NY, USA, 1986.
- Leitão, A.; Costa, C.; Rodrigues, A. Studies on the Impregnation Step of the Merox Process. Chem. Eng. Sci. 1987, 42, 2291–2299. [Google Scholar] [CrossRef]
- Ferraudi, G.; Arguello, G.A.; Ali, H.; van Lier, J.E. Types I y II Sensitized Photooxidation of Aminoacid by Phthalocyanines: A flash Photochemical Study. Photochem. Photobiol. 1988, 47, 657–660. [Google Scholar] [CrossRef]
- Savitsky, A.P.; Lapatin, K.V.; Golubeva, N.A.; Poroshina, Y.M.; Chernyaeva, E.B.; Stepanova, N.V.; Solovieva, L.I.; Lukyanets, E.A. pH Dependence of Fluorescence and Absorbance Spectra of Free Sulphonated Aluminium Phthalocyanine and its Conjugate with Monoclonal Antibodies. J. Photochem. Photobiol. B Biol. 1992, 13, 327–333. [Google Scholar] [CrossRef]
- Lee, S.K.; Okura, I. Optical Sensor for Oxygen Using a Porphyrin-doped Sol-Gel Glass. Analyst 1997, 122, 81–84. [Google Scholar] [CrossRef]
- Rakow, N.A.; Suslick, K.S. A Colorimetric Sensor Array for Odour Visualization. Nature 2000, 406, 710–712. [Google Scholar] [CrossRef]
- Feng, L.; Musto, C.J.; Kemling, J.W.; Lim, S.H.; Zhong, W.; Suslick, K.S. Colorimetric Sensor Array for Determination and Identification of Toxic Industrial Chemicals. Anal. Chem. 2010, 82, 9433–9440. [Google Scholar] [CrossRef]
- Dunbar, A.D.F.; Brittle, S.; Richardson, T.H.; Hutchinson, J.; Hunter, C.A. Detection of Volatile Organic Compounds Using Porphyrin Derivatives. J. Phys. Chem. B 2010, 114, 11697–11702. [Google Scholar]
- Worsfold, O.; Dooling, C.M.; Richardson, T.H.; Vysotsky, M.O.; Tregonning, R.; Hunter, C.A.; Malins, C. Nitrogen Dioxide Sensing Characteristics at Elevated Temperature of Sol-gel Glass Thin Films Containing Substituted Porphyrin Dyes. J. Mater. Chem. 2001, 11, 399–403. [Google Scholar] [CrossRef]
- Balaji, T.; Sasidharan, M.; Matsunaga, H. Optical Sensor for the Visual Detection of Mercury Using Mesoporous Silica Anchoring Porphyrin Moiety. Analyst 2005, 130, 1162–1167. [Google Scholar] [CrossRef]
- Zhang, H.; Sun, Y.; Ye, K.; Zhang, P.; Wang, Y. Oxygen Sensing Materials Based on Mesoporous Silica MCM-41 and Pt(II)-porphyrin Complexes. J. Mater. Chem. 2005, 15, 3181–3186. [Google Scholar] [CrossRef]
- Livage, J.; Henry, M.; Sanchez, C. Sol-gel Chemistry of Transition Metal Oxides. Prog.Solid State Chem. 1988, 18, 259–341. [Google Scholar] [CrossRef]
- Livage, J. Chimie Douce: From Shake-and-bake Processing to Wet Chemistry. New J. Chem. 2001, 25, 1. [Google Scholar] [CrossRef]
- Sanchez, C.; Rozes, L.; Ribot, F.; Laberty-Robert, C.; Grosso, D.; Sassoye, C.; Boissiere, C.; Nicole, L. "Chimie douce”: A Land of Opportunities for the Designed Construction of Functional Inorganic and Hybrid Organic-inorganic Nanomaterials. C. R. Chim. 2010, 13, 3–39. [Google Scholar] [CrossRef]
- Brinker, C.J.; Scherer, G. Sol-Gel Science: The Physics and Chemistry of Sol-Gel Processing; Academic Press: San Diego, CA, USA, 1990. [Google Scholar]
