Preparative Separation of Spirobisnaphthalenes from Endophytic Fungus Berkleasmium sp. Dzf12 by High-Speed Counter-Current Chromatography


Abstract
:1. Introduction
2. Results and Discussion
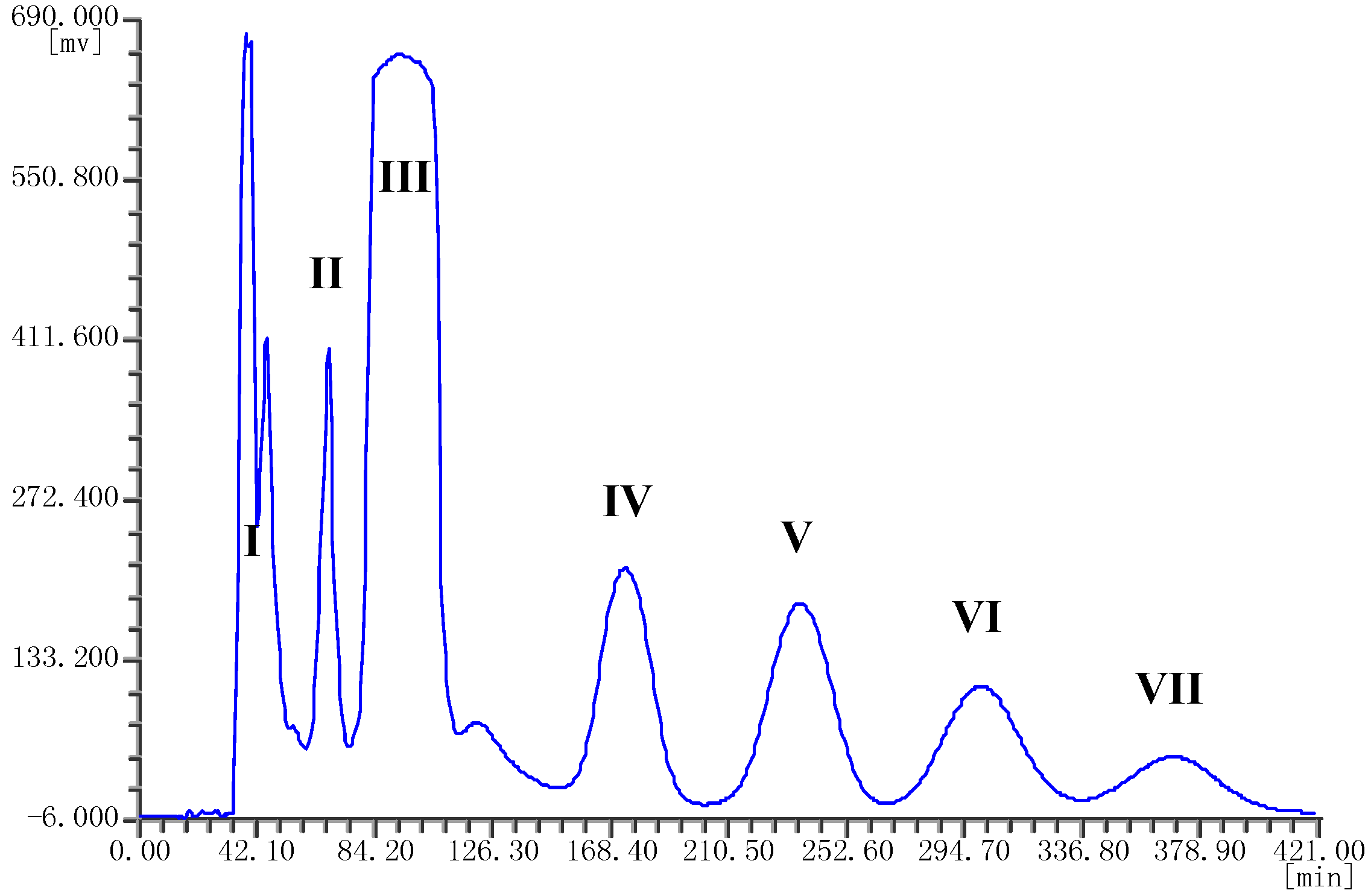
2.1. HPLC Analysis of the Crude Extract

2.2. Selection of Two-Phase Solvent System for HSCCC

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Ratio (v/v) | K value | ||||
|---|---|---|---|---|---|---|
| Peak a | Peak b | Peak c+d | Peak e | Peak f | ||
| 1 | 1.0:3.0:2.0:2.0 | 1.72 | 1.75 | 1.01 | 0.42 | 0.13 |
| 2 | 1.5:3.0:2.0:2.0 | 2.11 | 2.32 | 1.74 | 0.77 | 0.46 |
| 3 | 1.5:3.0:2.5:2.0 | 1.95 | 2.16 | 1.53 | 0.89 | 0.66 |
| 4 | 1.5:3.0:1.5:2.0 | 2.01 | 2.19 | 1.61 | 0.64 | 0.45 |
| 5 | 1.0:3.0:3.0:2.0 | 1.89 | 2.07 | 1.47 | 0.46 | 0.32 |
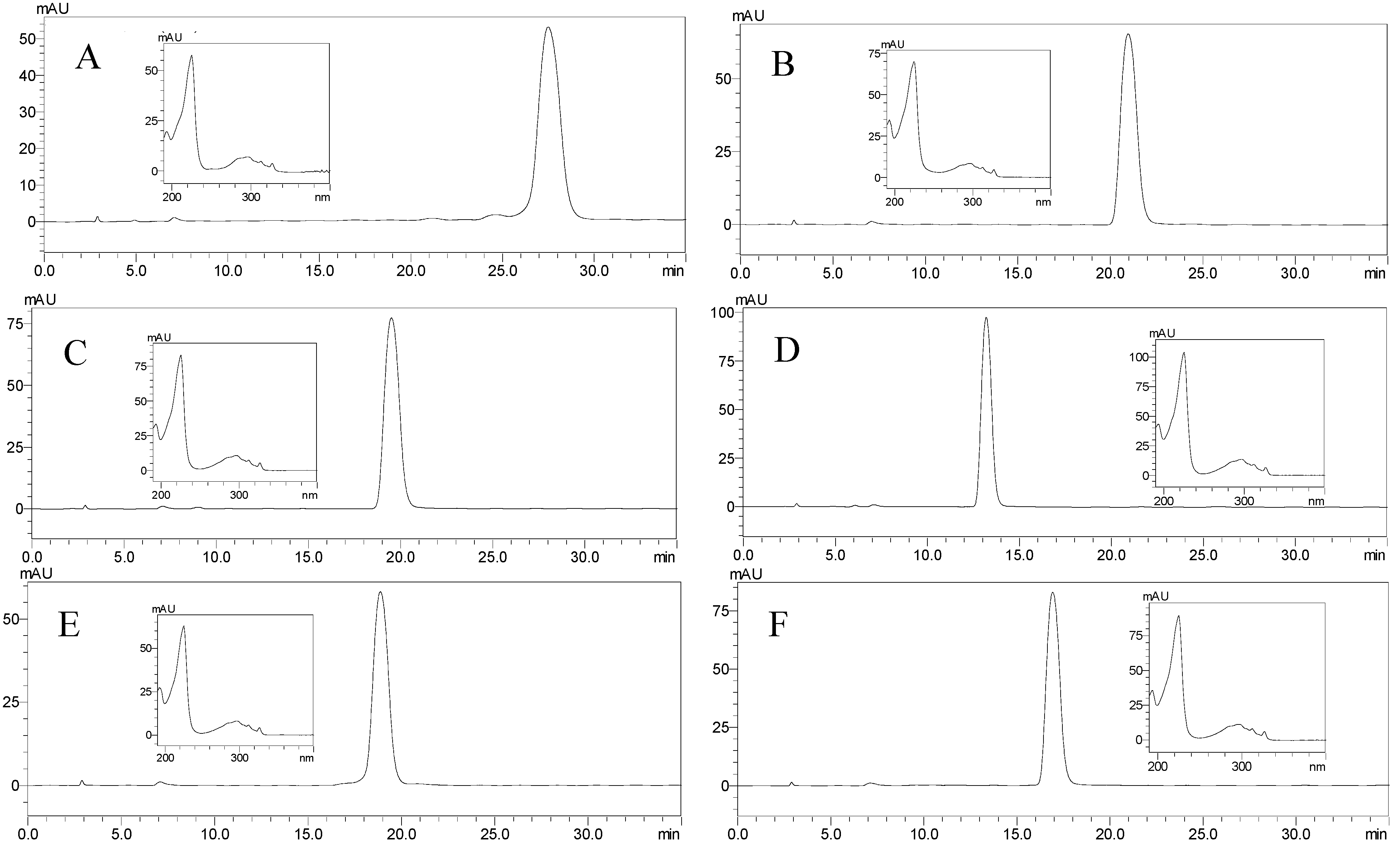
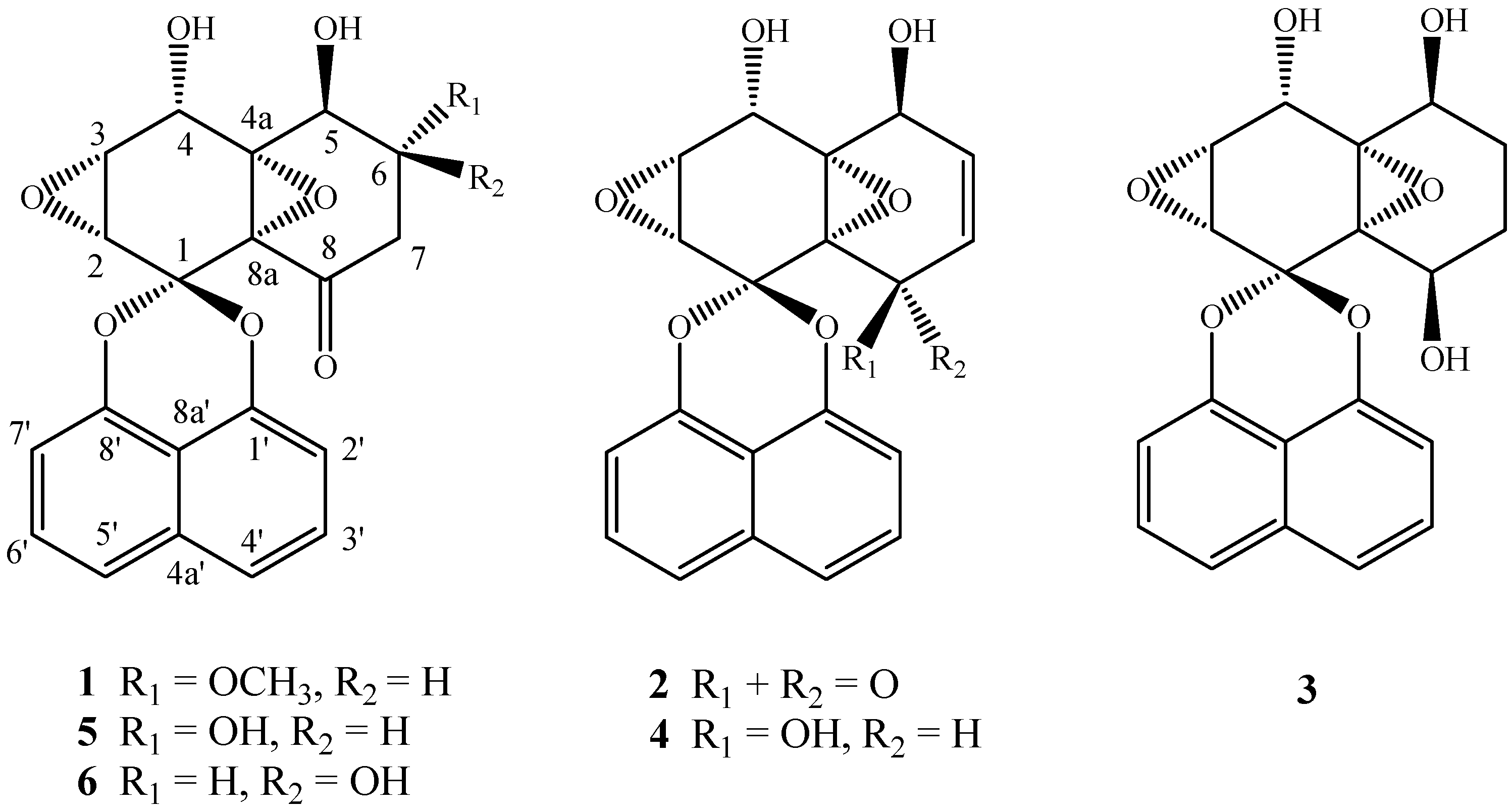
2.3. Separation of Spirobisnaphthalenes by HSCCC and Structural Identification



3. Experimental
3.1. General Analytical Methods
3.2. Chemicals and Reagents
3.3. Preparaton of Crude Sample
3.4. Solvent Systems for HSCCC
3.5. HSCCC Separation Procedure

3.6. Analysis and Identificaton of HSCCC Peak Fractions
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Cai, Y.-S.; Guo, Y.-W.; Krohn, K. Structure, bioactivities, biosynthetic relationships and chemical synthesis of the spirodioxynaphthalenes. Nat. Prod. Rep. 2010, 27, 1840–1870. [Google Scholar] [CrossRef]
- Zhou, L.; Zhao, J.; Shan, T.; Cai, X.; Peng, Y. Spirobisnaphthalenes from fungi and their biological activities. Mini Rev. Med. Chem. 2010, 10, 977–989. [Google Scholar] [CrossRef]
