3. Experimental
All reactions were performed using standard glassware and IKA num heating plates. Reactions utilizing air-sensitive reagents were performed in dried glassware under a nitrogen atmosphere. Solvents were used as received without further purifications. All reagents, if not otherwise specified, were used as received and, if necessary, stored under inert gas. Oxcarbazepine was supplied by Trifarma SpA, Ceriano Laghetto, Italy. For thin-layer chromatography (TLC) analysis Macherey-Nagel Polygram® sil G/UV 254 pre-coated plates were used. Column chromatography was performed on silica gel 60A (70–200 µm) (Carlo Erba Reagents, Arese, Italy). Melting point (mp) determinations were performed by using a Gallenkamp melting point apparatus (Weiss-Gallenkamp, Loughborough, UK). 1H-NMR (400 MHz) and 13C-NMR (100 MHz) were measured on a AV400 spectrometer (Bruker Corporation, Billerica, MA, USA). Chemical shifts (δ) are expressed in parts per million (ppm) and coupling constants are given in hertz (Hz). Splitting patterns are indicated as follows: s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, br = broad, dd = doublet-doublet, td = triplet-doublet. Chemical ionization mass spectra (+ve mode) (CI+-MS) were performed on a Finnigan-MAT TSQ70 instrument with isobutane as the reactant gas. Elemental analyses were performed on a Perkin Elmer Series II CHNS/O Analyzer 2400.
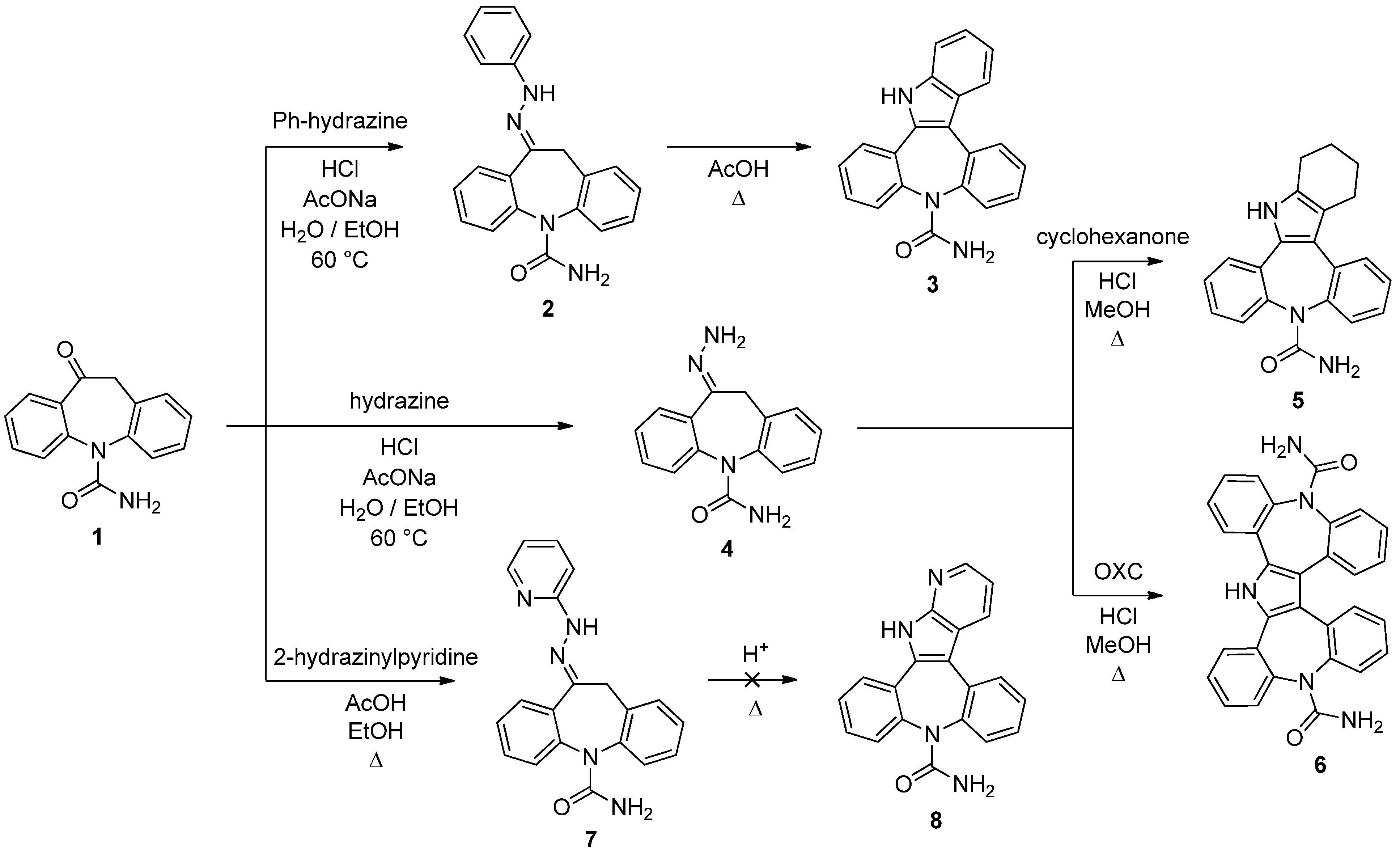
10-(Phenylhydrazono)-10,11-dihydro-5H-dibenzo[b,f]azepine-5-carboxamide (
2). The compound was prepared following the procedure present in the literature and the spectroscopic data were in accordance with those reported [
8]. Yield: 88%.
Dibenzo[2,3:6,7]azepino[4,5‑b]indole‑9(14H)-carboxamide (3). A well stirred solution of 2 (410 mg, 1.20 mmol) in acetic acid (5 mL) was brought to reflux for 1 h. The reaction progress was monitored by TLC analysis (SiO2, AcOEt, Rf 2 = 0.39, Rf 3 = 0.25). The mixture was then cooled to room temperature and added dropwise to a stirred ice/water solution (150 mL). The precipitate was filtered, washed with water and dried under vacuum to afford 3 as green solid (309 mg, 79%): 1H-NMR (DMSO-d6) δ 11.65 (s, 1H), 7.94 (m, 2H), 7.80 (m, 1H), 7.52 (m, 3H), 7.41 (m, 3H), 7.34 (m, 1H), 7.21 (t, J = 7.3 Hz, 1H), 7.14 (t, J = 7.5 Hz, 1H), 5.20 (br. s, 2H). 13C-NMR (DMSO-d6) δ 157.2, 141.3, 140.0, 137.6, 135.0, 134.2, 130.9, 130.1, 129.8, 129.6, 128.7, 128.6, 128.2, 128.1, 127.5, 126.4, 123.4, 120.8, 120.1, 113.1, 112.6; m.p. 216–219 °C dec.; MS (CI+): m/z = 326 [MH]+, 283 [(M-HNCO)H]+; Anal. Calcd. for C21H15N3O: C, 77.52; H, 4.65; N, 12.91; Found: C, 77.31; H, 4.87; N, 13.01.