- Klein, L.C. Sol-Gel Technology; Noyes Publications: Park Ridge, NJ, USA, 1988. [Google Scholar]
- Wang, S.H.; Campbell, C.; Hench, L.L. Ultrastructre Processing of Ceramics, Glasses and Composites; Hench, L.L., Ulrich, D.R., Eds.; Wiley: New York, NY, USA, 1988. [Google Scholar]
- Hench, L.L.; Wang, S.H.; Nogues, J.L. Sol-Gel Processing of Large Silica Optics, Multifunctional, Materials. In Proceedings of 4th International Conference on Ultrastructure Processing of Ceramics, Glasses and Composites, Tucson, AZ, USA, 20–24 February, 1989; Ggunshor, R.L., Ed.;
- Hench, L.L.; West, J.K. The Sol-gel Process. Chem. Rev. 1990, 90, 33–72. [Google Scholar] [CrossRef]
- Aelion, R.; Loebel, A.; Eirich, F. Hydrolysis of Ethyl Silicate. J. Am. Chem. Soc. 1950, 72, 5705–5712. [Google Scholar] [CrossRef]
- Fitremann, J.; Doeuff, S.; Sanchez, C. Propriétés Optiques de Gels à Base de TiO2, ZrO2, Al2O3 et SiO2 Dopés par de la Rhodamine 640. Ann. Chim. 1990, 15, 421–432. [Google Scholar]
- Sakka, S.; Kamiya, K. The Sol-gel Transition in the Hydrolysis of Metal Alkoxides in Relation to the Formation of Glass Fibers and Films. J. Non Cryst. Solids 1982, 48, 31–46. [Google Scholar] [CrossRef]
- Scheefer, D.W.; Martin, J.E.; Wiltzius, P.W. Kinetics of Aggregation and Gelation; Family, F., Landau, D.P., Eds.; North Holland: Amsterdam, The Netherlands, 1984. [Google Scholar]
- Brinker, C.J.; Keefer, K.D.; Schaefer, D.W.; Ashley, C.S. Sol-gel Transition in Simple Silicates. J. Non Cryst. Solids 1982, 48, 47–64. [Google Scholar] [CrossRef]
- Brinker, C.J.; Keefer, K.D.; Schaefer, D.W.; Assink, R.A.; Kay, B.D.; Ashley, C.S. Sol-gel Transition in Simple Silicates II. J. Non Cryst. Solids 1984, 63, 45–59. [Google Scholar] [CrossRef]
- Wang, S.H.; Campbell, C.; Hench, L.L. Ultrastructre Processing of Ceramics,Glasses and Composites; Hench, L.L., Ulrich, D.R., Eds.; Wiley: New York, NY, USA, 1988; p. 145. [Google Scholar]
- Orcel, G.; Hench, L.L.; Artaki, I.; Jonas, J.; Zerda, T.W. Effect of Formamide Additive on the Chemistry of Silica Sol-gels II. Gel Structure. J. Non Cryst. Solids 1988, 105, 223–231. [Google Scholar] [CrossRef]
- Hench, L.L.; Orcel, G. Physical-chemical and Biochemical Factors in Silica Sol-gels. J. Non Cryst. Solids 1986, 82, 1–10. [Google Scholar] [CrossRef]
- Yamane, M.; Inoue, S.; Yasumori, A. Sol-gel Transition in the Hydrolysis of Silicon Methoxide. J. Non Cryst. Solids 1984, 63, 13–21. [Google Scholar] [CrossRef]
- Sakka, S.; Kamiya, K. Glasses from Metal Alcoholates. J. Non Cryst. Solids 1980, 42, 403–421. [Google Scholar] [CrossRef]
- Debsikdar, J.C. Effect of the Nature of the Sol-gel Transition on the Oxide Content and Microstructure of Silica Gel. Adv. Ceram Mater. 1986, 1, 93–98. [Google Scholar]