- Krohn, K.; Michel, A.; Florke, U.; Aust, H.-J.; Draeger, S.; Schulz, B. Palmarumycins CP1–CP4 from Coniothyrium palmarum: Isolation, structure elucidation, and biological activity. Liebigs Ann. Chem. 1994, 1994, 1093–1097. [Google Scholar] [CrossRef]
- Krohn, K.; Michel, A.; Florke, U.; Aust, H.-J.; Draeger, S.; Schulz, B. Palmarumycins C1–C16 from Coniothyrium sp.: Isolation, structure elucidation, and biological activity. Liebigs Ann. Chem. 1994, 1994, 1099–1108. [Google Scholar] [CrossRef]
- Hu, H.; Guo, H.; Li, E.; Liu, X.; Zhou, Y.; Che, Y. Decaspirones F-I, bioactive secondary metabolites from the saprophytic fungus Helicoma viridis. J. Nat. Prod. 2006, 69, 1672–1675. [Google Scholar] [CrossRef]
- Schlingmann, G.; West, R.R.; Milne, L.; Pearce, C.; Carter, G. Diepoxins, novel fungal metabolites with antibiotic activity. Tetrahedron Lett. 1993, 34, 7225–7228. [Google Scholar] [CrossRef]
- Cai, X.; Shan, T.; Li, P.; Huang, Y.; Xu, L.; Zhou, L.; Wang, M.; Jiang, W. Spirobisnaphthalenes from the endophytic fungus Dzf12 of Dioscorea zingiberensis and their antimicrobial activities. Nat. Prod. Commun. 2009, 4, 1469–1472. [Google Scholar]
- Bode, H.B.; Walker, M.; Zeeck, A. Secondary metabolites by chemical screening, 42. Cladospirones B to I from Sphaeropsidales sp. F–24′707 by variation of culture conditions. Eur. J. Org. Chem. 2000, 2000, 3185–3193. [Google Scholar] [CrossRef]
- Seephonkai, P.; Isaka, M.; Kittakoop, P.; Palittapongarnpim, P.; Kamchonwongpaisan, S.; Tanticharoen, M.; Thebtaranonth, Y. Evaluation of antimycobacterial, antiplasmodial and cytotoxic activities of preussomerins isolated from the lichenicolous fungus Microsphaeropsis sp. BCC 3050. Planta Med. 2002, 68, 45–48. [Google Scholar] [CrossRef]