10-Hydrazinylidene-10,11-dihydro-5H-dibenzo[b,f]azepine-5-carboxamide (
4). The compound was prepared following the procedure present in the literature and the spectroscopic data were in accordance with those reported [
8]. Yield: 45%.
2,3,4,14-Tetrahydrodibenzo[2,3:6,7]azepino[4,5b]indole-9(1H)-carboxamide (5). Compound 4 (100 mg, 0.38 mmol) and cyclohexanone (45 µL, 0.44 mmol) were dissolved in methanol (15 mL). 37% aq. HCl (47 µL, 0.21 mmol) was added and the reaction mixture was heated at 68 °C for 30 min under stirring. The reaction progress was monitored by TLC analysis (SiO2, AcOEt, Rf 4 = 0.04, Rf 5 = 0.22). After cooling to room temperature and removing part of the solvent under reduced pressure, the solution was dropped into stirred ice/water (50 mL). The precipitate was filtered, washed with water and dried under vacuum to gave 5 as yellow solid (85 mg, 69%): 1H-NMR (DMSO-d6) δ 11.20 (s, 1H), 7.55–7.28 (m, 8H), 5.43 (br. S, 2H), 2.90 (m, 1H), 2.72–2.50 (m, 3H), 1.97 (m, 2H), 1.75 (m, 1H), 1.60 (m, 1H); 13C-NMR (DMSO-d6) δ 157.1, 139.3, 139.2, 134.8, 131.8, 130.3, 130.2, 130.1, 128.1, 128.1, 128.0, 127.7, 127.3, 126.9, 126.5, 119.1, 115.8, 24.5, 24.1, 23.5, 23.4; m.p. 268–269 °C; MS (CI+): m/z = 330 [MH]+, 287 [(M-HNCO)H]+; Anal. Calcd. for C21H19N3O: C, 76.57; H, 5.81; N, 12.76; O, 4.86; Found: C, 76.38; H, 5.93; N, 12.49.
5H-Tetrabenzo[b,b',f,f']pyrrolo[2,3-d:4,5‑d']bisazepine-5,14(19H)-dicarboxamide (6). Compound 1 (111 mg, 0.44 mmol) and 4 (117 mg, 0.44 mmol) were dissolved in hot methanol (15 mL) and the mixture was stirred at 68 °C for 10 min. 37% aq. HCl (47 µL, 0.21 mmol) was then added and the solution was left at reflux under stirring for 2 h. The reaction progress was monitored by TLC analysis (SiO2, AcOEt/MeOH 95:5, Rf 4 = 0.12, Rf 1 = 0.37, Rf 6 = 0.11). After 30 min the product started to precipitate as white solid. When the reaction was complete the mixture was cooled to r.t., 6 was recovered in a pure form by filtration and dried in vacuum (124 mg, 58%): 1H-NMR (DMSO-d6) δ 12.18 (s, 1H), 7.86 (m, 2H), 7.59–7.53 (m, 4H), 7.45 (m, 4H), 7.34 (m, 4H), 7.15 (m, 2H), 5.77 (br. s, 4H); 13C-NMR (DMSO-d6) δ 157.2, 141.1, 140.1, 134.0, 131.6, 130.8, 130.3, 130.0, 129.9, 129.1, 128.2, 128.1, 128.0, 127.9, 119.5; m.p. 336–337 °C; MS (CI+): m/z = 484 [MH]+, 441 [(M-HNCO)H]+; Anal. Calcd. for C30H21N5O2: C, 74.52; H, 4.38; N, 14.48; Found: C, 74.69; H, 4.27; N, 14.37.
10-(2-Pyridinylhydrazono)-10,11-dihydro-5H-dibenzo[b,f]azepine-5-carboxamide (7). A mixture of 1 (500 mg, 2.0 mmol) and 2-hydrazinopyridine (654 mg, 6.0 mmol) in ethanol (20 mL) and three drops of acetic acid was heated at 78 °C for 1 h under stirring. The reaction progress was monitored by TLC analysis (SiO2, AcOEt, Rf 1 = 0.26, Rf 7 = 0.07). After allowing to cool to room temperature, the precipitate was filtered, washed with ethanol and dried in vacuum to give (7) as yellow solid (410 mg, 60%): 1H-NMR (DMSO-d6) δ 10.24 (br. s, 1H), 8.20 (d, J = 4.7 Hz, 1H), 7.95 (m, 1H), 7.69 (m, 1H), 7.43–7.24 (m, 8H), 6.85 (m, 1H), 5.96 (br. s, 2H), 4.32 (d, J = 17.6 Hz, 1H), 3.94 (d, J = 17.6 Hz, 1H); 13C-NMR (DMSO-d6) δ 158.7, 156.9, 148.5, 144.1, 142.5, 141.8, 138.9, 135.5, 134.6, 130.8, 130.0, 129.7, 129.0, 128.9, 128.8, 128.7, 128.0, 116.6, 108.1, 33.9; m.p. 230–231 °C; MS (CI+): m/z = 327 [MH]+; Anal. Calcd. for C19H15N3O3: C, 69.96; H, 4.99; N, 20.40; Found: C, 69.75; H, 5.13; N 20.29.
10-(Hydroxyimino)-10,11-dihydro-5H-dibenzo[b,f]azepine-5-carboxamide (
9). The compound was prepared following the procedure present in the literature and the spectroscopic data were in accordance with those reported [
8]. Yield = 94%.
2-Phenyl-8H-[1,3]oxazolo[4,5-d]dibenzo[b,f]azepine-8-carboxamide (10). 9 (100 mg, 0.37 mmol), pyridine (30 μL, 0.37 mmol) and benzoyl chloride (108 μL, 0.93 mmol) were added to chlorobenzene (5 mL). The mixture was stirred for 30 min at r.t., then the temperature was rise to 132 °C and the reaction was stirred for 30 min. The reaction progress was monitored by TLC analysis (SiO2, CH2Cl2/EtOH 95:5, Rf 9 = 0.13, Rf 10 = 0.22). After cooling to r.t. the solvent was removed under reduced pressure, water (20 mL) was added to the residue and the aqueous phase was extracted with AcOEt (3 × 15 mL). The combined organic layers were washed with H2O (2 × 20 mL), dried (Na2SO4) and the solvent removed under vacuum. Flash column chromatography (SiO2, CH2Cl2/EtOH 95/5, Rf = 0.22) gave 10 as white solid (52 mg, 40%): 1H-NMR (CDCl3) δ 8.27–8.24 (m, 2H), 8.18–8.16 (m, 1H), 7.88 (dd, J = 7.6, 1.5 Hz, 1H), 7.67–7.50 (m, 9H), 4.61 (br. s, 2H); 13C-NMR (CDCl3) δ 161.7, 157.7, 137.7, 137.6, 137.1, 131.3, 130.8, 130.5, 130.4, 130.3, 130.3, 129.5, 129.3, 129.1, 128.7, 128.7, 127.8, 127.4, 127.2, 125.8; m.p. 230–232 °C; MS (CI+): m/z = 354 [MH]+, 382 [M+C2H5]+, 337 [(M-NH2)H]+, 310 [(M-HNCO)H]+; Anal. Calcd. for C22H15N3O2: C, 74.78; H, 4.28; N, 11.89; Found: C, 74.85; H, 4.22; N, 11.79.