- Avnir, D.; Levy, D.; Reisfield, R. The Nature of the Silica Cage as Reflected by Spectral Changes and Enhanced Photostability of Trapped Rhodamine 6G. J. Phys. Chem. 1984, 88, 5956–5959. [Google Scholar] [CrossRef]
- Levy, D.; Reisfeld, R.; Avnir, D. Fluorescence of Europium(III) Trapped in Silica Gel-glass as a Probe for Cation Binding and for Changes in Cage Symmetry During Gel Dehydration. Chem. Phys. Lett. 1984, 109, 593–597. [Google Scholar]
- Kaufman, V.R.; Avnir, D.; Pines-Rojanski, D.; Huppert, D. Water Consumption During the Early Stages of the Sol-gel Tetramethylorthosilicate Polymerization as Probed by Excited State Proton Transfer. J. Non Cryst. Solids 1988, 99, 379–386. [Google Scholar] [CrossRef]
- Levy, D.; Einhorn, S.; Avnir, D. Applications of the Sol-gel Process for the Preparation of Photochromic Information-recording Materials: Synthesis, Properties, Mechanisms. J. Non Cryst. Solids 1989, 113, 137–145. [Google Scholar] [CrossRef]
- Campostini, R.; Carturan, G.; Ferrari, M.; Montagna, M.; Pilla, O. Luminescence of Eu3+ Ions during Thermal Densification of SiO2 Gel. J. Mater. Res. 1992, 7, 745–753. [Google Scholar] [CrossRef]
- Pouxviel, J.C.; Dunn, B.; Zink, J.I. Fluorescence Study of Aluminosilicate Sols and Gels Doped with Hydroxy Trisulfonated Pyrene. J. Phys. Chem. 1989, 93, 2134–2139. [Google Scholar] [CrossRef]
- Canva, M.; Georges, P.; Brun, A.; Larrue, D.; Zarzycki, J. Impregnated SiO2 Gels Used as Dye Laser Matrix Hosts. J. Non Cryst. Solids 1992, 147-148, 636–640. [Google Scholar] [CrossRef]
- Miller, J.M.; Dunn, B.; Valentine, J.S.; Zink, J.I. Synthesis Conditions for Encapsulating Cytochrome c and Catalase in SiO2 Sol-gel Materials. J. Non Cryst. Solids 1996, 202, 279–289. [Google Scholar] [CrossRef]
- Dave, B.C.; Dunn, B.; Valentine, J.S.; Zink, J.I. Nanoconfined Proteins and Enzymes: Sol-Gel Based Biomolecular Materials. Adv. Chem. Ser. 1996, 622, 351–365. [Google Scholar]
- Menaa, B.; Torres, C.; Herrero, M.; Rives, V.; Gilbert, A.R.W.; Eggers, D.K. Protein Adsorption to Organically-modified Silica Glass Leads to a Different Structure than Sol-gel Encapsulation. Biophys. J. 2008, 95, L51–L53. [Google Scholar] [CrossRef]
- Menaa, B.; Miyagawa, Y.; Takahashi, M.; Herrero, M.; Rives, V.; Menaa, F.; Eggers, D.K. Bioencapsulation of Apomyoglobin in Nanoporous Organosilica Sol-gel glasses: Influence of the Siloxane Network on the Conformation and Stability of a Model Protein. Biopolymers 2009, 91, 895–906. [Google Scholar] [CrossRef]
- Menaa, B.; Menaa, F.; Aiolfi-Guimaraes, C.; Sharts, O. Silica-based Nanoporous Sol-gel Glasses: From Bioencapsulation to Protein Folding Studies. Int. J. Nanotechnol. 2010, 7, 1–45. [Google Scholar] [CrossRef]
- Campostrini, R.; Carturan, G.; Caniato, R.; Piovan, A.; Filippini, R.; Innocenti, G.; Cappelletti, E.M. Immobilization of Plant Cells in Hybrid Sol-gel Materials. J. Sol Gel Sci. Technol. 1996, 7, 87–97. [Google Scholar] [CrossRef]
