Virtual Screening and Structure-Based Discovery of Indole Acylguanidines as Potent β-secretase (BACE1) Inhibitors

Abstract
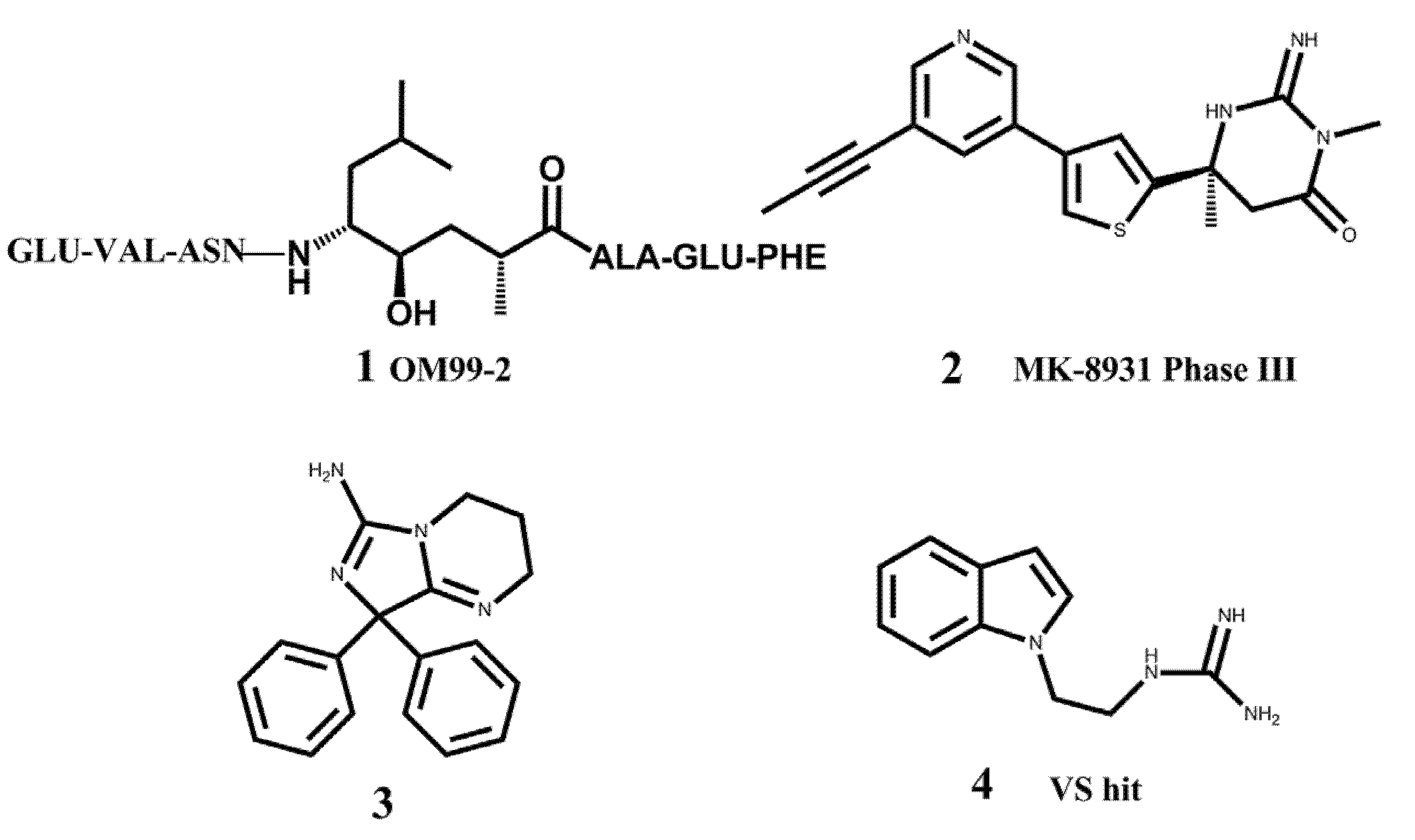
:1. Introduction
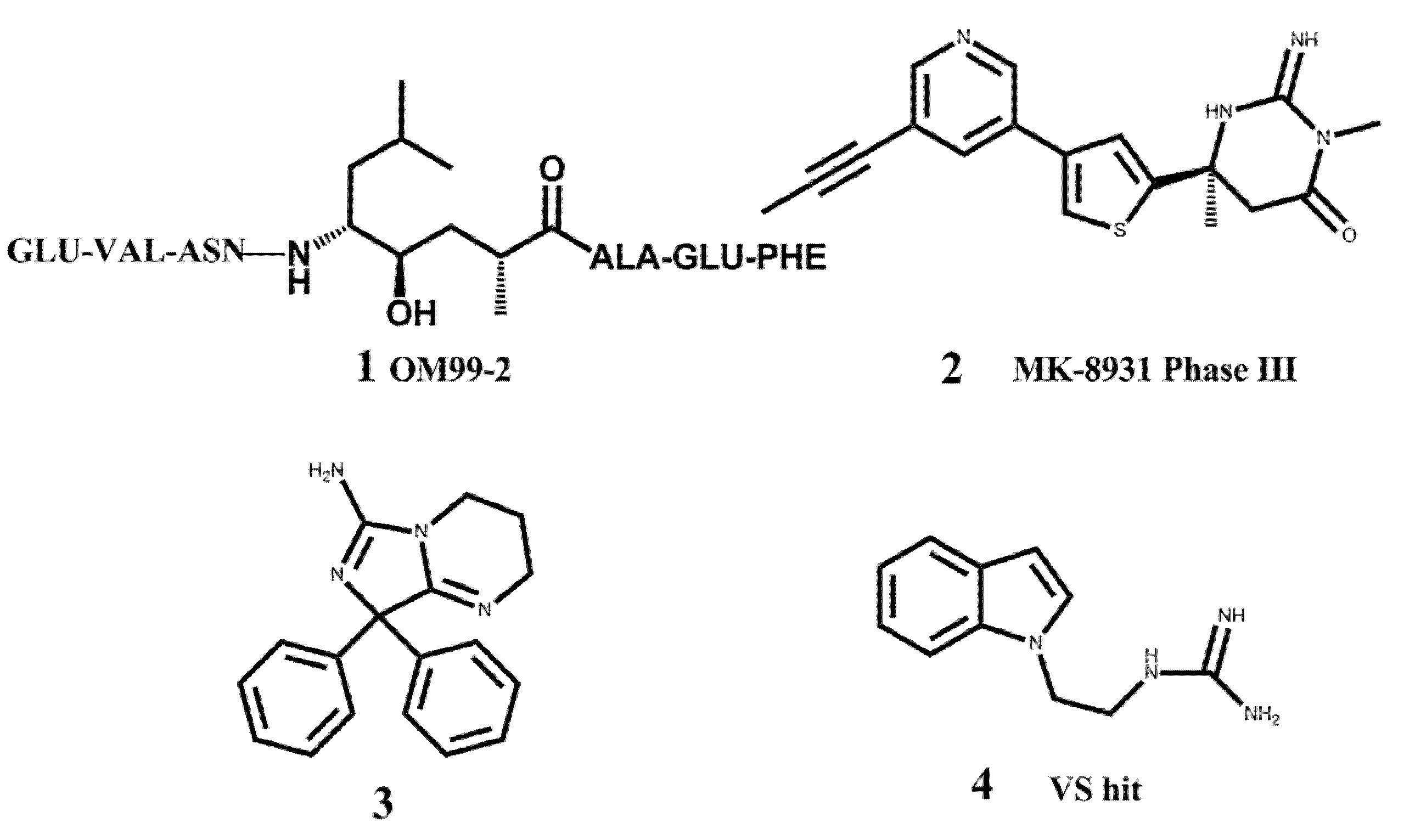
2. Results and Discussion
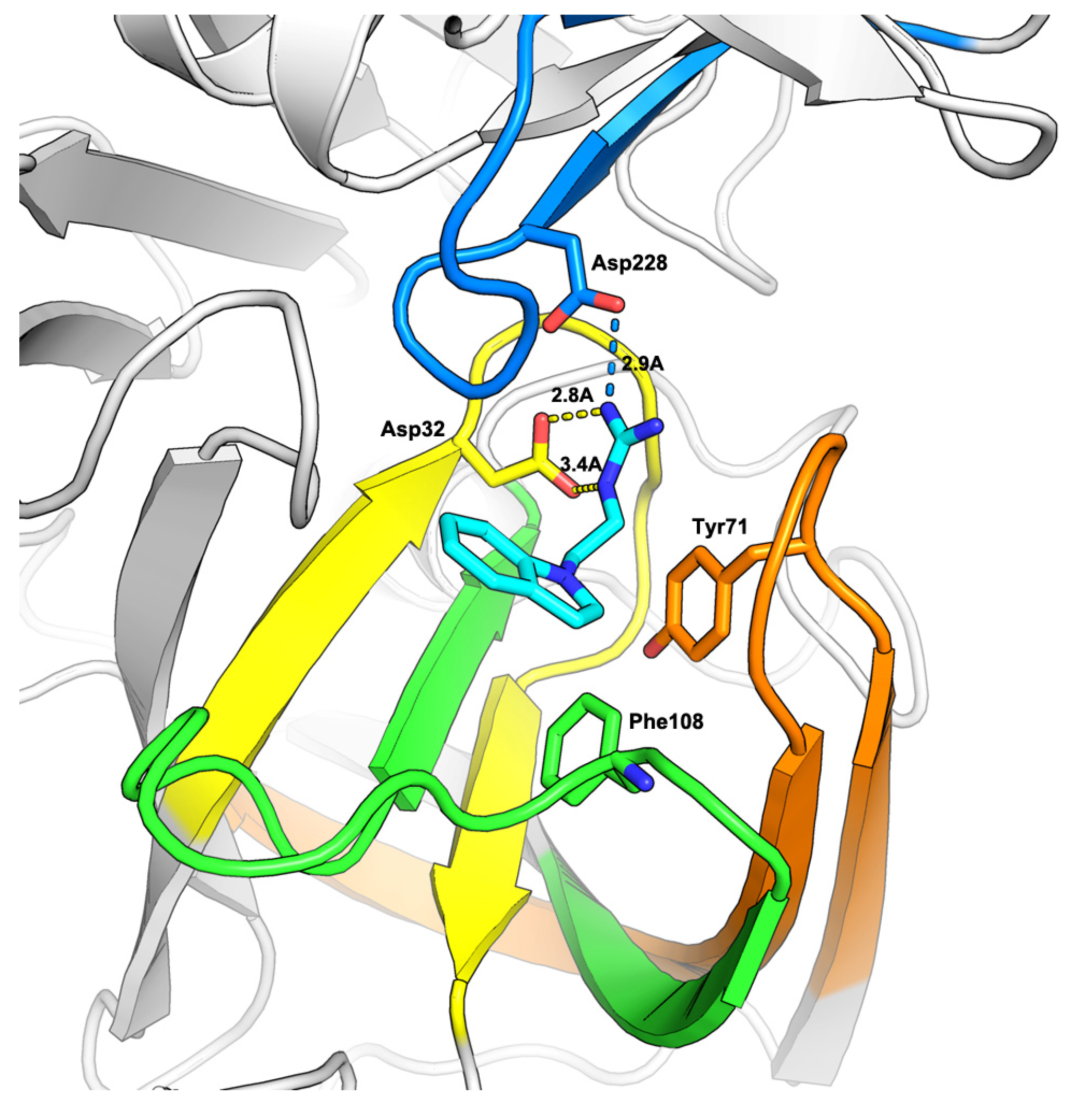
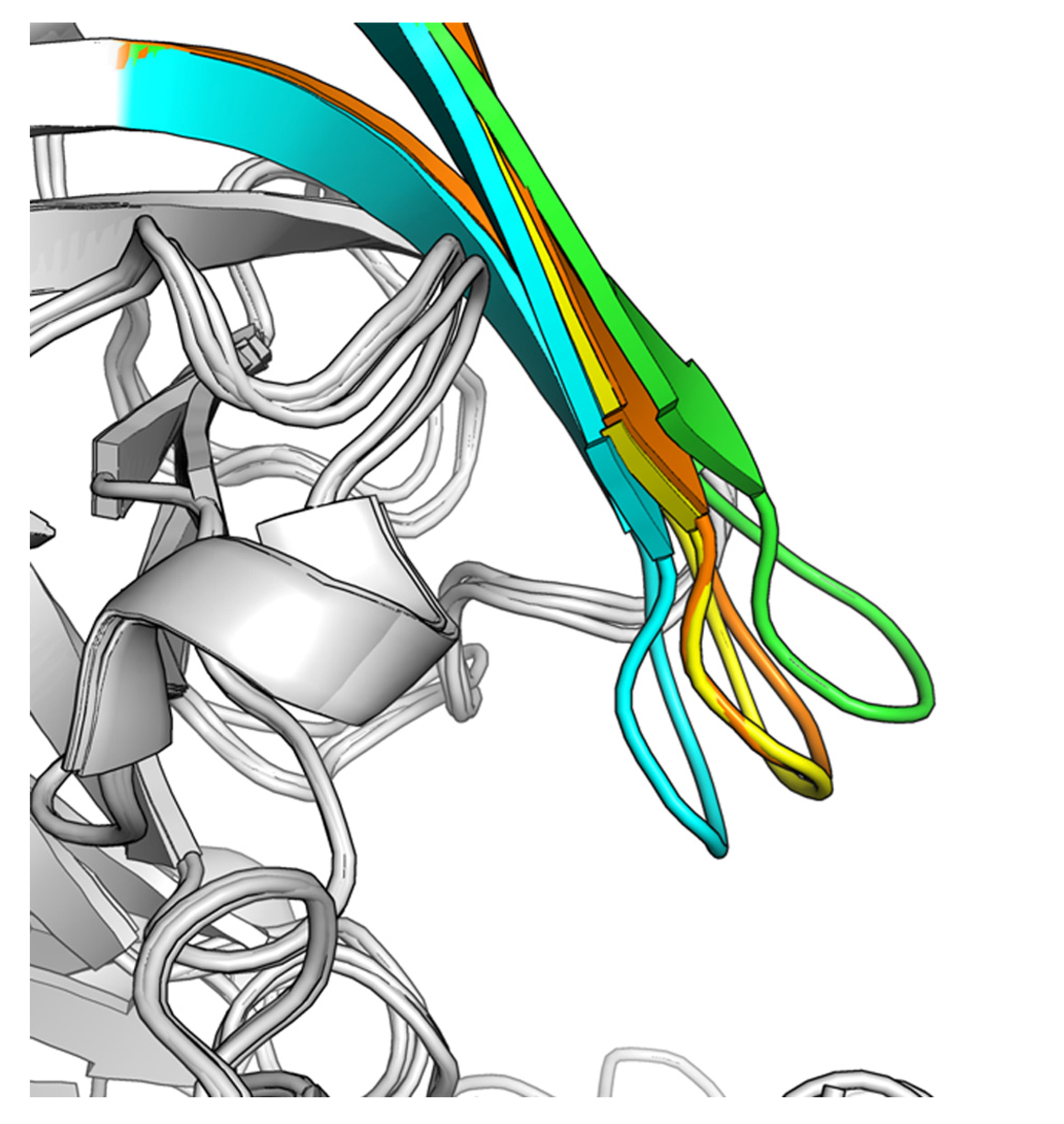
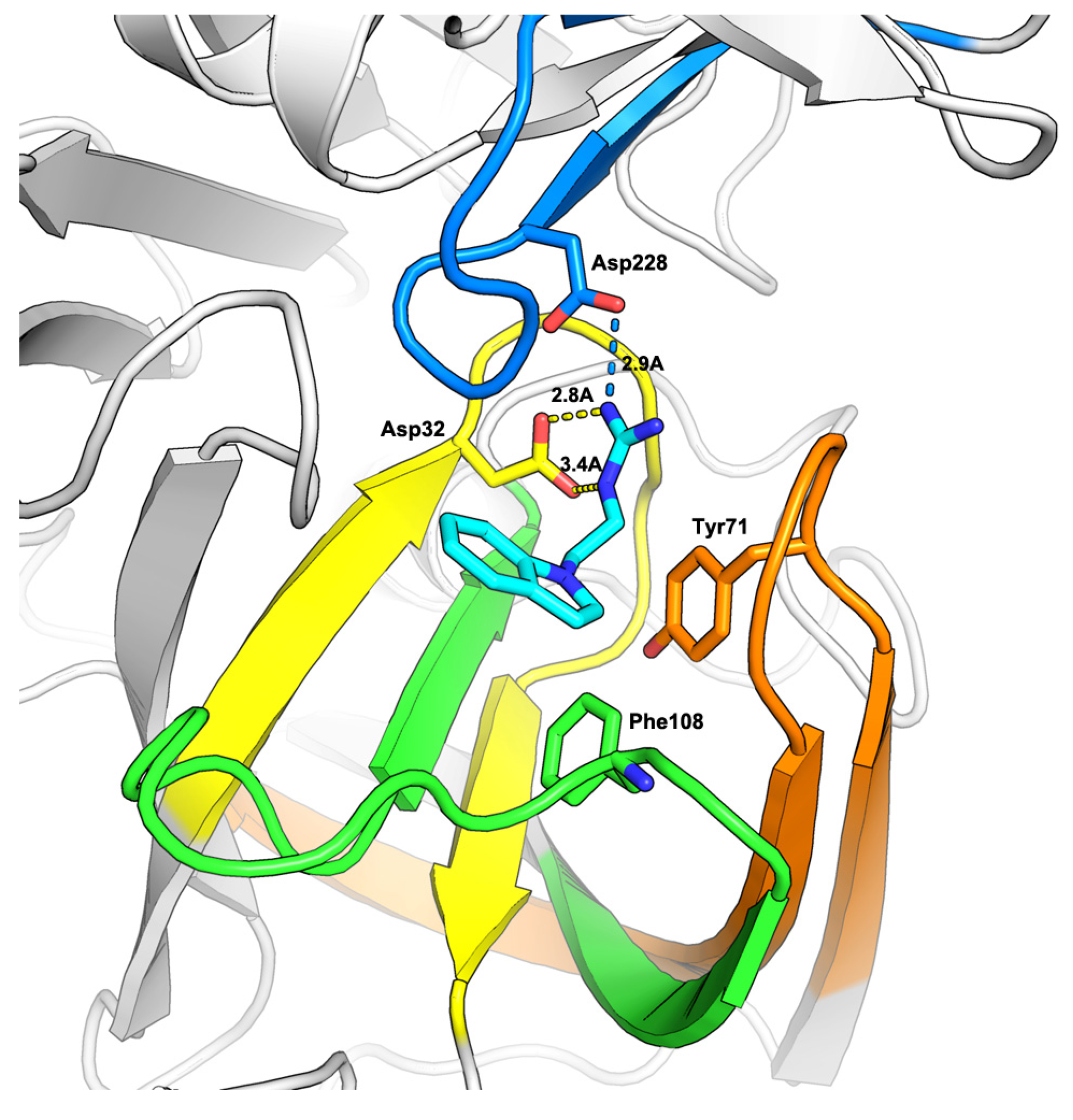
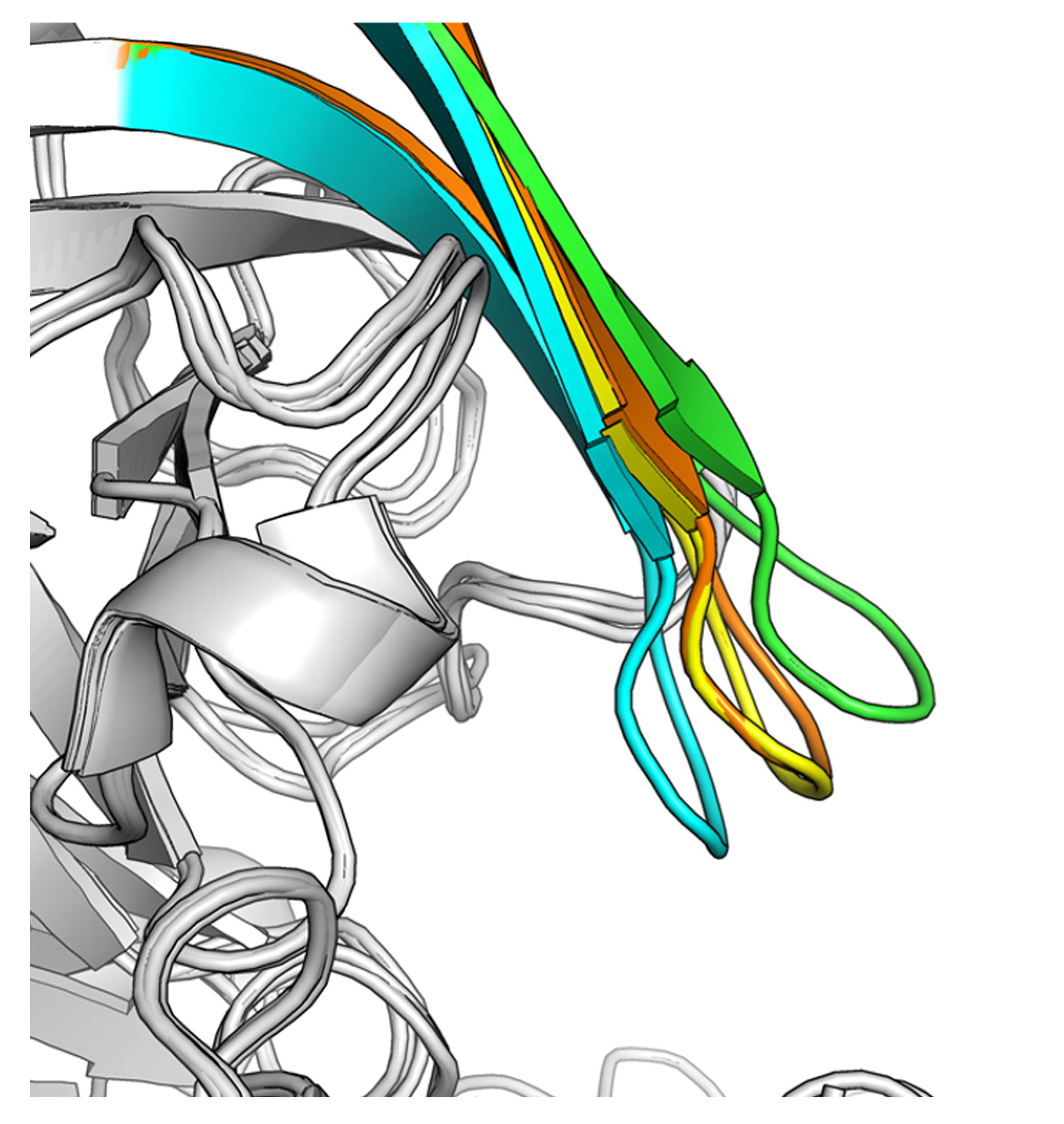
2.1. Virtual Screening
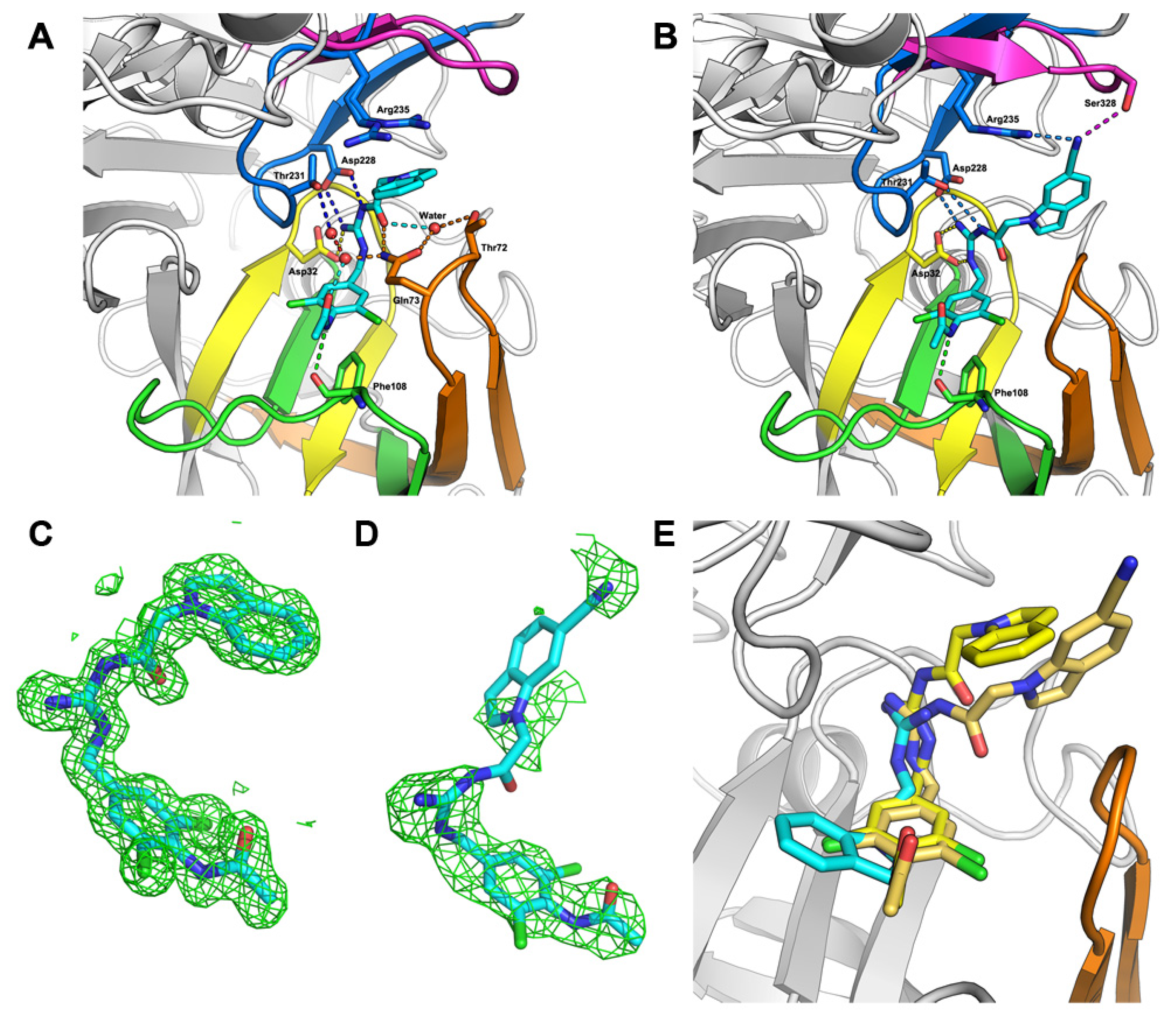
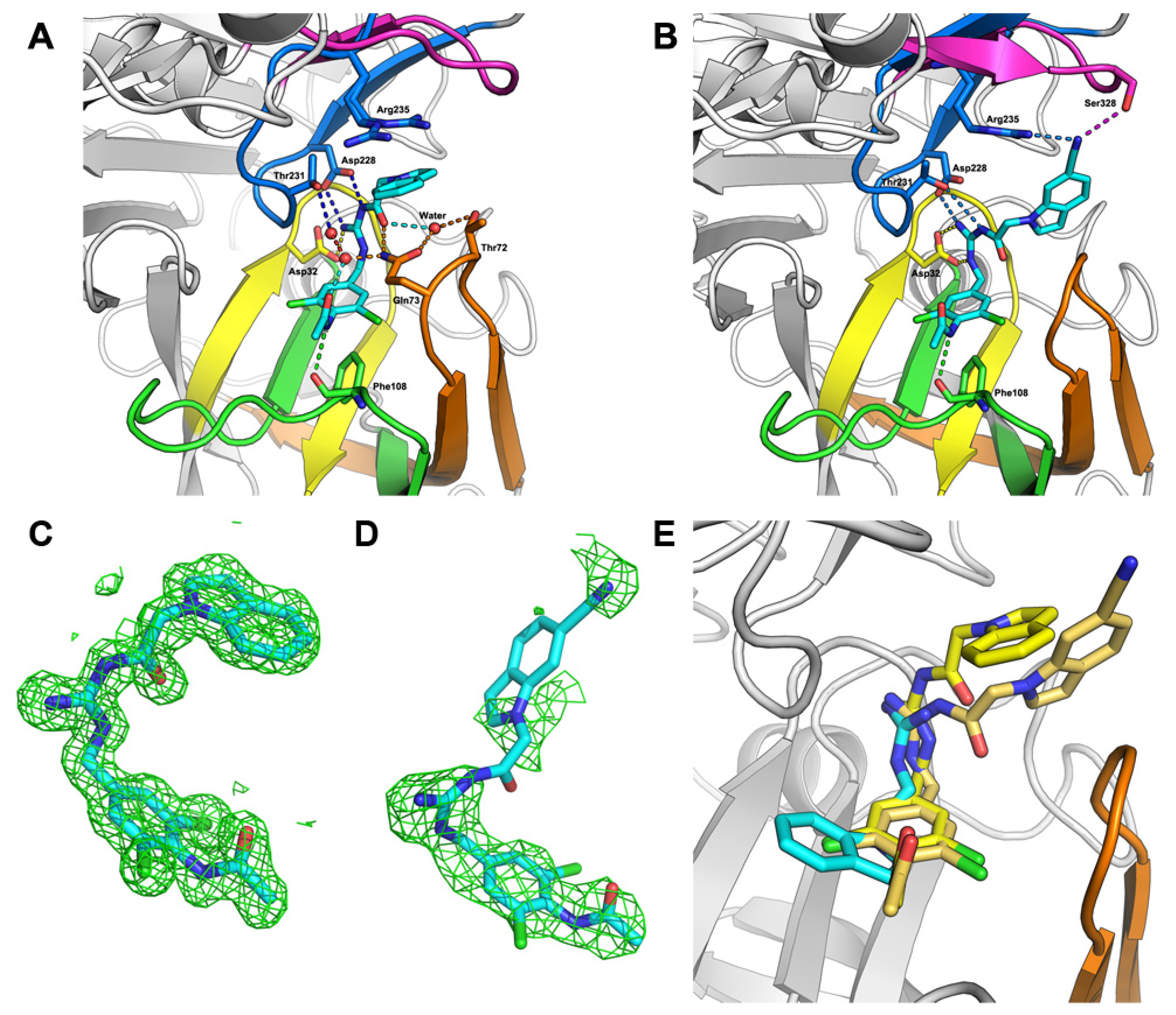
2.2. Hit-to-Lead Optimization



| Comp. | R1 | Inhibition ratio (100 μM) | IC50 (μM) |
|---|---|---|---|
| 5 | H | 77.00 | ND a |
| 12 |  | 40.69 | ND |
| 13 |  | 85.24 | ND |
| 14 |  | 58.75 | ND |
| 15 |  | 52.38 | ND |
| 16 |  | 66.51 | ND |
| 17 |  | 100 | 8.750 ± 1.075 |
| 18 |  | 35.3 | ND |
| 19 |  | 100 | 1.010 ± 0.091 |
| 20 |  | 46.8 | ND |
| 21 |  | 92.2 | 1.720 ± 0.210 |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Comp. | R2 | Inhibition % (100 µM) | IC50 (µM) |
|---|---|---|---|
| 19 |  | 100 | 1.010 ± 0.091 |
| 22 |  | 108.7 | 0.106 ± 0.013 |
| 23 |  | 100 | 0.079 ± 0.008 |
| 24 |  | 99.5 | 0.057 ± 0.004 |
| 25 |  | 100 | 0.044 ± 0.005 |
| 26 |  | 100 | 0.450 ± 0.058 |
| 27 |  | 103.2 | 0.224 ± 0.033 |
| 28 |  | 100 | 0.840 ± 0.101 |
3. Experimental
3.1. Chemistry
3.1.1. General Methods
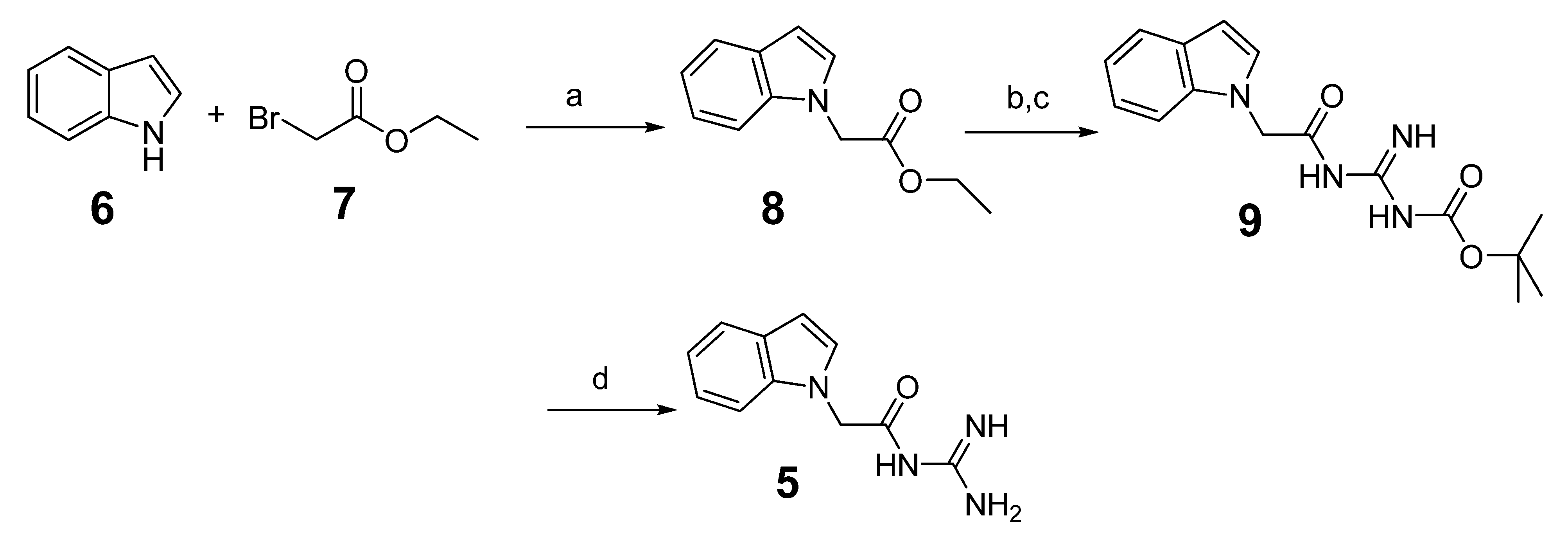
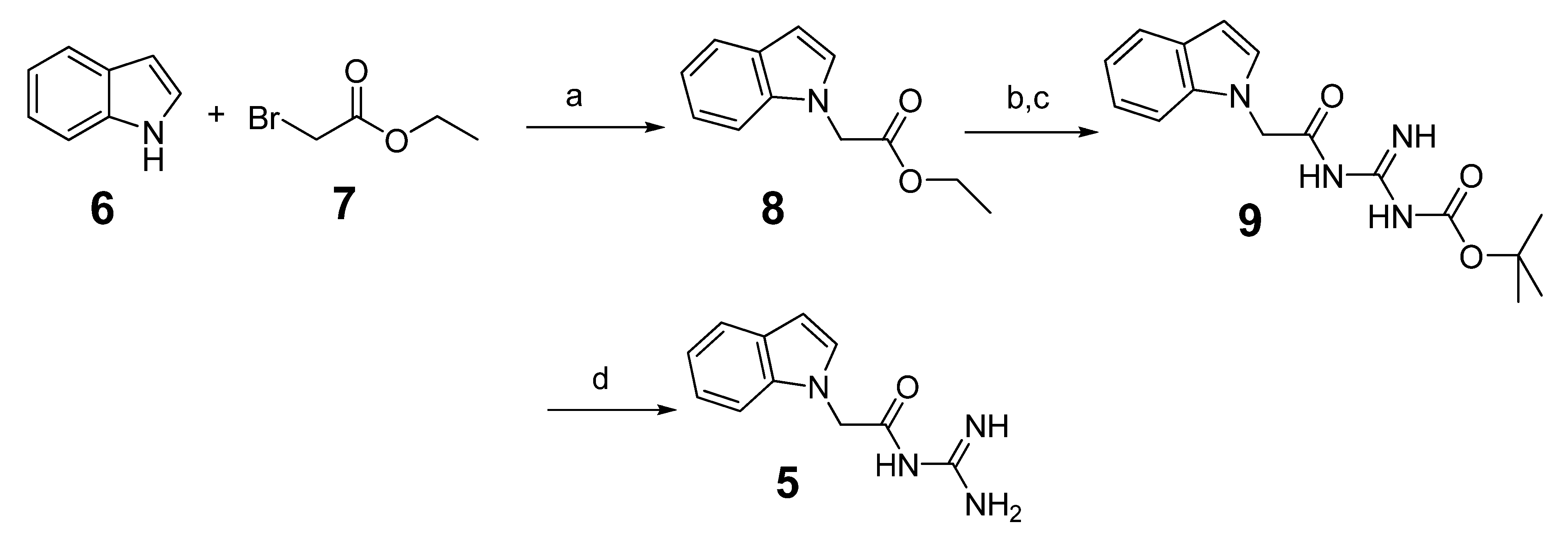
3.1.2. Preparation of N-Carbamimidoyl-2-(1H-indol-1-yl)acetamide (5)
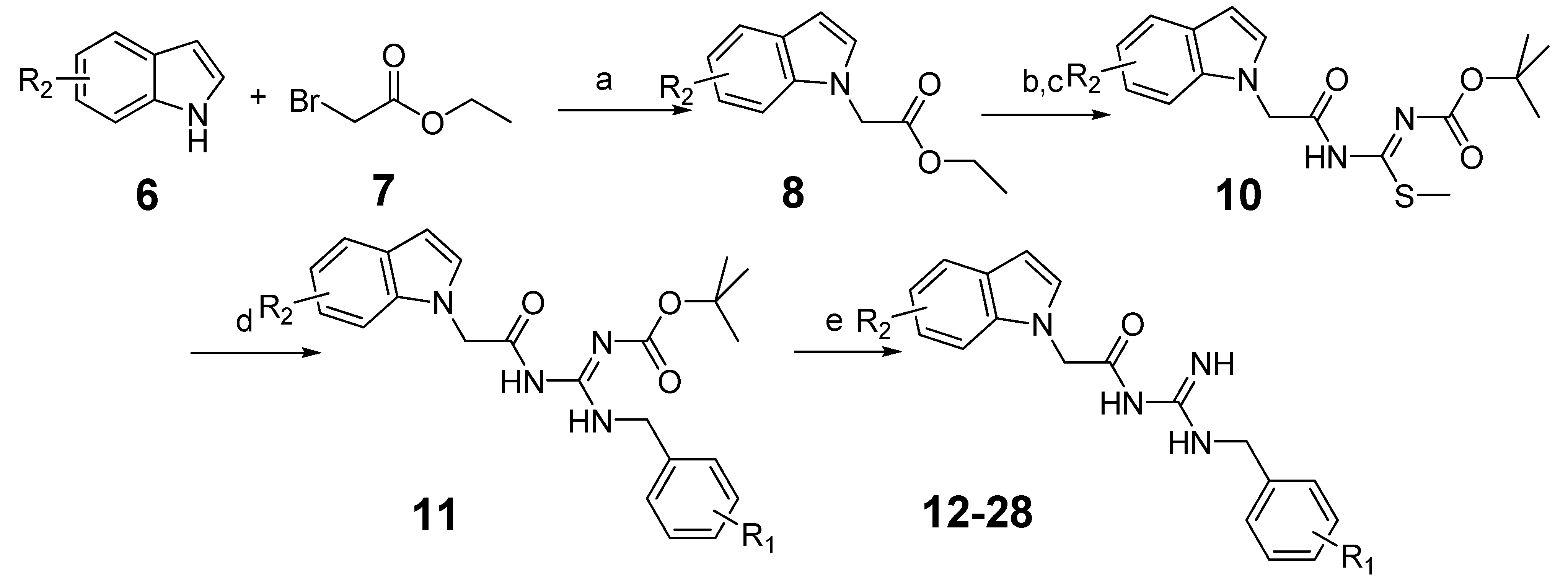
3.1.3. General Procedure for the Preparation of Indole Acylguanidine Analogs 12–28
3.2. Virtual Screening
3.3. Fluorescence Resonance Energy Transfer (FRET) Based Enzymatic Assay