Methyl-8-carbamoyl-1,8-dihydropyrrolo[2,3-d]dibenzo[b,f]azepine-3-carboxylate (11a). The oxime 9 (200 mg, 0.75 mmol) was dissolved in a mixture of CH3CN (5 mL) and DMF (1 mL). Triethyl amine (104 μL, 0.75 mmol) and methyl propiolate (67 μL, 0.75 mmol) were then added and the reaction was left at r.t. for 1 h under stirring. The reaction progress was monitored by TLC analysis (SiO2, AcOEt, Rf 9 = 0.22, Rf 11a = 0.10). Acetonitrile was removed under reduced pressure and the DMF residue was dropped into water (20 mL). The brown precipitate was collected by filtration, washed with H2O and dried under vacuum to gave 11a in a pure form (141 mg, 57%): 1H-NMR (DMSO-d6) δ 12.33 (s, 1H), 7.76–7.23 (m, 9H), 5.59 (br. s, 2H), 3.74 (s, 3H); 13C-NMR (DMSO-d6) δ 165.2, 156.7, 141.1, 140.1, 132.8, 131.9, 131.2, 130.0, 129.9, 129.5, 129.4, 128.4, 128.3, 128.2, 127.4, 127.3, 121.1, 113.4, 51.6; m.p. 198–199 °C; MS (CI+): m/z = 333 [MH]+; Anal. Calcd. for C19H15N3O3: C, 68.46; H, 4.54; N, 12.61; Found: C, 68.41; H, 4.63; N 12.49.
3-(Phenylcarbonyl)dibenzo[b,f]pyrrolo[2,3-d]azepine-8(1H)-carboxamide (11b). The oxime 9 (100 mg, 0.37 mmol) was dissolved in a mixture of CH3CN (2.5 mL) and DMF (0.5 mL). Triethyl amine (52 μL, 0.37 mmol) and phenyl propynone (49 mg, 0.37 mmol) were then added and the reaction was left at r.t. for 1 h under stirring. The reaction progress was monitored by TLC analysis (SiO2, AcOEt, Rf 9 = 0.22, Rf 11b = 0.13). Acetonitrile was removed under reduced pressure and the DMF residue was dropped into water (10 mL). The yellow precipitate was collected by filtration, washed with H2O and dried under vacuum to gave 11b in a pure form (101 mg, 71%): 1H-NMR (DMSO-d6) δ 12.41 (s, 1H), 7.82 (d, J = 7.4 Hz, 1H), 7.76–7.08 (m, 13H), 5.58 (br. s, 2H);13C-NMR (DMSO-d6) δ 191.1, 156.9, 141.0, 140.2, 139.8, 135.5, 133.4, 132.7, 131.5, 130.2, 130.1, 129.9, 129.5, 129.4, 129.1, 128.9, 128.8, 128.3, 128.2, 128.1, 127.6, 127.4, 122.7, 121.4; m.p. 265–266 °C; MS (CI+): m/z = 380 [MH]+, 337 [(M-HNCO)H]+; Anal. Calcd. for C24H17N3O2: C, 75.97; H, 4.52; N, 11.08; Found: C, 75.78; H, 4.55; N, 11.21.
3-[(4-Nitrophenyl)carbonyl]dibenzo[b,f]pyrrolo[2,3-d]azepine-8(1H)-carboxamide (11c). The oxime 9 (200 mg, 0.75 mmol) was dissolved in a mixture of CH3CN (4 mL) and DMF (1 mL). Triethylamine (104 μL, 0.75 mmol) and p-nitrophenyl propynone (131 mg, 0.75 mmol) were then added and the reaction was left at r.t. for 1 h under stirring. The reaction progress was monitored by TLC analysis (SiO2, AcOEt, Rf 9 = 0.22, Rf 11c = 0.13). Acetonitrile was removed under reduced pressure and the DMF residue was dropped into water (20 mL). The brown precipitate was collected by filtration, washed with H2O and dried under vacuum to gave 11c in a pure form (249 mg, 78%): 1H-NMR (DMSO-d6) δ 12.56 (br. s, 1H), 8.27–8.23 (m, 2H), 8.02–7.98 (m, 2H), 7.70–7.77 (m, 1H), 7.60 (s, 1H), 7.55–7.52 (m, 1H), 7.48–7.41 (m, 3H), 7.30–7.24 (m, 2H), 7.10 (dt, J = 7.6, 1.2 Hz, 1H) 5.63 (br. s, 2H); 13C-NMR (DMSO-d6) δ 189.1, 156.5, 149.4, 145.2, 140.8, 139.7, 132.7, 131.6, 131.2, 130.8, 130.4, 129.8, 129.3, 128.0, 127.9, 127.2, 127.1, 123.8, 121.7, 120.9; m.p. 188–189 °C; MS (CI+): m/z = 425 [MH]+, 382 [(M-HNCO)H]+; Anal. Calcd. for C21H15N4O4: C, 67.92; H, 3.80; N, 13.20; Found: C, 68.02; H, 3.93; N, 13.05.
10-(Carbamoylhydrazono)-10,11-dihydro-5H-dibenzo[b,f]azepine-5-carboxamide (
12). The compound was prepared following the procedure present in the literature and the spectroscopic data were in accordance with those reported [
8]. Yield = 85%.
8H-[1,2,3]Thiadiazolo[4,5-d]dibenzo[b,f]azepine-8-carboxamide (13). Thionyl chloride (5 mL) was cooled to −10 °C with an ice/NaCl bath, 12 (496 mg, 1.60 mmol) was slowly added and the reaction mixture was left rise to r.t. under stirring. The reaction progress was monitored by TLC analysis (SiO2, CH2Cl2/EtOH 95:5, Rf 12 = 0.07, Rf 13 = 0.35). When the reaction was complete (after about 2 h), the mixture was added drop wise to water (100 mL) and the pH was set to 7 adding an aqueous solution of NH3. The yellow precipitate was collected and dried under vacuum to afford 13 in a pure form (392 mg, 83%): 1H-NMR (CDCl3) δ 8.36 (d, J = 8.0 Hz, 1H), 7.72–7.59 (m, 6H), 7.46 (t, J = 7.5 Hz, 1H), 4.50 (br. s, 2H); 13C-NMR (CDCl3) δ 156.6, 155.9, 150, 140.6, 140.2, 132.4, 132.0, 131.5, 130.8, 130.5, 130.4, 129.5, 129.2, 129.2, 127.3; m.p. 231–232 °C; MS (CI+): m/z = 295 [MH]+, 323 [M+C2H5]+, 252 [(M-HNCO)H]+; Anal. Calcd. for C15H10N4OS: C, 61.21; H, 3.42; N, 19.04; Found: C, 61.28; H, 3.54; N, 19.11.