- Nassif, N.; Bouvet, O.; Rager, M.N.; Roux, C.; Coradin, T.; Livage, J. Living Bacteria in Silica Gels. Nat. Mater. 2002, 1, 42–44. [Google Scholar] [CrossRef]
- Nassif, N.; Roux, C.; Coradin, T.; Rager, M.N.; Bouvet, O.M.M.; Livage, J. A Sol-gel Matrix to Preserve the Viability of Encapsulated Bacteria. J. Mater. Chem. 2003, 13, 203–208. [Google Scholar] [CrossRef]
- Avnir, D.; Coradin, T.; Lev, O.; Livage, J. Recent Bio-applications of Sol-gel Materials. J. Mater. Chem. 2006, 16, 1013–1030. [Google Scholar] [CrossRef]
- Dickey, F.H. The Preparation of Specific Adsorbens. Proc. Natl. Acad. Sci.USA 1949, 35, 227–229. [Google Scholar] [CrossRef]
- Rama, N.K.; Anderson, M.T.; Brinker, C.J. Template-Based Approaches to the Preparation of Amorphous, Nanoporous Silicas. Chem. Mater. 1996, 8, 1682–1701. [Google Scholar] [CrossRef]
- Fuqua, P.D.; Manssur, K.; Alvares, D.; Marder, S.R.; Perry, J.W.; Dunn, B. Synthesis and Nonlinear Optical Properties of Sol-gel Material Containing Phthalocyanines. In Proceedings of the SPIE Conference Sol-Gel Optics-II, Bellingham, WA, USA, 9 December 1992; Mackenzie, J.D., Ed.; 1758.
- Fuqua, P.D.; Dunn, B.; Zink, J.I. Dimer Formation in phthalocyanine-doped sol-gel materials. In Proceedings of the SPIE Conference Sol-Gel Optics III, San Diego, CA, USA, 13 October 1994; 2210.
- Fuqua, P.D.; Dunn, B. Optical Properties and Dimer Formation in Copper Phthalocyanine-doped Sol-gel Materials. J. Sol Gel Sci. Technol. 1998, 11, 241–250. [Google Scholar] [CrossRef]
- Fuqua, P.D. Second and Third Order Nonlinear Optical Properties of Organic Dye Dipped Sol-gel Materials. Ph.D. Thesis, UCLA, Los Angeles, CA, USA, 1993. [Google Scholar]
- Litrán, R.; Blanco, E.; Ramírez-del-Solar, M.; Esquivias, L. Trapping Copper Phthalocyanine in a Silica Sono-Xerogel. J. Sol Gel Sci. Technol. 1997, 8, 985–990. [Google Scholar]
- Kamitani, K.; Uo, M.; Inoue, H.; Makishima, A. Synthesis and Spectroscopy of TPP’s-doped Silica Gels by the Sol-gel process. J. Sol Gel Sci. Technol. 1993, 1, 85–92. [Google Scholar] [CrossRef]
- Wang, X.J.; Yates, L.M., III; Knobbe, E.T. Study of Nonlinear Absorption in Metalloporphyrin-Doped Sol-gel Materials. J. Lumin. 1994, 60-61, 469–473. [Google Scholar] [CrossRef]
- Ciuffi, K.; Sacco, H.C.; Valim, J.B.; Manso, C.M.C.P.; Serra, O.A.; Nascimento, O.R.; Vidoto, E.A.; Iamamoto, Y. Polymeric Organic-inorganic Hybrid Material Containing Iron(III) Porphyrin Using Sol-gel Process. J. Non Crystal. Solids 1999, 247, 146–152. [Google Scholar] [CrossRef]
- Ribeiro, A.O.; Biazzotto, J.C.; Serra, O.A. A Phthalocyanine Covalently Bonded to a Silica Network by a Sol-gel Process. J. Non Cryst. Solids 2000, 273, 198–202. [Google Scholar] [CrossRef]