3.4. BACE1 Protein Expression and Crystallization
3.4.1. Protein Purification and Crystallization
3.4.2. Structure Determination and Refinement
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Anders, W.; Linus, J.; John, B.; Martin, P.; Bengt, W. The worldwide economic impact of dementia 2010. Alzheimers Dement. 2013, 9, 1–11. [Google Scholar] [CrossRef]
- Citron, M. Strategies for disease modification in Alzheimer’s disease. Nat. Rev. Neurosci. 2004, 5, 677–685. [Google Scholar] [CrossRef]
- Hardy, J.; Selkoe, D.J. The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef]
- Vassar, R.; Bennett, B.D.; Babu-Khan, S.; Kahn, S.; Mendiaz, E.A.; Denis, P.; Teplow, D.B.; Ross, S.; Amarante, P.; Loeloff, R.; et al. Beta-secretase cleavage of Alzheimer’s amyloid precursor protein by the transmembrane aspartic protease BACE. Science 1999, 286, 735–741. [Google Scholar] [CrossRef]
- Wolfe, M.S.; Xia, W.; Ostaszewski, B.L.; Diehl, T.S.; Kimberly, W.T.; Selkoe, D.J. Two transmembrane aspartates in presenilin-1 required for presenilin endoproteolysis and γ-secretase activity. Nature 1999, 398, 513–517. [Google Scholar] [CrossRef]
- Beher, D.; Graham, S.L. Protease inhibitors as potential disease-modifying therapeutics for Alzheimer’s disease. Expert Opin. Invest. Drugs 2005, 14, 1385–1409. [Google Scholar] [CrossRef]
- Durham, T.B.; Shepherd, T.A. Progress toward the discovery and development of efficacious BACE inhibitors. Curr. Opin. Drug Discovery Dev. 2006, 9, 776–791. [Google Scholar]
- Luo, Y.; Bolon, B.; Kahn, S.; Bennett, B.D.; Babu-Khan, S.; Denis, P.; Fan, W.; Kha, H.; Zhang, J.; Gong, Y.; et al. Mice deficient in BACE1, the Alzheimer’s beta-secretase, have normal phenotype and abolished beta-amyloid generation. Nat. Neurosci. 2001, 4, 231–232. [Google Scholar] [CrossRef]