8H-[1,2,3]Selenadiazolo[4,5-d]dibenzo[b,f]azepine-8-carboxamide (14). 12 (470 mg, 1.52 mmol) was dissolved in acetic acid (5 mL) and SeO2 (202 mg, 1.82 mmol) was added portionwise while stirring. The temperature was rise to 118 °C and the mixture was left under stirring for 30 min. The reaction progress was monitored by TLC analysis (SiO2, CH2Cl2/EtOH 95:5, Rf 12 = 0.07, Rf 14 = 0.37). After cooling to r.t. water (100 mL) was added, the aqueous phase was extracted with ethyl acetate (3 × 40 mL), the combined organic layers were dried (Na2SO4) and the solvent was removed under reduced pressure to afford 14 as pink solid (250 mg, 48%): 1H-NMR (DMSO-d6) δ 8.15 (d, J = 7.7 Hz, 1H), 7.69–7.64 (m, 5H), 7.55 (m, 1H), 7.42 (m, 1H), 5.87 (br. s, 2H); 13C-NMR (DMSO-d6) δ 158.2, 156.7, 156.2, 142.1, 141.4, 141.4, 135.2, 132.8, 131.4, 131.1, 130.9, 130.4, 130.3, 129.3, 129.0; m.p. 233–234 °C; MS (CI+): m/z = 345 [M(82Se)H]+ (8.7%), 343 [M(80Se)H]+ (49.6%), 341 [M(78Se)H]+ (23.7%), 340 [M(77Se)H]+ (7.6%), 339 [M(76Se)H]+ (9.3%), 317 [(M(82Se)-N2)H]+, 315 [(M(80Se)-N2)H]+, 313 [(M(78Se)-N2)H]+, 312 [(M(77Se)-N2)H]+, 311 [(M(76Se)-N2)H]+; Anal. Calcd. for C15H10N4OSe: C, 52.80; H, 2.95; N, 16.42; Found: C, 52.74; H, 3.02; N, 16.26.
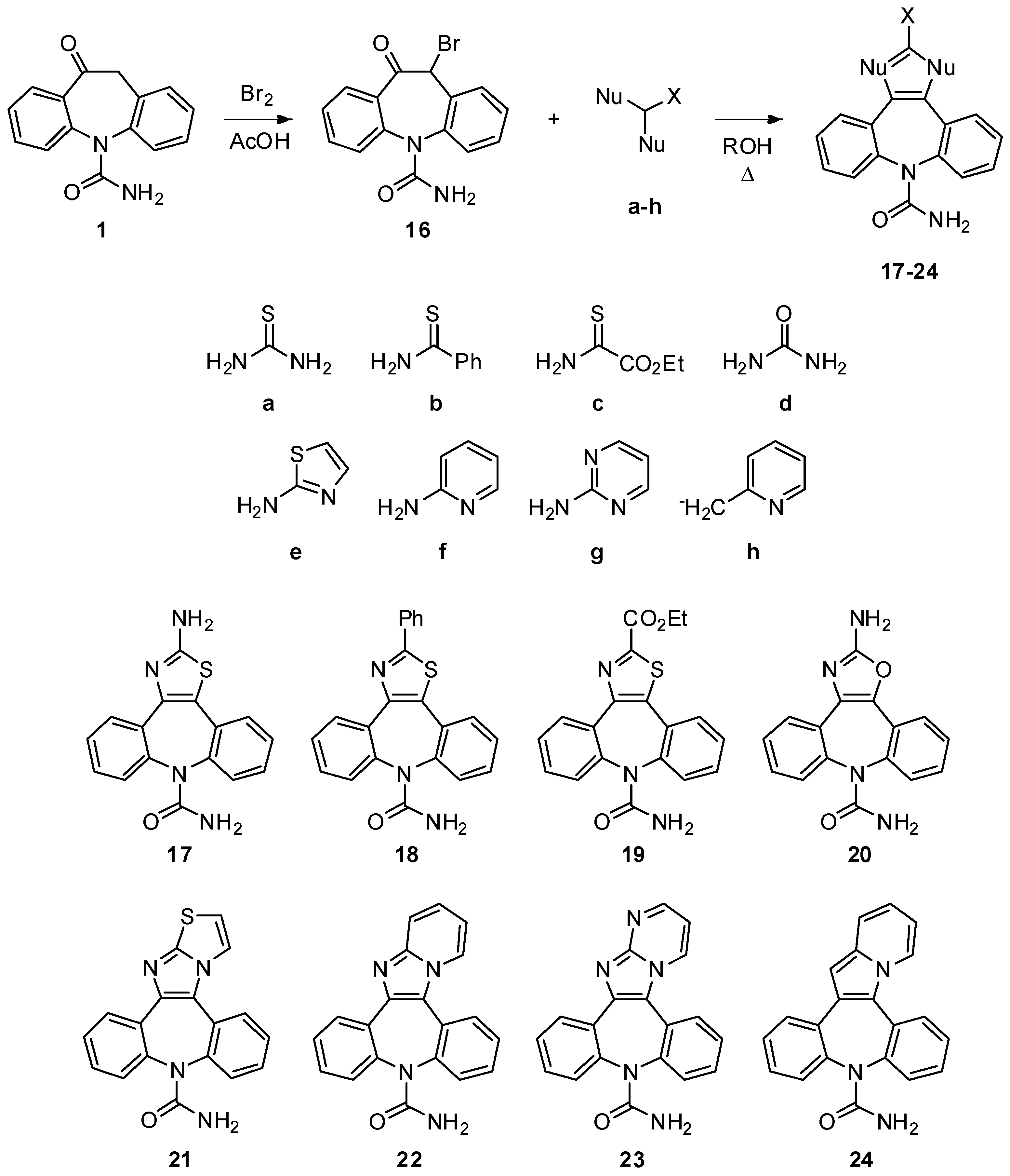
10-Bromo-11-oxo-10,11-dihydro-5H-dibenzo[b,f]azepine-5-carboxamide (
16). The compound was prepared following the procedure present in the literature and the spectroscopic data were in accordance with those reported [
26]. Yield = 92%.