- Sacco, H.C.; Ciuffi, K.J.; Biazzotto, J.C.; Mello, C.; de Oliveira, D.C.; Vidoto, E.A.; Nascimento, O.R.; Serra, O.A.; Iamamoto, Y. Ironporphyrins Trapped Sol-gel Glasses: A Chemometric Approach. J. Non Cryst. Solids 2001, 284, 174–182. [Google Scholar] [CrossRef]
- Biazzotto, J.C.; Vidoto, E.A.; Nascimento, O.R.; Iamamoto, Y.; Serra, O.A. Iron(III)-tetra-o-ureaphenylporphyrinosilica Obtained by a Sol-gel Process: A Study of EPR, Surface Area and Catalytic Activity. J. Non Cryst. Solids 2002, 304, 101–108. [Google Scholar] [CrossRef]
- Neri, C.R.; Calefi, P.S.; Iamamoto, Y.; Serra, O.A. Luminiscent Hybrid Porphyrinosilica Obtained by Sol-gel Chemistry. Mater. Res. São Carlos 2003, 6, 71–74. [Google Scholar]
- Beck, J.S.; Vartuli, J.C.; Roth, W.J.; Leonowicz, M.E.; Kresge, C.T.; Schmitt, K.D.; Chu, C.T.W.; Olson, D.H.; Sheppard, E.W.; McCullen, S.B.; et al. A New Family of Mesoporous Molecular Sieves Prepared with Liquid Crystal Templates. J. Am. Chem. Soc. 1992, 114, 10834–10843. [Google Scholar]
- Kresge, C.T.; Leonowicz, M.E.; Roth, W.J.; Vartuli, J.C.; Beck, J.S. Ordered Mesoporous Molecular Sieves Synthesized by a Liquid Crystal Template Mechanism. Nature 1992, 359, 710–712. [Google Scholar]
- Huo, Q.; Margolese, D.I.; Ciesla, U.; Demuth, D.G.; Feng, P.; Gier, T.E.; Sieger, P.; Firouzi, A.; Chmelka, B.F.; Schüth, F.; et al. Organization of Organic Molecules with Inorganic Molecular Species into Nanocomposite Biphase Arrays. Chem. Mater. 1994, 6, 1176–1191. [Google Scholar] [CrossRef]
- Zhao, D.; Feng, J.; Huo, Q.; Melosh, N.; Fredrickson, G.H.; Chmelka, B.F.; Stucky, G.D. Triblock Copolymer Syntheses of MesoporousSilica with Periodic 50 to 300 Angstrom Pores. Science 1998, 279, 548–552. [Google Scholar] [CrossRef]
- Zhao, D.; Huo, Q.; Feng, J.; Chmelka, B.F.; Stucky, G.D. Nonionic Triblock and Star Diblock Copolymer and Oligomeric Surfactant Syntheses of Highly Ordered, Hydrothermally Stable, Mesoporous Silica Structures. J. Am. Chem. Soc. 1998, 120, 6024–6036. [Google Scholar] [CrossRef]
- Chao, M.C.; Wang, D.S.; Lin, H.P.; Mou, C.Y. Control of Single Crystal Morphology of SBA-1 Mesoporous Silica. J. Mater. Chem. 2003, 13, 2853–2854. [Google Scholar] [CrossRef]
- Naik, B.; Ghosh, N.N. A Review on Chemical Methodologies for Preparation of Mesoporous Silica and Alumina Based Materials. Recent Pat. Nanotechnol. 2009, 3, 213–224. [Google Scholar] [CrossRef]
- Inagaki, S.; Fukushima, Y.; Kuroda, K. Synthesis of Highly Ordered Mesoporous Materials from a Layered Polysilicate. J. Chem. Soc. Chem. Commun. 1993, 8, 680–682. [Google Scholar]
- Inagaki, S.; Guan, S.; Ohsuna, T.; Terasaki, O. An Ordered Mesoporous Organosilica Hybrid Material with a Crystal-like Wall Structure. Nature 2002, 416, 204–307. [Google Scholar]
- Hatton, B.; Landskron, K.; Whitnall, W.; Perovic, D.; Ozin, G. Past, Present, and Future of Periodic Mesoporous Organosilicas—The PMOs. Acc. Chem. Res. 2005, 38, 305–312. [Google Scholar] [CrossRef]
