Celastrol Induces Apoptosis in Gefitinib-Resistant Non-Small Cell Lung Cancer Cells via Caspases-Dependent Pathways and Hsp90 Client Protein Degradation

Abstract
:1. Introduction
2. Results and Discussion
2.1. Celastrol is More Effective in Decreasing Cell Viability of Gefitinib-Resistant H1650 and H1975 Cells


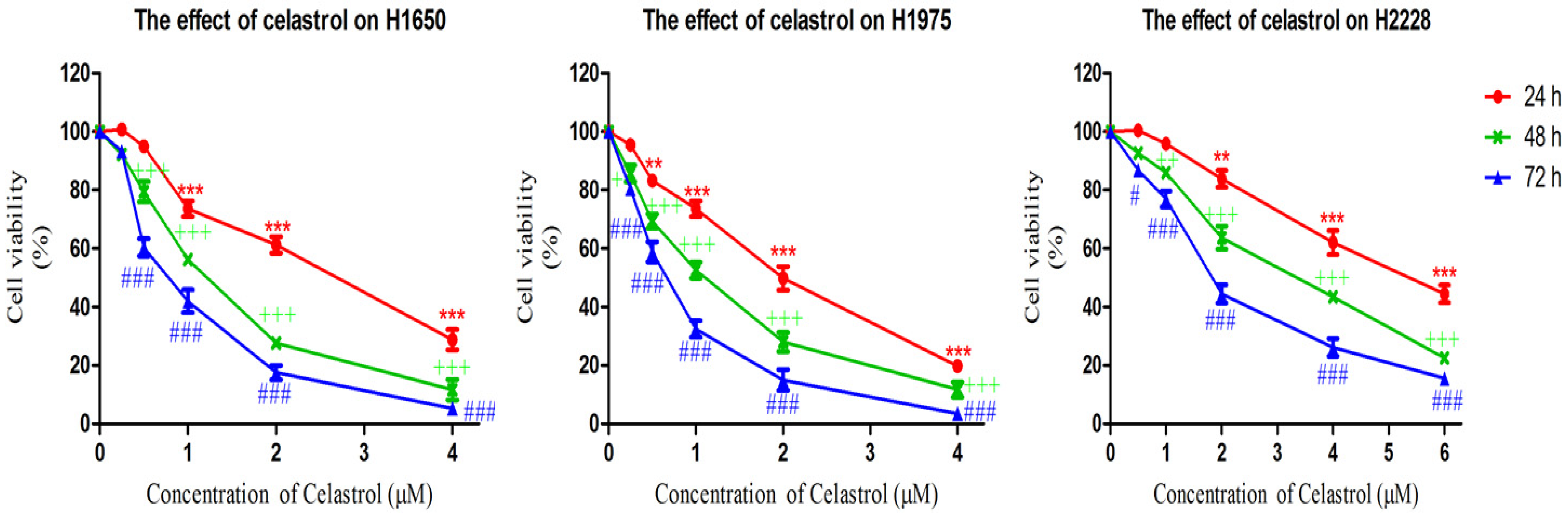{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell line | IC50 Value (μM) | ||
|---|---|---|---|
| 24 h | 48 h | 72 h | |
| H1650 | 2.78 ± 0.77 | 1.18 ± 0.32 | 0.79 ± 0.27 |
| H1975 | 2.03 ± 0.48 | 1.00 ± 0.18 | 0.60 ± 0.15 |