2-Amino-8H-dibenzo[b,f][1,3]thiazolo[4,5-d]azepine-8-carboxamide (17). Compound 16 (300 mg, 0.91 mmol) and thiourea (205 mg, 2.69 mmol) were dissolved in ethanol (13 mL). The well stirred reaction mixture was brought to reflux for 3 h. The reaction progress was monitored by TLC analysis (SiO2, AcOEt/EtOH 9:1, Rf 17 = 0.28). After cooling to r.t., the reaction mixture was dropped into water (100 mL), the white precipitate was collected and dried under vacuum to afford 17 in a pure form (120 mg, 43%). 1H-NMR (DMSO-d6) δ 7.78 (d, J = 7.5 Hz, 1H), 7.50–7.36 (m, 9H), 5.65 (br. s, 2H); 13C-NMR (DMSO-d6) δ 167.5, 157.0, 140.4, 139.9, 132.9, 131.4, 130.4, 130.3, 130.1, 129.1, 128.7, 128.6, 128.3, 120.6; m.p. 215–217 °C; MS (CI+): m/z = 309 [MH]+, 266 [(M-HNCO)H]+; Anal. Calcd. for C16H12N4OS: C, 62.32; H, 3.92; N, 18.17; Found: C, 62.43; H, 3.97; N, 17.98.
2-Amino-8H-dibenzo[b,f][1,3]oxazolo[4,5-d]azepine-8-carboxamide (20). 16 (200 mg, 0.60 mmol) and urea (181 mg, 3.02 mmol) were dissolved in n-butyl alcohol (3 mL), under a nitrogen atmosphere. The well stirred reaction mixture was brought to reflux for 16 h. The reaction progress was monitored by TLC analysis (SiO2, CH2Cl2/EtOH 95:5, Rf 16 = 0.33, Rf 20 = 0.03). After cooling to r.t., an aqueous solution of NaHCO3 was added to the reaction mixture and the solvent was removed under reduced pressure. Flash column chromatography (SiO2, CH2Cl2/EtOH 9:1, Rf = 0.16) afforded 20 as white solid (56 mg, 32%): 1H-NMR (CDCl3) δ 7.80 (m, 1H), 7.54–7.33 (m, 7H), 5.79 (br. s, 2H), 5.04 (br. s, 2H); 13C-NMR (CDCl3) δ 161.1, 158.1, 137.0, 136.0, 130.9, 130.1, 129.8, 129.6, 129.0, 128.9, 128.6, 127.5, 127.0, 124.2; m.p. 224–226 °C; MS (CI+): m/z = 293 [MH]+, 250 [(M-HNCO)H]+; Anal. Calcd. for C16H12N4O2: C, 65.75; H, 4.14; N, 19.17; Found: C, 65.44; H, 4.11; N, 19.28.
10H-Dibenzo[b,f]thiazolo[1',2':1,2]imidazo[4,5-d]azepine-10-carboxamide (21). Compound 16 (200 mg, 0.60 mmol) and 2-aminothiazole (121 mg, 1.21 mmol) were dissolved in n-butyl alcohol (20 mL). The well stirred reaction mixture was brought to reflux for 6 h. The reaction progress was monitored by TLC analysis (SiO2, CH2Cl2/EtOH 95:5, Rf 16 = 0.33, Rf 2‑aminothiazole = 0.11, Rf 21 = 0.06). After cooling to r.t., water was added to the reaction mixture and the solvent was removed under reduced pressure. Flash column chromatography (SiO2, CH2Cl2/EtOH 9:1, Rf = 0.27) gave 21 as ochre solid (113 mg, 56%): 1H-NMR (DMSO-d6) δ 8.33 (d, J = 4.4 Hz, 1H), 7.92 (m, 1H), 7.82 (m, 1H), 7.61 (m, 1H), 7.56–7.45 (m, 7H), 5.62 (br. s, 2H); 13C-NMR (DMSO-d6) δ 169.8, 157.1, 151.6, 143.8, 139.9, 139.4, 132.9, 131.1, 130.2, 129.9, 129.5, 128.9, 128.7, 128.3, 128.2, 125.4, 120.3, 115.5; m.p. 257–258 °C; MS (CI+): m/z = 333 [MH]+, 290 [(M-HNCO)H]+; Anal. Calcd. for C18H12N4OS: C, 65.04; H, 3.64; N, 16.86; Found: C, 65.19; H, 3.59; N, 16.82.
10H-Dibenzo[b,f]pyrido[1',2':1,2]imidazo[4,5-d]azepine-10-carboxamide (22). Compound 16 (300 mg, 0.91 mmol) and 2-aminopyridine (128 mg, 1.36 mmol) were dissolved in n-butyl alcohol (30 mL). The well stirred reaction mixture was brought to reflux for 1 h. The reaction progress was monitored by TLC analysis (SiO2, CH2Cl2/EtOH 95:5, Rf 16 = 0.33, Rf 2-aminopyridine = 0.18, Rf 22 = 0.06). After cooling to r.t., water was added to the reaction mixture and the solvent was removed under reduced pressure. Flash column chromatography (SiO2, CH2Cl2/EtOH 9:1, Rf = 0.32) gave 22 as pink solid (157 mg, 53%): 1H-NMR (CDCl3) δ 8.71 (d, J = 7.0 Hz, 1H), 7.97–6.94 (m, 11H), 5.45 (br. s, 2H); 13C-NMR (DMSO-d6) δ 156.9, 146.4, 143.1, 141.9, 141.2, 135.4, 133.0, 131.1, 130.2, 129.9, 129.6, 129.0, 128.8, 128.5, 128.3, 126.9, 125.9, 125.5, 118.5, 112.9; m.p. 255–256 °C; MS (CI+): m/z = 327 [MH]+, 284 [(M-HNCO)H]+; Anal. Calcd. for C20H14N4O: C, 73.61; H, 4.32; N, 17.17; Found: C, 73.69; H, 4.28; N, 17.24.
10H-Dibenzo[b,f]pyrimido[1',2':1,2]imidazo[4,5-d]azepine-10-carboxamide (23). Compound 16 (100 mg, 0.30 mmol) and 2-aminopyrimidine (29 mg, 0.30 mmol) were dissolved in n-butyl alcohol (5 mL), under nitrogen atmosphere. The well stirred reaction mixture was brought to reflux for 8 h. The reaction progress was monitored by TLC analysis (SiO2, CH2Cl2/EtOH 8:2, Rf 16 = 0.71, Rf 2-amino-pyrimidine = 0.36, Rf 23 = 0.28). After cooling to r.t., an aqueous solution of K2CO3 (0.30 mmol, 10 mL) was added to the reaction mixture and the solvent was removed under reduced pressure. Flash column chromatography (SiO2, CH2Cl2/EtOH 9:1, Rf = 0.21) afforded 23 as white solid (33 mg, 34%): 1H-NMR (DMSO-d6) δ 9.29 (m, 1H), 8.72 (m, 1H), 8.06 (m, 1H), 7.97 (m, 1H), 7.69–7.49 (m, 6H), 7.44 (m, 1H), 5.69 (br. s, 2H); 13C-NMR (DMSO-d6) δ 152.5, 134.6, 131.2, 130.8, 130.2, 130.0, 129.2, 128.8, 128.7, 126.0, 110.5; m.p. 259–261 °C dec.; MS (CI+): m/z = 328 [MH]+, 285 [(M-HNCO)H]+; Anal. Calcd. for C19H13N5O: C, 69.71; H, 4.00; N, 21.39; Found: C, 69.60; H, 4.13; N, 21.33.