- Inagaki, S.; Mizoshita, N.; Taniab, T. Synthesis, Properties, and Applications of Periodic Mesoporous Organosilicas Prepared from Bridged Organosilane Precursors. Chem. Soc. Rev. 2011, 40, 789–800. [Google Scholar] [CrossRef]
- Lio, C.J.; Li, S.G.; Pang, W.Q.; Che, C.M. Ruthenium Porphyrin Encapsulated in Modified Mesoporous Molecular Sieve MCM-41 for Alkane Oxidation. Chem. Commun. 1997, 1, 65–66. [Google Scholar]
- Sung-suh, H.M.; Luan, Z.; Kevan, L. Photoionization of Porphyrins in Mesoporous Siliceous MCM-41, AlMCM-41, and TiMCM-41 Molecular Sieves. J. Phys. Chem. B 1997, 101, 10455–10463. [Google Scholar] [CrossRef]
- Xu, W.; Guo, H.; Akins, D.L. Aggregation of Tetrakis(p-sulfonatophenyl)porphyrin within Modified Mesoporous MCM-41. J. Phys. Chem. B 2001, 105, 1543–1546. [Google Scholar]
- Tanamura, Y.; Uchida, T.; Teramae, N.; Kikuchi, M.; Kusaba, K.; Onodera, Y. Ship-in-a-bottle Synthesis of Copper Phthalocyanine Molecules within Mesoporous Channels of MCM-41 by a Chemical Vapor Deposition Method. Nano Lett. 2001, 1, 387–389. [Google Scholar] [CrossRef]
- Lu, X.B.; Wang, H.; He, R. Aluminum Phthalocyanine Complex Covalently Bonded to MCM-41 Silica as Heterogeneous Catalyst for the Synthesis of Cyclic Carbonates. J. Mol. Catal. A Chem. 2002, 186, 33–42. [Google Scholar] [CrossRef]
- Debecker, D.P.; Mutin, P.H. Non-hydrolytic sol-gel routes to heterogeneous catalysts. Chem. Soc. Rev. 2012, 41, 3624–3650. [Google Scholar] [CrossRef]
- Sui, R.; Charpentier, P. Synthesis of Metal Oxide Nanostructures by Direct Sol-Gel Chemistry in Supercritical Fluids. Chem. Rev. 2012, 112, 3057–3082. [Google Scholar]
- Monton, M.R.N.; Forsberg, E.M.; Brennan, J.D. Tailoring Sol-Gel-Derived Silica Materials for Optical Biosensing. Mater. Chem. 2012, 24, 796–811. [Google Scholar] [CrossRef]
- Li, Z.; Barnes, J.C.; Bosoy, A.; Stoddart, J.F.; Zink, J.I. Mesoporous silica nanoparticles in biomedical applications. Chem. Soc. Rev. 2012, 41, 2590–2605. [Google Scholar] [CrossRef]
- Chaudhuri, R.G.; Paria, S. Core/Shell Nanoparticles: Classes, Properties, Synthesis Mechanisms, Characterization, and Applications. Chem. Rev. 2012, 112, 2373–2433. [Google Scholar] [CrossRef]
- Goutterman, M. Study of the Effects of Substitution on the Absorption Spectra of Porphin. J. Chem. Phys. 1959, 30, 1139–1161. [Google Scholar] [CrossRef]
- Goutterman, M. The Porphyrins. In Physical Chemistry, Part A; Dolphin, D., Ed.; Academic Press: New York, NY, USA, 1979. [Google Scholar]
- García-Sánchez, M.A.; Campero, A. Aggregation Properties of Metallic Tetrasulphophtalocyanines Encapsulated in Sol-Gel Materials. J. Sol Gel Sci. Technol. 1998, 37, 651–655. [Google Scholar]
- García-Sánchez, M.A.; Campero, A. Aggregation properties of metallic tetrasulfophthalocyanines embedded in sol-gel silica. Polyhedron 2000, 19, 2383–2386. [Google Scholar] [CrossRef]