- Luo, Y.; Bolon, B.; Damore, M.A.; Fitzpatrick, D.; Liu, H.; Zhang, J.; Yan, Q.; Vassar, R.; Citron, M. BACE1 (beta-secretase) knockout mice do not acquire compensatory gene expression changes or develop neural lesions over time. Neurobiol. Dis. 2003, 14, 81–88. [Google Scholar] [CrossRef]
- Willem, M.; Garratt, A.N.; Novak, B.; Citron, M.; Kaufmann, S.; Rittger, A.; DeStrooper, B.; Saftig, P.; Birchmeier, C.; Haass, C. Control of peripheral nerve myelination by the β-secretase BACE1. Science 2006, 314, 664–666. [Google Scholar] [CrossRef]
- Wang, H.; Song, L.; Laird, F.; Wong, P.C.; Lee, H.K. BACE1 knock-outs display deficits in activity-dependent potentiation of synaptic transmission at mossy fiber to CA3 synapses in the hippocampus. J. Neurosci. 2008, 28, 8677–8681. [Google Scholar] [CrossRef]
- Hom, R.K.; Fang, L.Y.; Mamo, S.; Tung, J.S.; Guinn, A.C.; Walker, D.; Davis, D.L.; Gailunas, A.F.; Thorsett, E.D.; Sinha, S.; et al. Design and synthesis of statine-based cell permeable peptidomimetic inhibitors of human β-secretase. J. Med. Chem. 2003, 46, 1799–1802. [Google Scholar] [CrossRef]
- Stachel, S.J.; Coburn, C.A.; Steele, T.G.; Jones, K.G.; Loutzenhiser, E.F.; Gregro, A.R.; Rajapakse, H.A.; Lai, M.T.; Crouthamel, M.C.; Xu, M.; et al. Structure-based design of potent and selective cell-permeable inhibitors of human β-secretase (BACE-1). J. Med. Chem. 2004, 47, 6447–6450. [Google Scholar] [CrossRef]
- Barrow, J.C.; Rittle, K.E.; Ngo, P.L.; Selnick, H.G.; Graham, S.L.; Pitzenberger, S.M.; McGaughey, G.B.; Colussi, D.; Lai, M.T.; Huang, Q.; et al. Design and synthesis of 2,3,5-Sub-stituted Imidazolidin-4-one inhibitors of BACE-1. ChemMedChem 2007, 2, 995–999. [Google Scholar] [CrossRef]