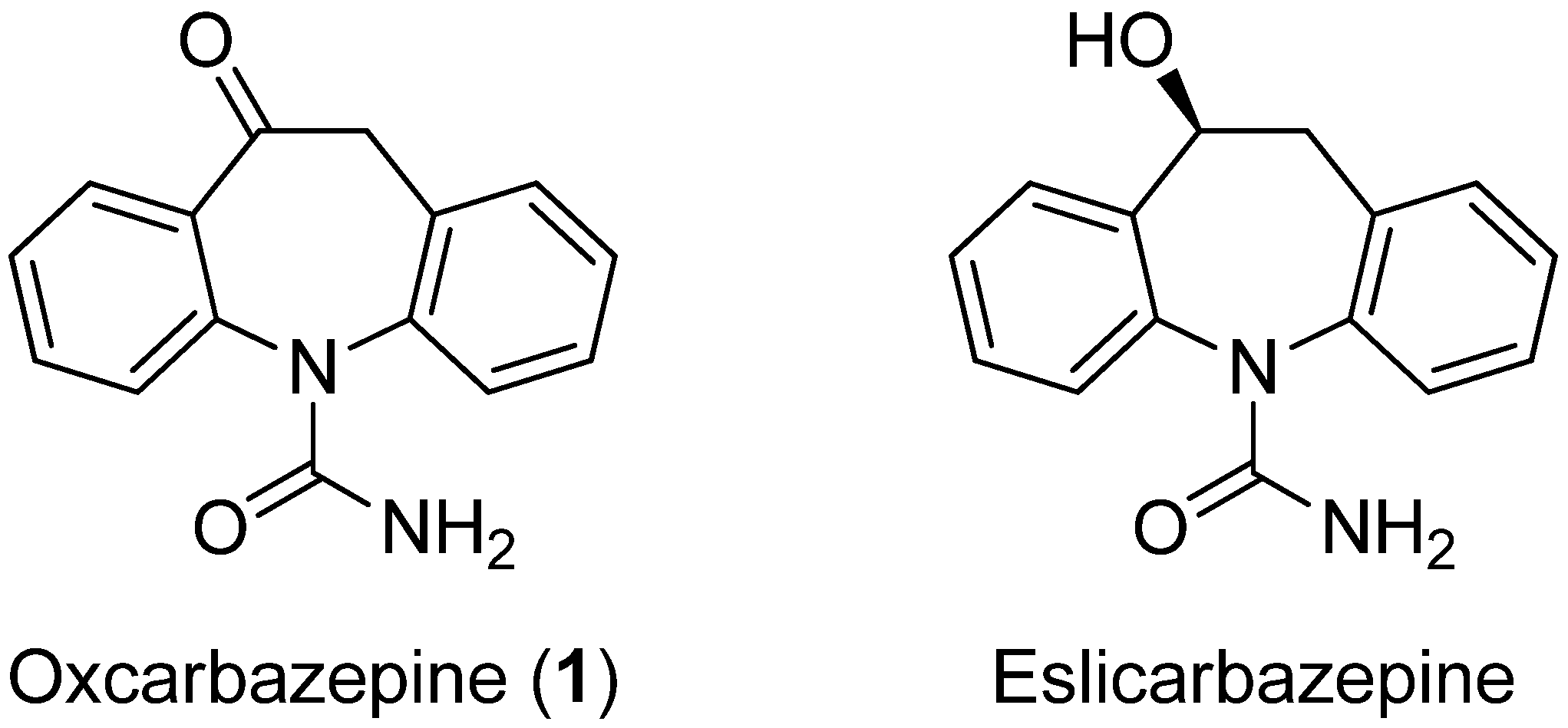
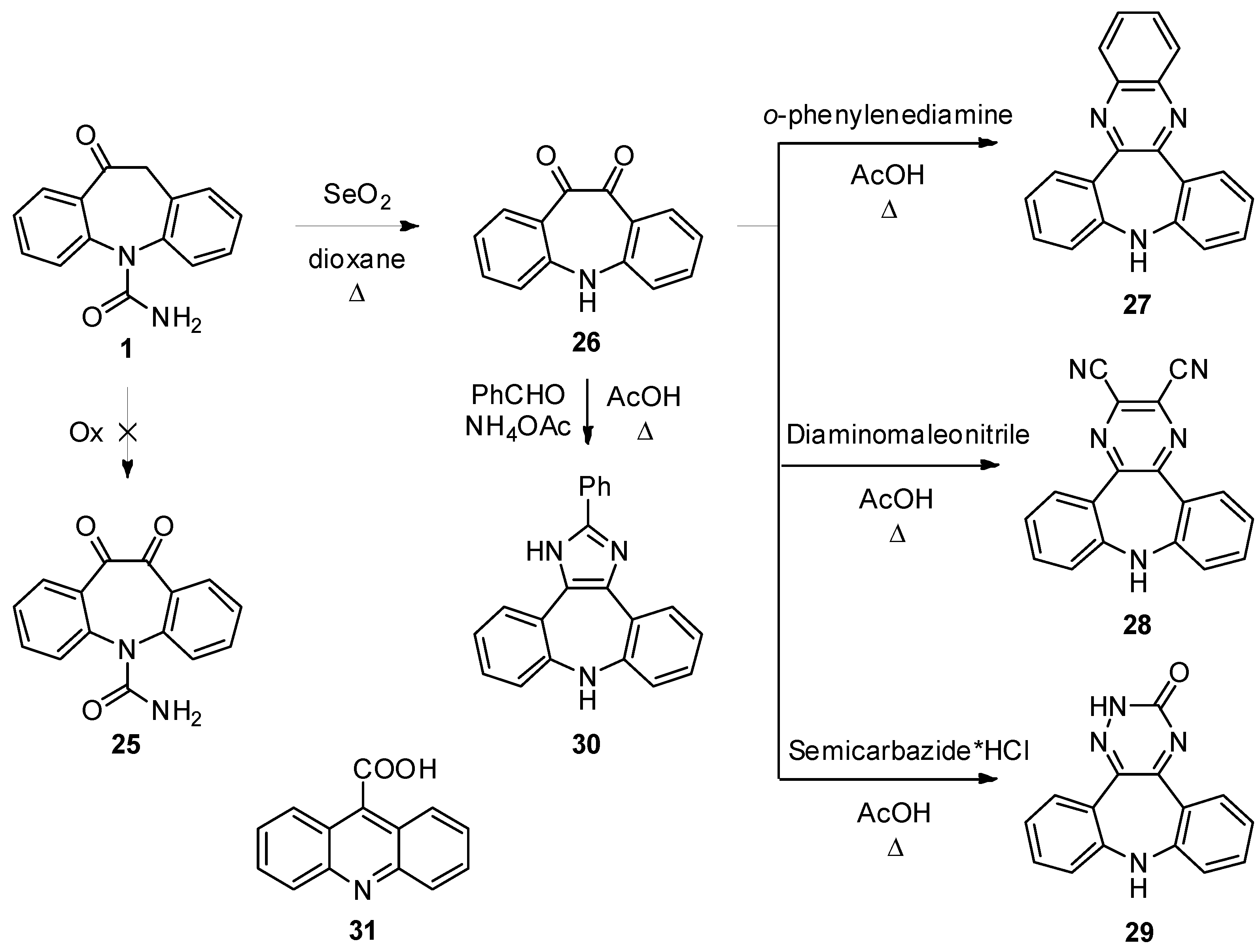
5H-Dibenzo[b,f]azepine-10,11-dione (
26). Oxcarbazepine (
1, 5.0 g, 19.82 mmol) and SeO
2 (5.4 g, 48.67 mmol) in dioxane (100 mL) were brought to reflux under vigorous stirring for 7 h. The reaction progress was monitored by TLC analysis (SiO
2, CH
2Cl
2/EtOH 95:5,
Rf 1 = 0.27,
Rf 26 = 0.56). After cooling to r.t. the orange precipitate was filtered and dried under vacuum to afford
26 (4.0 g, 90%): the spectroscopic data were in accordance with those reported in the literature [
30].
10H-Quinoxalino[2,3-d]dibenzo[b,f]azepine (
27). Compound
26 (100 mg, 0.45 mmol) and 1,2-diaminobenzene (49 mg, 0.45 mmol) in AcOH (2 mL) were refluxed for 1 h under stirring. The reaction progress was monitored by TLC analysis (SiO
2, CH
2Cl
2/EtOH 95:5,
Rf 26 = 0.54,
Rf 1,2-diaminobenzene = 0.23,
Rf 27 = 0.69). The reaction mixture was left cooling to r.t. and added dropwise to stirred ice/water (25 mL), the yellow precipitate was collected by filtration, washed with water and dried under vacuum to afford
27 (64 mg, 49%): the spectroscopic data were in accordance with those reported in the literature [
30].
9H-Dibenzo[b,f]pyrazino[2,3-d]azepine-2,3-dicarbonitrile (28). Compound 26 (100 mg, 0.45 mmol) and diaminomalonitrile (49 mg, 0.45 mmol) in AcOH (2 mL) were refluxed for 1 h under stirring. The reaction progress was monitored by TLC analysis (SiO2, CH2Cl2/EtOH 95:5, Rf 26 = 0.54, Rf 28 = 0.73). The reaction mixture was left cooling to r.t., the dark red precipitate was collected by filtration and dried under