| H2228 | >6 | 3.28 ± 0.74 | 1.95 ± 0.45 |
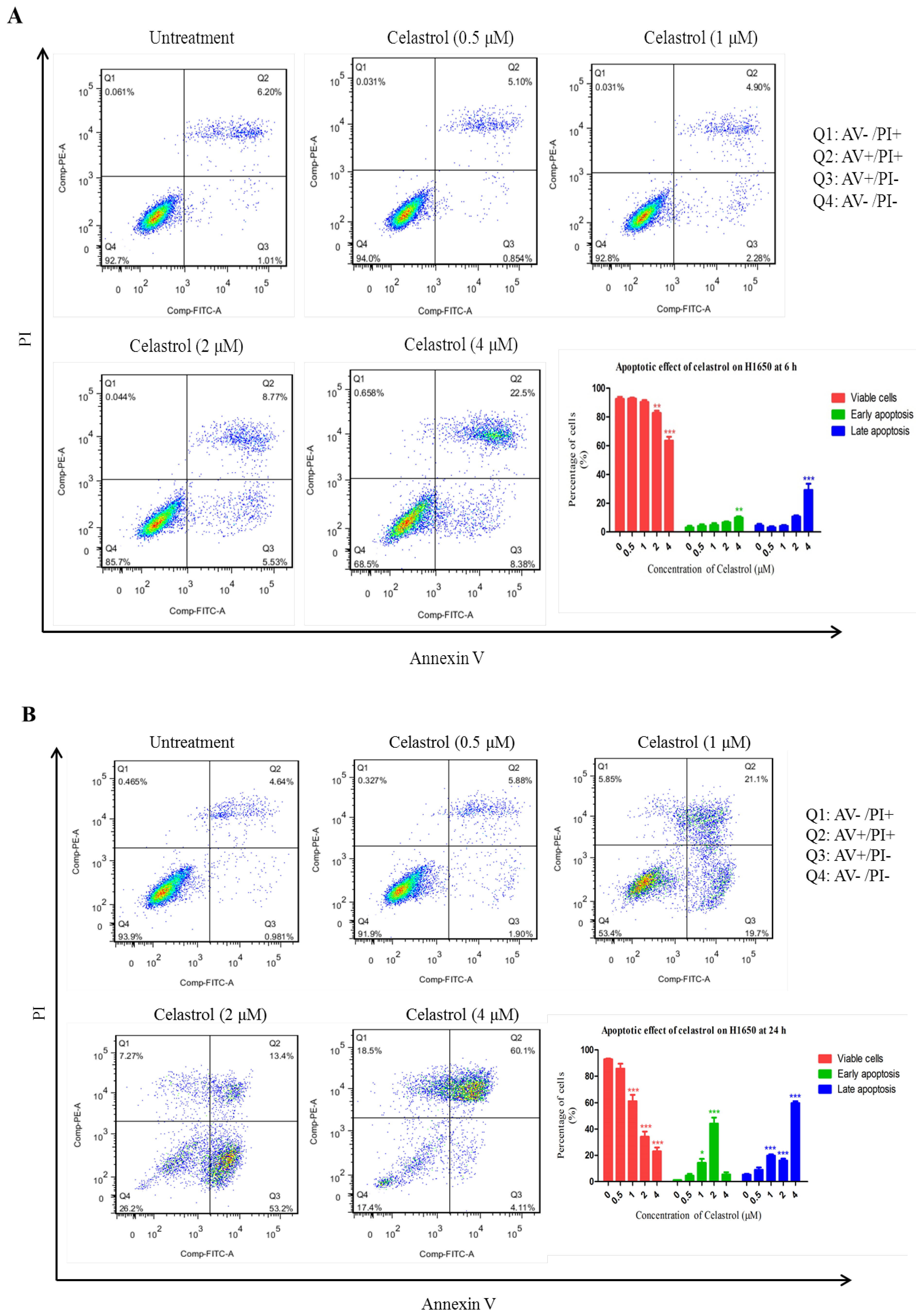
2.2. Celastrol Induced Apoptosis in H1650

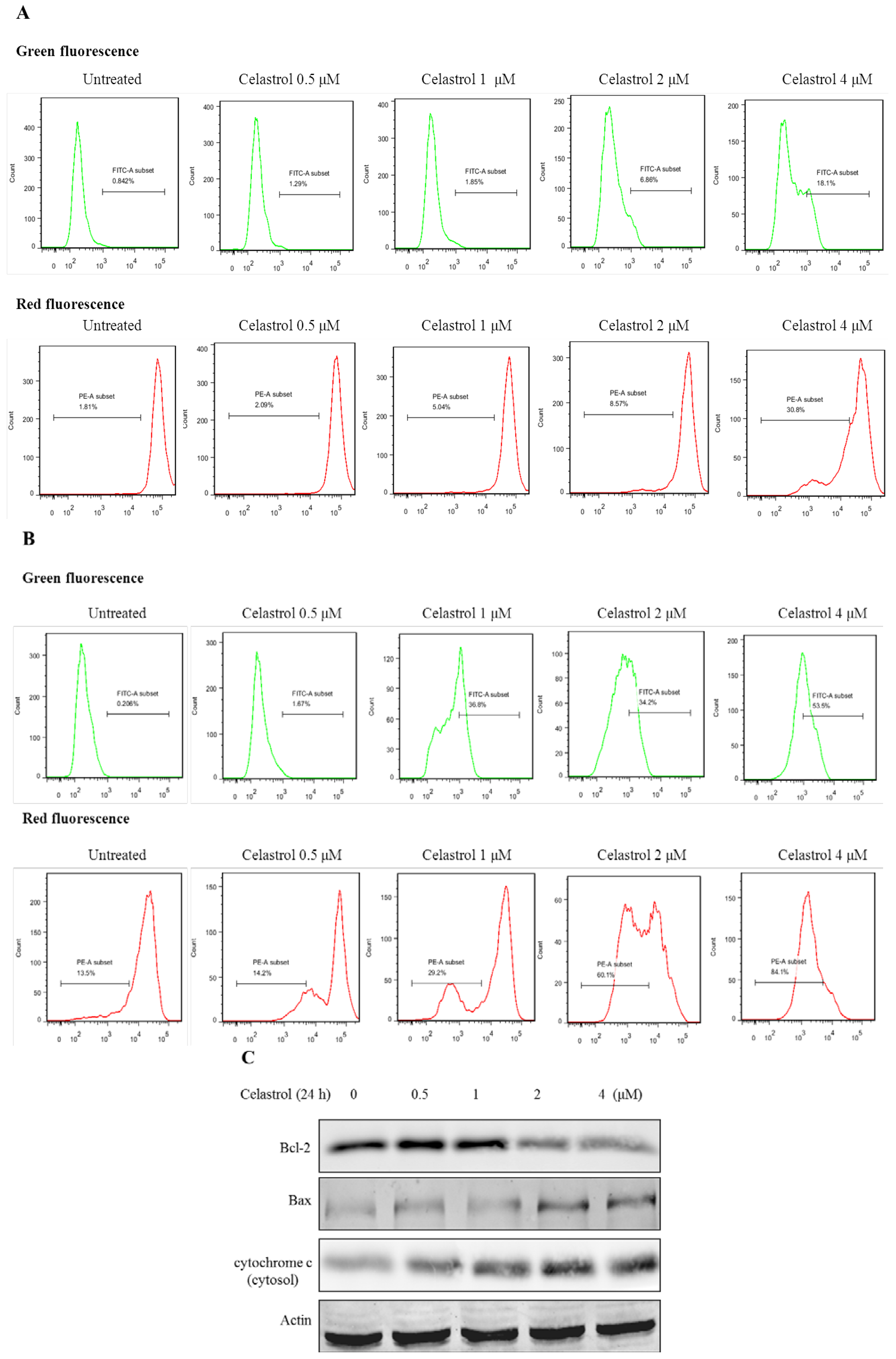
2.3. Celastrol Induced Apoptosis via Mitochondria-Dependent Pathway

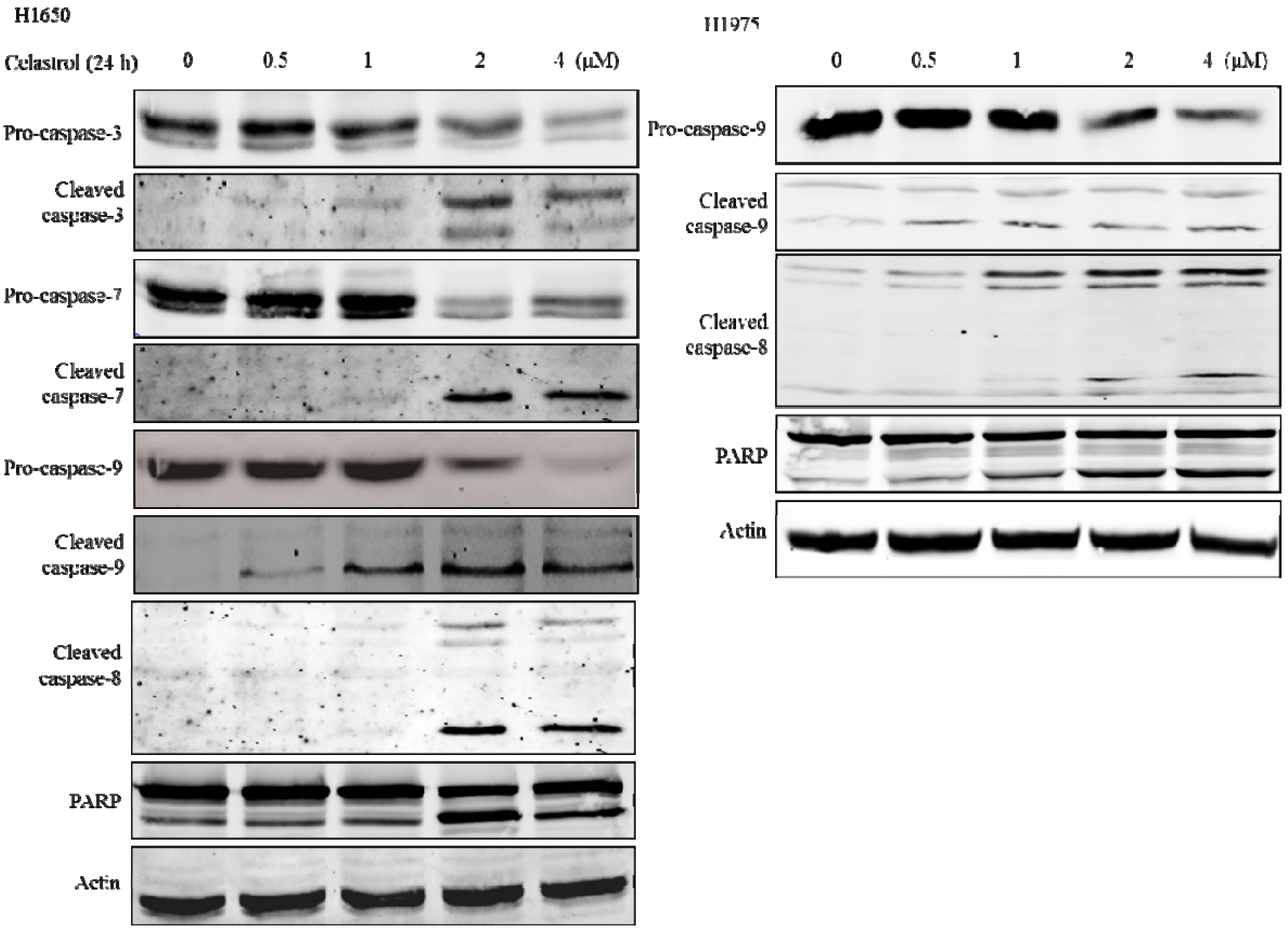
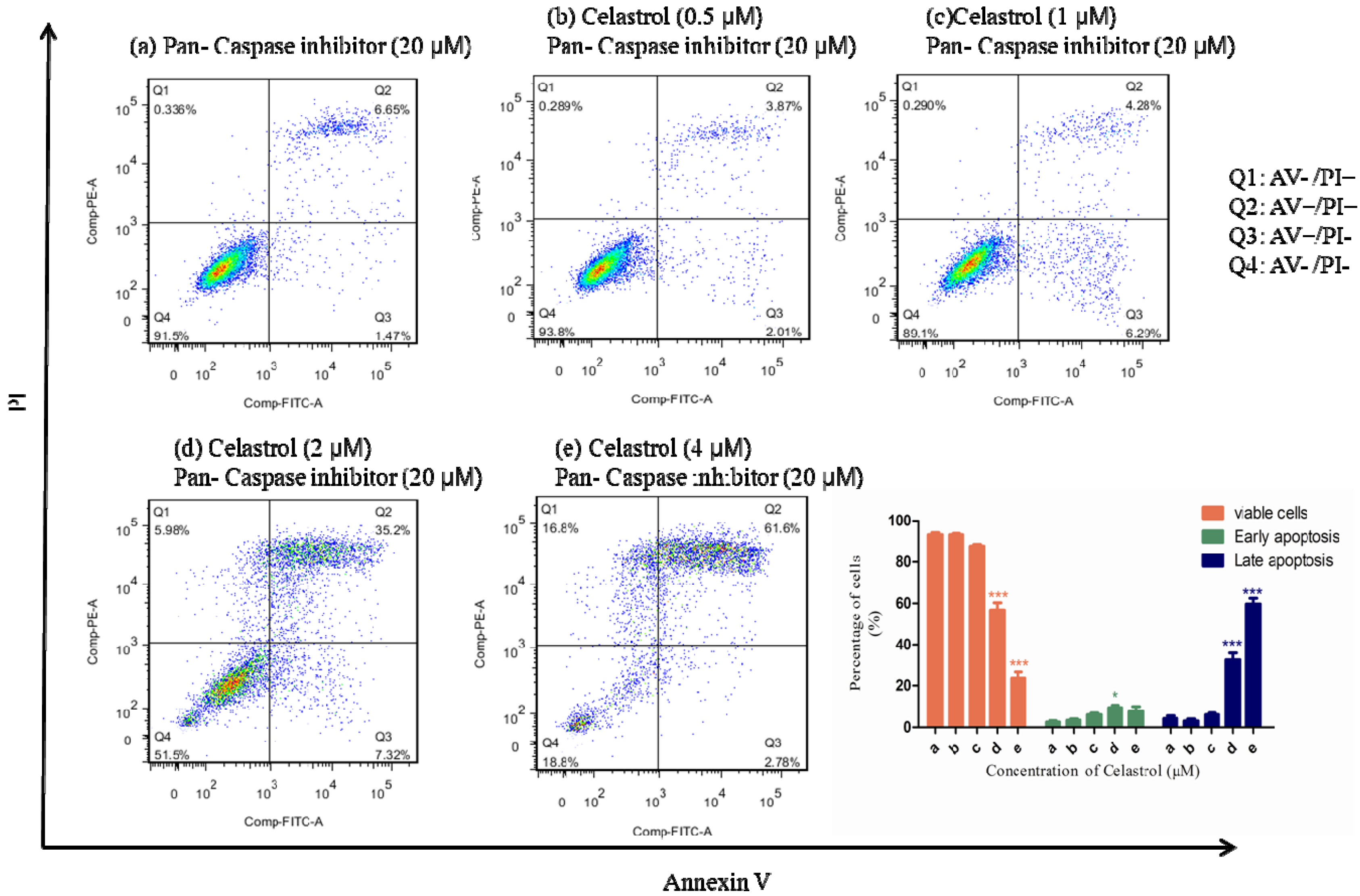
2.4. Caspases were Significantly Activated by the Treatment of Celastrol and Inhibition of Caspases could Partially Attenuate the Apoptotic Effect Induced by Celastrol


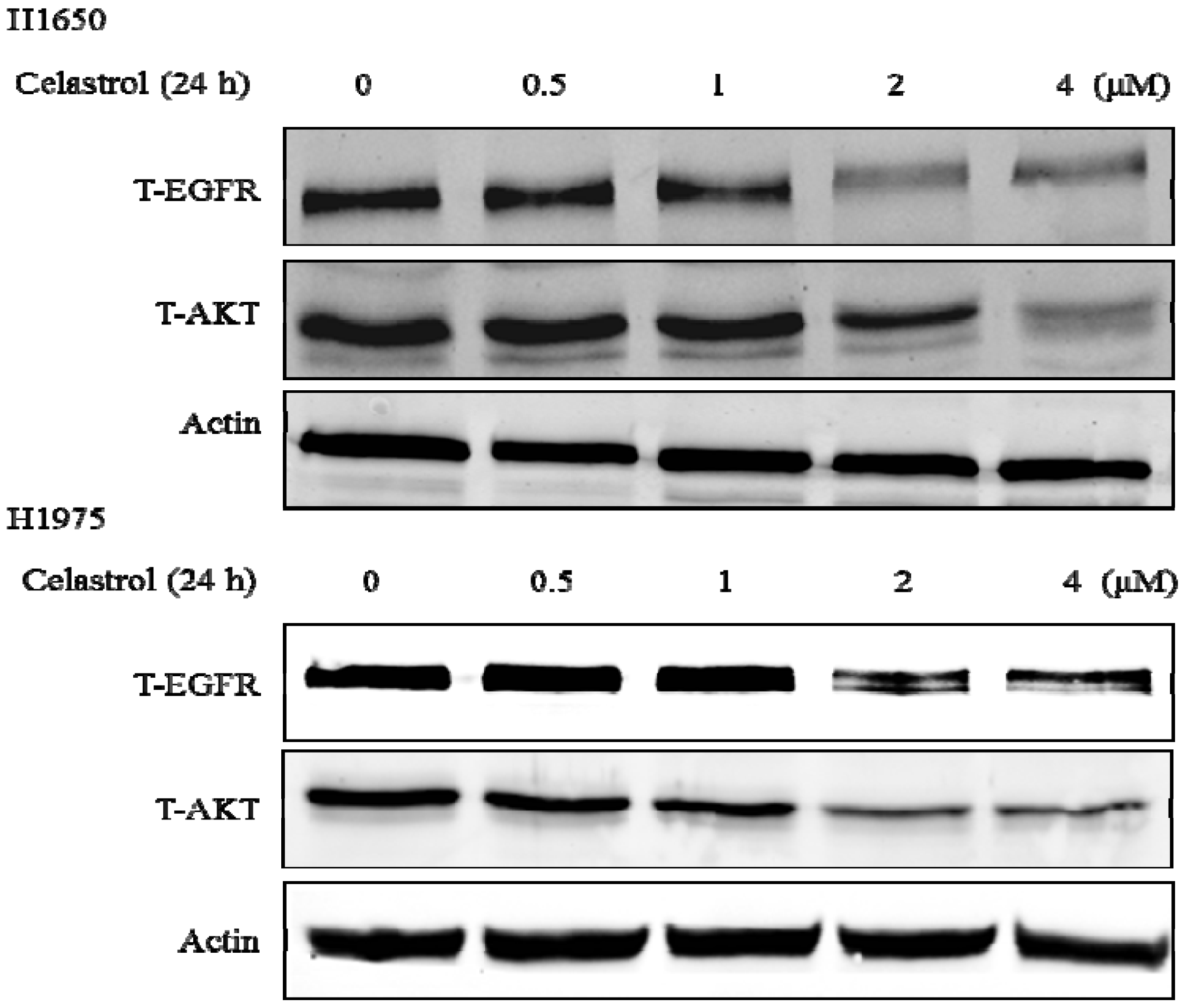
2.5. Celastrol Down-Regulated Hsp90 Client Proteins

3. Experimental