- Charrier, N.; Clarke, B.; Cutler, L.; Demont, E.; Dingwall, C.; Dunsdon, R.; East, P.; Hawkins, J.; Howes, C.; Hussain, I.; et al. Second generation of hydroxyethylamine BACE-1 inhibitors: Optimizing potency and oral bioavailability. J. Med. Chem. 2008, 51, 3313–3317. [Google Scholar] [CrossRef]
- Hom, R.K.; Gailunas, A.F.; Mamo, S.; Fang, L.Y.; Tung, J.S.; Walker, D.E.; Davis, D.; Thorsett, E.D.; Jewett, N.E.; Moon, J.B.; et al. Design and synthesis of hydroxyethylene-based peptidomimetic inhibitors of human β-secretase. J. Med. Chem. 2004, 47, 158–164. [Google Scholar] [CrossRef]
- Hanessian, S.; Yun, H.; Hou, Y.; Yang, G.; Bayrakdarian, M.; Therrien, E.; Moites sier, N.; Roggo, S.; Veenstra, S.; Tintelnot-Blomley, M.; et al. Structure-based design, synthesis, and memapsin 2 (BACE) inhibitory activity of carbocyclic and heterocyclic peptidomimetics. J. Med. Chem. 2005, 48, 5175–5190. [Google Scholar] [CrossRef]
- Hu, J.; Cwi, C.L.; Smiley, D.L.; Timm, D.; Erickson, J.A.; McGee, J.E.; Yang, H.C.; Mendel, D.; May, P.C.; Shapiro, M.; et al. Design and synthesis of statine-containing BACE inhibitors. Bioorg. Med. Chem. Lett. 2003, 13, 134335–134339. [Google Scholar]
- Back, M.; Nhylen, J.; Kvarnstrom, I.; Apelgren, S.; Borkakoti, N.; Jansson, K.; Lindberg, J.; Nystrom, S.; Hallberg, A.; Rosenquist, A.; et al. Design, synthesis and SAR of potent statine-based BACE-1 inhibitors: Exploration of P1 phenoxy and benzyloxy residues. Bioorg. Med. Chem. 2008, 16, 9471–9486. [Google Scholar] [CrossRef]