- Dong, J.Y.; Song, H.C.; Li, J.H.; Tang, Y.S.; Sun, R.; Wang, L.; Zhou, Y.P.; Wang, L.M.; Shen, K.Z.; Wang, C.R.; et al. Ymf 1029A-E, preussomerin analogues from the fresh-water-derived fungus YMF 1.01029. J. Nat. Prod. 2008, 71, 952–956. [Google Scholar] [CrossRef]
- Martinez-Luis, S.; Della-Togna, G.; Coley, P.D.; Kursar, T.A.; Gerwick, W.H.; Cubilla-Rios, L. Antileishmanial constituents of the Panamanian endophytic fungus Edenia sp. J. Nat. Prod. 2008, 71, 2011–2014. [Google Scholar] [CrossRef]
- Chu, M.; Truumees, I.; Patel, M.G.; Gullo, V.P.; Blood, C.; King, I.; Pai, J.-K.; Puar, M.S. A novel class of antitumor metabolites from the fungus Nattrassia Mangiferae. Tetrahedron Lett. 1994, 35, 1343–1346. [Google Scholar]
- Singh, S.B.; Zink, D.L.; Liesch, J.M.; Ball, R.G.; Goetz, M.A.; Bolessa, E.A.; Giacobbe, R.A.; Silverman, K.C.; Bills, G.F.; Pelaez, F.; et al. Preussomerins and deoxypreussomerins: Novel inhibitors of ras farnesyl-protein transferase. J. Org. Chem. 1994, 59, 6296–6302. [Google Scholar] [CrossRef]
- Sakemi, S.; Inagaki, T.; Kaneda, K.; Hirai, H.; Iwata, E.; Sakakibara, T.; Yamauchi, Y.; Norcia, M.; Wondrack, L.M.; Sutcliffe, J.A.; et al. CJ-12,371 and CJ-12,372, two novel DNA gyrase inhibitors. Fermentation, isolation, structural elucidation and biological activities. J. Antibiot. 1995, 48, 134–142. [Google Scholar] [CrossRef]
- Chu, M.; Truumees, I.; Patel, M.; Blood, C.; Das, P.R.; Puar, M.S. Sch 50673 and Sch 50676, two novel antitumor fungal metabolites. J. Antibiot. 1995, 48, 329–331. [Google Scholar] [CrossRef]
- Wipf, P.; Hopkins, T.D.; Jung, J.-K.; Rodriguez, S.; Birmingham, A.; Southwick, E.C.; Lazo, J.S.; Powis, G. New inhibitors of the thioredoxin-thioredoxin reductase system based on a naphthoquinone spiroketal natural product lead. Bioorg. Med. Chem. Lett. 2001, 11, 2637–2641. [Google Scholar] [CrossRef]