3.1. Materials
3.2. Cell Lines and Cell Culture
3.3. 3-(4, 5-Dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide (MTT) Assay
3.4. Assessment of Apoptosis Levels by Annexin V/PI Staining
3.5. Analysis of Mitochondrial Membrane Potential
3.6. Western Blot Analysis
3.7. Statistical Analysis
4. Conclusions
Acknowledgments
Conflictts of Interest
References
- Sung, B.; Park, B.; Yadav, V.R.; Aggarwal, B.B. Celastrol, a triterpene, enhances TRAIL-induced apoptosis through the down-regulation of cell survival proteins and up-regulation of death receptors. J. Biol. Chem. 2010, 285, 11498–11507. [Google Scholar] [CrossRef]
- Tao, X.; Cush, J.J.; Garret, M.; Lipsky, P.E. A phase I study of ethyl acetate extract of the chinese antirheumatic herb Tripterygium wilfordii hook F in rheumatoid arthritis. J. Rheumatol. 2001, 28, 2160–2167. [Google Scholar]
- Li, H.; Zhang, Y.Y.; Huang, X.Y.; Sun, Y.N.; Jia, Y.F.; Li, D. Beneficial effect of tripterine on systemic lupus erythematosus induced by active chromatin in BALB/c mice. Eur. J. Pharmacol. 2005, 512, 231–237. [Google Scholar] [CrossRef]