- Clark, D.E. Rapid calculation of polar molecular surface area and its application to the prediction of transport phenomena. 2. Prediction of blood -brain barrier penetration. J. Pharm. Sci. 1999, 88, 815–821. [Google Scholar] [CrossRef]
- Pardridge, W.M. Blood-brain delivery. Drug Discovery Today 2007, 12, 54–61. [Google Scholar]
- Godemann, R.; Madden, J.; Krämer, J.; Smith, M.; Fritz, U.; Hesterkamp, T.; Barker, J.; Höppner, S.; Hallett, D.; Cesura, A.; Ebneth, A.; Kemp, J. Fragment-based discovery of BACE1 inhibitors using functional assays. Biochemistry 2009, 48, 10743–10751. [Google Scholar]
- Congreve, M.; Aharony, D.; Albert, J.; Callaghan, O.; Campbell, J.; Carr, R.A.; Chessari, G.; Cowan, S.; Edwards, P.D.; Frederickson, M.; et al. Application of fragment screening by X-ray crystallography to the discovery of aminopyridines as inhibitors of beta-secretase. J. Med. Chem. 2007, 50, 1124–1132. [Google Scholar] [CrossRef]
- Geschwindner, S.; Olsson, L.L.; Albert, J.S.; Deinum, J.; Edwards, P.D.; Beer, T.; Folmer, R.H. Discovery of a novel warhead against beta-secretase through fragment-based lead generation. J. Med. Chem. 2007, 50, 5903–5911. [Google Scholar] [CrossRef]
- Wang, Y.S.; Strickland, C.; Voigt, J.H.; Kennedy, M.E.; Beyer, B.M.; Senior, M.M.; Smith, E.M.; Nechuta, T.L.; Madison, V.S.; Czarniecki, M.; et al. Application of fragment-based NMR screening, X-ray crystallography, Structure-based design, and focused chemical library design to identify novel microM leads for the development of nM BACE-1 (beta-site APP cleaving enzyme 1) inhibitors. J. Med. Chem. 2010, 53, 942–950. [Google Scholar]
- Baker, M. Fragment-based lead discovery grows up. Nat. Rev. Drug Discov. 2013, 12, 5–7. [Google Scholar]