- Powis, G.; Wipf, P.; Lynch, S.M.; Birmingham, A.; Kirkpatrick, D.L. Molecular pharmacology and antitumor activity of palmarumycin-based inhibitors of thioredoxin reductase. Mol. Cancer Ther. 2006, 5, 630–636. [Google Scholar]
- Li, Y.; Shan, T.; Mou, Y.; Li, P.; Zhao, J.; Zhao, W.; Peng, Y.; Zhou, L.; Ding, C. Enhancement of palmarumycin C12 and C13 production in liquid culture of endophytic fungus Berkleasmium sp. Dzf12 by oligosaccharides from its host plant Dioscorea zingiberensis. Molecules 2012, 17, 3761–3773. [Google Scholar] [CrossRef]
- Zhao, J.; Zheng, B.; Li, Y.; Shan, T.; Mou, Y.; Lu, S.; Li, P.; Zhou, L. Enhancement of diepoxin ζ production by yeast extract and its fractions in liquid culture of Berkleasmium-like endophytic fungus Dzf12 from Dioscorea zingiberensis. Molecules 2011, 16, 847–856. [Google Scholar] [CrossRef]
- Zhao, J.; Li, Y.; Shan, T.; Mou, Y.; Zhou, L. Enhancement of diepoxin ζ production with in situ resin adsorption in mycelial liquid culture of the endophytic fungus Berkleasmium sp. Dzf12 from Dioscorea zingiberensis. World J. Microbiol. Biotechnol. 2011, 27, 2753–2758. [Google Scholar] [CrossRef]
- Li, Y.; Li, P.; Mou, Y.; Zhao, J.; Shan, T.; Ding, C.; Zhou, L. Enhancement of diepoxin ζ production in liquid culture of endophytic fungus Berkleasmium sp. Dzf12 by polysaccharides from its host plant Dioscorea zingiberensis. World J. Microbiol. Biotechnol. 2012, 28, 1407–1413. [Google Scholar] [CrossRef]
- Mou, Y.; Luo, H.; Mao, Z.; Shan, T.; Sun, W.; Zhou, K.; Zhou, L. Enhancement of palmarumycins C12 and C13 production in liquid culture of endophytic fungus Berkleasmium sp. Dzf12 after treatments with metal ions. Int. J. Mol. Sci. 2013, 14, 979–998. [Google Scholar] [CrossRef]