- García Sánchez, M.A.; Campero, A.; Avilés, M.L. Decomposition of Metal Tetrasulphophthalocyanines Incorporated in SiO2 gels. J. Non Cryst. Solids 2005, 351, 962–969. [Google Scholar] [CrossRef]
- Pedersen, C.J. Reversible Oxidation of Phthalocyanines. J. Org. Chem. 1957, 22, 127–132. [Google Scholar] [CrossRef]
- Mathur, S.C. Simple LCAO MO Calculations for Hydrogen Phthalocyanine. J. Chem. Phys. 1966, 45, 3470–3471. [Google Scholar] [CrossRef]
- Borisenkova, S.A.; Denisova, E.P.; Batanova, E.A.; Girenko, E.G.; Kaliya, O.L.; Lukyanets, E.A.; Vorozhtsov, G.N. Kinetics and Mechanism of Destruction of Cobalt Phthalocyanine Derivatives in Aqueous Solutions. J. Porphyr. Phthalocyanines 2000, 4, 684–688. [Google Scholar] [CrossRef]
- Bonnett, R.; Martínez, G. Photobleaching studies on Azabenzoporphyrins and Related Systems: A Comparison of the Photobleaching of the Zinc (II) Complexes of the Tetrabenzoporphyrin, 5-Azadibenzo[b,g]Porphyrin and Phthalocyanine Systems. J. Porphyr. Phthalocyanines 2000, 4, 544–550. [Google Scholar] [CrossRef]
- Ishii, K.; Siine, M.; Kikukawa, Y.; Kobayashi, N.; Shiragami, T.; Matsumoto, J.; Yasuda, M.; Suzuki, H.; Yokoi, H. Silica Gel-Supported Photofunctional Silicon Phthalocyanine Complexs: Photodesorption of Molecular Oxygen by Singlet Oxygen Generation. Chem. Phys. Lett. 2007, 448, 264–267. [Google Scholar] [CrossRef]
- García-Sánchez, M.A. Síntesis y Caracterización de Complejos Macrocíclicos Mixtos Lantanoides. M.Sc. Thesis, Universidad Autónoma Metropolitana, México D.F., México, 1993. [Google Scholar]
- Stern, A.; Wenderlein, H. Über die Lichlt-Absorption der Porphyrine IV. Z. Physik. Chem. 1936, A175, 405–437. [Google Scholar]
- Dorough, G.D.; Miller, J.R.; Huennekens, F.M. Spectra of the Metallo-Derivatives of α,β,γ,δ-Tetraphenylporphine. J. Am. Chem. Soc. 1951, 73, 4315–4320. [Google Scholar] [CrossRef]
- García-Sánchez, M.A.; Campero, A. Insertion of Lanthanide Porphyrins in Silica Gel. J. Non Cryst. Solids 2001, 296, 50–56. [Google Scholar] [CrossRef]
- Rothemund, P. Porphyrin Studies. III. The Structure of the Porphine Ring System. J. Am. Chem. Soc. 1939, 61, 2912–2915. [Google Scholar] [CrossRef]
- García-Sánchez, M.A.; Tello-Solís, S.R.; Sosa-Fonseca, R.; Campero, A. Fluorescent Porphyrins Trapped in Monolithic SiO2 gels. J. Sol Gel Sci. Technol. 2006, 37, 93–97. [Google Scholar] [CrossRef]
- García-Sánchez, M.A.; de la Luz, V.; Estrada-Rico, M.L.; Murillo-Martínez, M.M.; Coahuila-Hernández, M.I.; Sosa-Fonseca, R.; Tello-Solís, S.R.; Rojas, F.; Campero, A. Fluorescent Porphyrins Covalently Bound to Silica Xerogel Matrices. J. Non Cryst. Solids 2009, 355, 120–125. [Google Scholar]
- Sing, K.S.W.; Everett, D.H.; Haul, R.A.W.; Moscou, L.; Pierotti, R.A.; Rouquerol, J.; Siemieniewska, T. Reporting Physisorption Data for Gas/Solid Systems With Special Reference to the Determination of Surface Area and Porosity. Pure Appl. Chem. 1985, 57, 603–619. [Google Scholar] [CrossRef]