- Hong, L.; Koelsch, G.; Lin, X.; Wu, S.; Terzyan, S.; Ghosh, A.K.; Zhang, X.C.; Tang, J. Structure of the protease domain of memapsin 2 (beta-secretase) complexed with inhibitor. Science 2000, 290, 150–153. [Google Scholar]
- Malamas, M.S.; Erdei, J.; Gunawan, I.; Barnes, K.; Johnson, M.; Hui, Y.; Turner, J.; Hu, Y.; Wagner, E.; Fan, K.; et al. Aminoimidazoles as potent and selective human beta-secretase (BACE1) inhibitors. J. Med. Chem. 2009, 52, 6314–6323. [Google Scholar] [CrossRef]
- Cole, D.C.; Manas, E.S.; Stock, J.R.; Condon, J.S.; Jennings, L.D.; Aulabaugh, A.; Chopra, R.; Cowling, R.; Ellingboe, J.W.; Fan, K.Y.; et al. Acylguanidines as small-molecule beta-secretase inhibitors. J. Med. Chem. 2006, 49, 6158–6161. [Google Scholar] [CrossRef]
- Gerritz, S.W.; Zhai, W.; Shi, S.; Zhu, S.; Toyn, J.H.; Meredith, J.E., Jr.; Iben, L.G.; Burton, C.R.; Albright, C.F.; Good, A.C.; et al. Acyl guanidine inhibitors of β-secretase (BACE-1): optimization of a micromolar hit to a nanomolar lead via iterative solid- and solution-phase library synthesis. J. Med. Chem. 2012, 55, 9208–9223. [Google Scholar] [CrossRef]
- McDonald, I.M.; Black, J.W.; Buck, I.M.; Dunstone, D.J.; Griffin, E.P.; Harper, E.A.; Hull, R.A.D.; Kalindjian, S.B.; Lilley, E.J.; et al. Optimization of 1,3,4-benzotriazepine-based CCK2 antagonists to obtain potent, orally active inhibitors of gastrin-mediated gastric acid secretion. J. Med. Chem. 2007, 50, 3101–3112. [Google Scholar] [CrossRef]
- Luker, T.; Bonnert, R.; Brough, S.; Cook, A.R.; Dickinson, M.R.; Mohammeda, R.T.; Sanganee, H.J.; Teague, S.; Thom, S.; Dougall, I.; et al. Substituted indole-1-acetic acids as potent and selective CRTh2 antagonists—Discovery of AZD1981. Bioorg. Med. Chem. Lett. 2011, 21, 6288–6292. [Google Scholar] [CrossRef]