- Zhao, J.; Shan, T.; Mou, Y.; Zhou, L. Plant-derived bioactive compounds produced by endophytic fungi. Mini Rev. Med. Chem. 2011, 11, 159–168. [Google Scholar] [CrossRef]
- Sannomiya, M.; Rodrigues, C.M.; Coelho, R.G.; dos Santos, L.C.; Hiruma-Lima, C.A.; Souza Brito, A.R.M.; Vilegas, W. Application of preparative high-speed counter-current chromatography for the separation of flavonoids from the leaves of Byrsonima crassa Niedenzu (IK). J. Chromatogr. A 2004, 1035, 47–51. [Google Scholar]
- Wang, J.; Gao, H.; Zhao, J.; Wang, Q.; Zhou, L.; Han, J.; Yu, Z.; Yang, F. Preparative separation of phenolic compounds from Halimodendron halodendron by high-speed counter-current chromatography. Molecules 2010, 15, 5998–6007. [Google Scholar] [CrossRef]
- Gao, H.; Luo, C.; Wang, L.; Wang, J.; Zheng, B.; Peng, Y.; Zhou, L. Preparative separation of 3-O-methylkaempferol from Caragana leucophloea by high-speed counter-current chromatography and its antimicrobial activity. J. Med. Plants Res. 2012, 6, 2081–2087. [Google Scholar]
- Tang, Q.; Liu, J.; Xue, J.; Ye, W.; Zhang, Z.; Yang, C. Preparative isolation and purification of two new isomeric diterpenoid alkaloids from Aconitum coreanum by high-speed counter-current chromatography. J. Chromatogr. B 2008, 872, 181–185. [Google Scholar] [CrossRef]
- Jerz, G.; Skotzki, T.; Fiege, K.; Winterhalter, P.; Wybraniec, S. Separation of betalains from berries of Phytolacca americana by ion-pair high-speed counter-current chromatography. J. Chromatogr. A 2008, 1190, 63–73. [Google Scholar] [CrossRef]