vacuum to afford 28 (86 mg, 65%): 1H-NMR (DMSO-d6) δ 8.13 (s, 1H), 7.70 (dd, J = 7.1, 1.4 Hz, 2H), 7.43 (td, J = 8.1, 1.5 Hz, 2H), 7.14 (t, J = 7.6 Hz, 2H), 7.11 (d, J = 8.1 Hz, 2H); 13C-NMR (DMSO-d6) δ 156.3, 153.6, 133.8, 132.6, 131.6, 126.7, 124.7, 121.4, 115.21; MS (CI+): m/z = 296 [MH]+; Anal. Calcd. for C18H9N5: C, 73.21; H, 3.07; N, 23.72; Found: C, 73.52; H, 3.04; N, 23.48.
2,9-Dihydro-3H-dibenzo[b,f][1,2,4]triazino[5,6-d]azepin-3-one (29). Semicarbazide hydrochloride (50 mg, 0.45 mmol) was dissolved in H2O (0.5 mL), this solution was added to a stirred solution of 26 (100 mg, 0.45) in AcOH (6 mL). The mixture was then refluxed for 1 h. The reaction progress was monitored by TLC analysis (SiO2, CH2Cl2/EtOH 95:5, Rf 26 = 0.54, Rf 29 = 0.25). When the reaction was complete, the solvent was removed under vacuum, water was added to the residue and the aqueous phase was extracted with AcOEt (3 × 10 mL). The combined organic layers were dried (Na2SO4) and the solvent was removed under reduced pressure. Flash column chromatography (SiO2, CH2Cl2/EtOH 95:5, Rf = 0.25) gave 29 as red solid (23 mg, 19%): 1H-NMR (DMSO-d6) δ 8.23 (br. s, 1H), 7.89 (dd, J = 7.9, 1.6 Hz, 1H), 7.52 (dd, J = 7.8, 1.5 Hz, 1H), 7.45 (td, J = 7.8, 1.6 Hz, 1H), 7.33 (td, J = 7.6, 1.5 Hz, 1H), 7.17-7.14 (m, 2H), 7.10-7.04 (m, 2H), 5.75 (s, 1H);13C-NMR (DMSO-d6) δ 167.5, 153.9, 151.4, 148.4, 141.7, 134.2, 131.8, 131.1, 129.9, 125.2, 124.3, 123.7, 122.9, 120.8, 120.2; m.p. 227–230 °C dec.; MS (CI+): m/z = 263 [MH]+; Anal. Calcd. for C15H10N4O: C, 68.69; H, 3.84; N, 21.36; Found: C, 68.52; H, 3.95; N, 21.24.