- Zhang, Z.S.; Larson, E.T.; Merritt, E.A.; Verlinde, C.L.M.J.; Fan, E.; Ojo, K.K.; He, P.Q.; Van, V.; Wesley, C.; et al. Benzoylbenzimidazole-based selective inhibitors targeting Cryptosporidium parvum and Toxoplasma gondii calcium-dependent protein kinase-1. Bioorg. Med. Chem. Lett. 2012, 22, 5264–5267. [Google Scholar] [CrossRef]
- Yu, M.; Pochapsky, S.; Snider, B. Synthesis of (±)-Bistellettadine A. Org. Lett. 2010, 12, 828–831. [Google Scholar] [CrossRef]
- Andreas, S.; Burkhard, K. Binding of a hemoregulatory tetrapeptide by a bis-guanidinium crown ether. Tetrahedron 2010, 66, 6019–6025. [Google Scholar] [CrossRef]
- Stefan, W.; Max, K.; Günther, B.; Armin, B.; Burkhard, K. N-Acyl-argininamides as NPY Y1 receptor antagonists: Influence of structurally diverse acyl substituents on stability and affinity. Bioorg. Med. Chem. 2010, 18, 6292–6304. [Google Scholar] [CrossRef]
- ZINC database. Available online: Available online: http:/zic.docking.org (accessed on 10 May 2012).
- ChemAxon Chemoinformatics Software, version 5.6. Available online: Available online: http://www.chemaxon.com/ (accessed on 7 September 2011).
- Schordinger Software, version 8.0. Available online: Available online: http://www.schrodinger.com/ (accessed on 10 November 2007).
- Xu, Y.; Li, M.J.; Greenblatt, H.; Chen, W.; Paz, A.; Dym, O.; Peleg, Y.; Chen, T.; Shen, X.; He, J.; et al. Flexibility of the flap in the active site of BACE1 as revealed by crystal structures and molecular dynamics simulations. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2012, 68, 13–25. [Google Scholar] [CrossRef]