- Gregg, S.J.; Sing, K.S.W. Adsorption, Surface Area and Porosity; Academic Press: London, UK, 1967. [Google Scholar]
- García-Sánchez, M.A.; de la Luz, V.; Coahuila-Hernández, M.I.; Rojas-González, F.; Tello-Solís, S.R.; Campero, A. Effects of the Structure of Entrapped Substituted Porphyrins on the Textural Characteristics of Silica Network. J. Photochem. Photobiol. A Chem. 2011, 223, 172–181. [Google Scholar] [CrossRef]
- De la Luz, V.; García-Sánchez, M.A.; Campero, A. Luminescent Porphyrinosilica Obtained by the Sol-Gel Method. J. Non Cryst. Solids 2007, 353, 2143–2149. [Google Scholar] [CrossRef]
- De la Luz, V. Porfirinas como Agentes de Ahormado en Materiales Hibridos Porosos. Ph.D. Tesis, Universidad Autónoma Metropolitana, México D.F., México, 2009. [Google Scholar]
- Peng, H.; Lu, Y. Squarely Mesoporous and Functional Nanocomposites by Self-Directed Assembly of Organosilane. Adv. Mater. 2008, 20, 797–800. [Google Scholar] [CrossRef]
- González-Santiago, B.; García-Sánchez, M.A. Macrocycle-Pore Network Interactions: Aluminum Tetrasulfophthalocyanine in Organically Modified Silica. J. Non Cryst. Solids 2011, 357, 3168–3175. [Google Scholar] [CrossRef]
- García-Sánchez, M.A. Introducción de Marcocioclos Orgánicos en Matrices inorgánicas por el Método Sol-Gel. Ph.D. Thesis, Universidad Autónoma Metropolitana, México D.F., México, 2008. [Google Scholar]
- Quiroz-Segoviano, R.I.Y.; Garcia-Sánchez, M.A.; Rojas-González, F. Cobalt Porphyrin Covalently Bonded to Organo Modified Silica Xerogels. J. Non Crystal. Solids 2012, 358, 2868–2876. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
García-Sánchez, M.A.; Rojas-González, F.; Menchaca-Campos, E.C.; Tello-Solís, S.R.; Quiroz-Segoviano, R.I.Y.; Diaz-Alejo, L.A.; Salas-Bañales, E.; Campero, A. Crossed and Linked Histories of Tetrapyrrolic Macrocycles and Their Use for Engineering Pores within Sol-Gel Matrices. Molecules 2013, 18, 588-653. https://doi.org/10.3390/molecules18010588
García-Sánchez MA, Rojas-González F, Menchaca-Campos EC, Tello-Solís SR, Quiroz-Segoviano RIY, Diaz-Alejo LA, Salas-Bañales E, Campero A. Crossed and Linked Histories of Tetrapyrrolic Macrocycles and Their Use for Engineering Pores within Sol-Gel Matrices. Molecules. 2013; 18(1):588-653. https://doi.org/10.3390/molecules18010588
Chicago/Turabian StyleGarcía-Sánchez, Miguel A., Fernando Rojas-González, E. Carmina Menchaca-Campos, Salvador R. Tello-Solís, R. Iris Y. Quiroz-Segoviano, Luis A. Diaz-Alejo, Eduardo Salas-Bañales, and Antonio Campero. 2013. "Crossed and Linked Histories of Tetrapyrrolic Macrocycles and Their Use for Engineering Pores within Sol-Gel Matrices" Molecules 18, no. 1: 588-653. https://doi.org/10.3390/molecules18010588