- Wang, J.; Wen, Y.; Chen, X.; Lin, Y.; Zhou, J.; Xie, Y.; Wang, H.; Jiang, H.; Zheng, W. Preparative separation of six antimycin A components from antimycin fermentation broth by high-speed counter-current chromatography. J. Chromatogr. A 2010, 1217, 5687–5692. [Google Scholar] [CrossRef]
- Gu, M.; Wang, X.; Su, Z.; Ouyang, F. One-step separation and purification of 3,4-dihydroxyphenyllactic acid, salvianolic acid B and protocatechualdehyde from Salvia miltiorrhiza Bunge by high-speed counter-current chromatography. J. Chromatogr. A 2007, 1140, 107–111. [Google Scholar] [CrossRef]
- Wu, H.; Su, Z.; Yang, Y.; Ba, H.; Aisa, H.A. Isolation of three sesquiterpene lactones from the roots of Cichorium glandulosum Boiss. et Huet. by high-speed counter-current chromatography. J. Chromatogr. A 2007, 1176, 217–222. [Google Scholar] [CrossRef]
- Peng, A.; Li, R.; Hu, J.; Chen, L.; Zhao, X.; Luo, H.; Ye, H.; Yuan, Y.; Wei, Y. Flow rate gradient high-speed counter-current chromatography separation of five diterpenoids from Triperygium wilfordii and scale-up. J. Chromatogr. A 2008, 1200, 129–135. [Google Scholar] [CrossRef]
- Zhao, J.; Mou, Y.; Shan, T.; Li, Y.; Lu, S.; Zhou, L. Preparative separation of helvolic acid from the endophytic fungus Pichia guilliermondii Ppf9 by high-speed counter-current chromatography. World J. Microbiol. Biotechnol. 2012, 28, 835–840. [Google Scholar] [CrossRef]
- Assimopoulou, A.N.; Sturm, S.; Stuppner, H.; Papageorgiou, V.P. Preparative isolation and purification of alkannin/shikonin derivatives from natural products by high-speed counter-current chromatography. Biomed. Chromatogr. 2009, 23, 182–198. [Google Scholar] [CrossRef]
- Ito, Y. Golden rules and pitfalls in selecting optimum conditions for high-speed counter-current chromatography. J. Chromatogr. A 2005, 1065, 145–168. [Google Scholar] [CrossRef]