- Kabsch, W. XDS. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010, 66, 125–132. [Google Scholar] [CrossRef]
- Collaborative-Computational-Project-Number-4. The CCP4 suite: Programs for protein crystallography. Acta Crystallogr., Sect. D: Biol. Crystallogr. 1994, 50, 760–763. [Google Scholar] [CrossRef]
- Adams, P.D.; Grosse-Kunstleve, R.W.; Hung, L.W.; Ioerger, T.R.; McCoy, A.J.; Moriarty, N.W.; Read, R.J.; Sacchettini, J.C.; Sauter, N.K.; Terwilliger, T.C. PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2002, 58, 1948–1954. [Google Scholar] [CrossRef]
- Emsley, P.; Cowtan, K. Coot: Model-building tools for molecular graphics. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2004, 60, 2126–2132. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds 5–28 are available from the authors.
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Zou, Y.; Li, L.; Chen, W.; Chen, T.; Ma, L.; Wang, X.; Xiong, B.; Xu, Y.; Shen, J. Virtual Screening and Structure-Based Discovery of Indole Acylguanidines as Potent β-secretase (BACE1) Inhibitors. Molecules 2013, 18, 5706-5722. https://doi.org/10.3390/molecules18055706
Zou Y, Li L, Chen W, Chen T, Ma L, Wang X, Xiong B, Xu Y, Shen J. Virtual Screening and Structure-Based Discovery of Indole Acylguanidines as Potent β-secretase (BACE1) Inhibitors. Molecules. 2013; 18(5):5706-5722. https://doi.org/10.3390/molecules18055706
Chicago/Turabian StyleZou, Yiquan, Li Li, Wuyan Chen, Tiantian Chen, Lanping Ma, Xin Wang, Bing Xiong, Yechun Xu, and Jingkang Shen. 2013. "Virtual Screening and Structure-Based Discovery of Indole Acylguanidines as Potent β-secretase (BACE1) Inhibitors" Molecules 18, no. 5: 5706-5722. https://doi.org/10.3390/molecules18055706