2-Phenyl-1,8-dihydrodibenzo[b,f]imidazo[4,5-d]azepine (30). Compound 26 (300 mg, 1.34 mmol), benzaldehyde (143 mg, 1.34 mmol) and ammonium acetate (268 mg, 3.48 mmol) were dissolved in AcOH (12 mL). The reaction mixture was brought to 80 °C for 1 h under stirring. The reaction progress was monitored by TLC analysis (SiO2, CH2Cl2/EtOH 95:5, Rf 26 = 0.54, Rf 30 = 0.28). The mixture was cooled to r.t and dropped into ice/water (50 mL). The aqueous suspension was extracted with AcOEt (3 × 20 mL) and the unreacted diketone was removed by filtration. The organic phase was dried (Na2SO4) and the solvent was removed under reduced pressure. Flash column chromatography (SiO2, CH2Cl2/EtOH 95:5, Rf = 0.28) gave 30 in a pure form (115 mg, 28%): 1H-NMR (DMSO-d6) δ 12.38 (br. s, 1H), 8.09 (m, 2H), 7.62 (m, 1H), 7.49 (m, 2H), 7.41 (m, 2H), 7.22 (s, 1H), 7.06 (m, 2H), 6.94–6.84 (m, 4H);13C-NMR (DMSO-d6) δ 148.3, 147.9, 147.8, 139.2, 131.1, 129.9, 129.6, 129.5, 129.5, 129.3, 129.1, 127.7, 127.3, 126.7, 126.2, 126.2, 123.2, 123.0, 122.9, 120.8, 120.5; m.p. 102–103 °C; MS (CI+): m/z = 310 [MH]+; Anal. Calcd. for C21H15N3: C, 81.53; H, 4.89; N, 13.58; Found: C, 81.58; H, 4.95; N, 13.46.
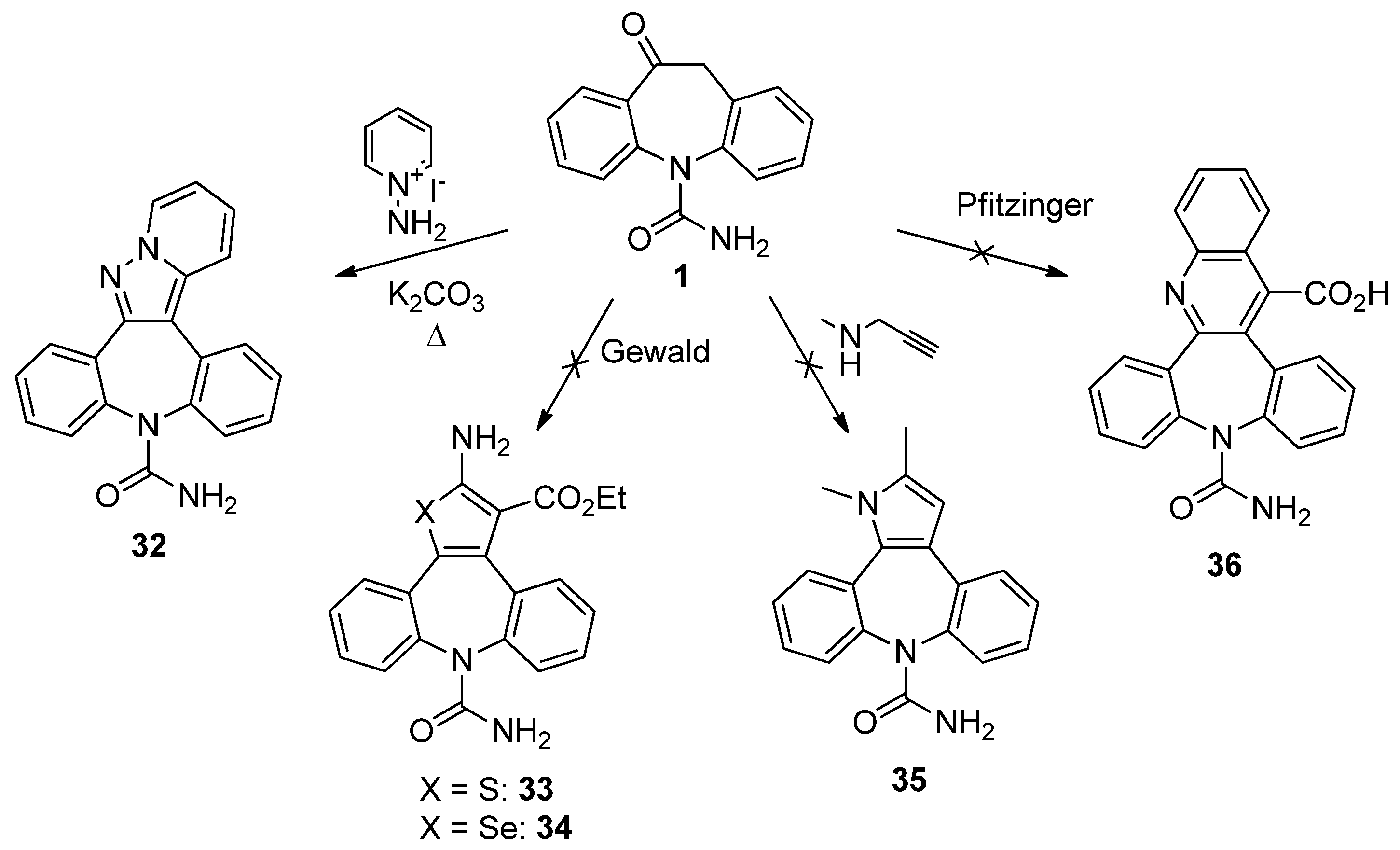
9H-Dibenzo[b,f]pyrido[1',2':1,5]pyrazolo[3,4-d]azepine-9-carboxamide (32). Oxcarbazepine (1, 300 mg, 1.19 mmol), 1-aminopyridinium iodide (317 mg, 1.43 mmol) and potassium carbonate (493 mg, 3.57 mmol) were dissolved in a mixture of H2O (20 mL) and i-PrOH (20 mL). The temperature was rise to 100 °C and the mixture was left under stirring for 2 h. The reaction progress was monitored by TLC analysis (SiO2, CH2Cl2/EtOH 95:5, Rf 1 = 0.27, Rf 32 = 0.18). Isopropanol was removed under reduced pressure and the aqueous residue was extracted with ethyl acetate (3 × 20 mL). The combined organic layers were dried (Na2SO4) and the solvent removed in vacuum. Flash column chromatography (SiO2, CH2Cl2/EtOH 9:1, Rf = 0.36) gave 32 as red solid (85 mg, 22%): 1H-NMR (DMSO-d6) δ 8.91 (d, J = 6.9 Hz, 1H), 8.07 (d, J = 9.0 Hz, 1H), 7.94 (m, 1H), 7.83 (m, 1H), 7.61–7.40 (m, 7H), 7.12 (t, J = 6.8 Hz, 1H), 5.60 (br. s, 2H); 13C-NMR (DMSO-d6) δ 157.1, 149.9, 142.7, 141.2, 138.0, 131.6, 131.5, 130.9, 130.7, 130.5, 130.2, 129.1, 128.9, 128.7, 128.4, 128.2, 126.3, 118.3, 114.7, 108.6; m.p. 254–255 °C; MS (CI+): m/z = 328 [MH]+, 284 [(M-HNCO)H]+; Anal. Calcd. for C20H14N4O: C, 73.61; H, 4.32; N, 17.17; Found: C, 73.49; H, 4.43; N, 17.10.

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}