- Zhao, W.-H.; Gao, C.-C.; Ma, X.-F.; Bai, X.-Y.; Zhang, Y.-X. The isolation of 1,2,3,4,6-penta-O-galloyl-beta-D-glucose from Acer truncatum Bunge by high-speed counter-current chromatography. J. Chromatogr. B 2007, 850, 523–527. [Google Scholar] [CrossRef]
- Li, F.-Y.; Gou, Z.-P.; Ma, X.-C.; Tian, Y.; Tian, G.; Wang, C.-Y.; Su, D.-H.; Liu, K.X.; Xin, X.-L. Preparative isolation and purification of two phyenylpropanoids from Daphne giraldii Nitsche by HSCCC. Chromatographia 2010, 71, 481–485. [Google Scholar] [CrossRef]
- Schlingmann, G.; Matile, S.; Berova, N.; Nakanishi, K.; Carter, G.T. Absolute stereochemistry of the diepoxins. Tetrahedron 1996, 52, 435–446. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the spirobisnaphthalenes are available from the authors.
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Shan, T.; Lu, S.; Luo, C.; Luo, R.; Mou, Y.; Wang, M.; Peng, Y.; Zhou, L. Preparative Separation of Spirobisnaphthalenes from Endophytic Fungus Berkleasmium sp. Dzf12 by High-Speed Counter-Current Chromatography. Molecules 2013, 18, 12896-12908. https://doi.org/10.3390/molecules181012896
Shan T, Lu S, Luo C, Luo R, Mou Y, Wang M, Peng Y, Zhou L. Preparative Separation of Spirobisnaphthalenes from Endophytic Fungus Berkleasmium sp. Dzf12 by High-Speed Counter-Current Chromatography. Molecules. 2013; 18(10):12896-12908. https://doi.org/10.3390/molecules181012896
Chicago/Turabian StyleShan, Tijiang, Shiqiong Lu, Chao Luo, Ruiya Luo, Yan Mou, Mingan Wang, Youliang Peng, and Ligang Zhou. 2013. "Preparative Separation of Spirobisnaphthalenes from Endophytic Fungus Berkleasmium sp. Dzf12 by High-Speed Counter-Current Chromatography" Molecules 18, no. 10: 12896-12908. https://doi.org/10.3390/molecules181012896