- Kiaei, M.; Kipiani, K.; Petri, S.; Chen, J.; Calingasan, N.Y.; Beal, M.F. Celastrol blocks neuronal cell death and extends life in transgenic mouse model of amyotrophic lateral sclerosis. Neurodegener Dis. 2005, 2, 246–254. [Google Scholar] [CrossRef]
- Allison, A.C.; Cacabelos, R.; Lombardi, V.R.; Alvarez, X.A.; Vigo, C. Celastrol, a potent antioxidant and anti-inflammatory drug, as a possible treatment for Alzheimer’s disease. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2001, 25, 1341–1357. [Google Scholar] [CrossRef]
- Kim, D.Y.; Park, J.W.; Jeoung, D.; Ro, J.Y. Celastrol suppresses allergen-induced airway inflammation in a mouse allergic asthma model. Eur. J. Pharmacol. 2009, 612, 98–105. [Google Scholar] [CrossRef]
- Pang, X.; Yi, Z.; Zhang, J.; Lu, B.; Sung, B.; Qu, W.; Aggarwal, B.B.; Liu, M. Celastrol suppresses angiogenesis-mediated tumor growth through inhibition of AKT/mammalian target of rapamycin pathway. Cancer Res. 2010, 70, 1951–1959. [Google Scholar] [CrossRef]
- Sethi, G.; Ahn, K.S.; Pandey, M.K.; Aggarwal, B.B. Celastrol, a novel triterpene, potentiates TNF-induced apoptosis and suppresses invasion of tumor cells by inhibiting NF-kappaB-regulated gene products and TAK1-mediated NF-kappaB activation. Blood 2007, 109, 2727–2735. [Google Scholar]
- Yang, H.; Chen, D.; Cui, Q.C.; Yuan, X.; Dou, Q.P. Celastrol, a triterpene extracted from the Chinese “Thunder of God Vine,” is a potent proteasome inhibitor and suppresses human prostate cancer growth in nude mice. Cancer Res. 2006, 66, 4758–4765. [Google Scholar] [CrossRef]
- Huang, Y.; Zhou, Y.; Fan, Y.; Zhou, D. Celastrol inhibits the growth of human glioma xenografts in nude mice through suppressing VEGFR expression. Cancer Lett. 2008, 264, 101–106. [Google Scholar] [CrossRef]
- Jemal, A.; Siegel, R.; Xu, J.; Ward, E. Cancer statistics, 2010. CA-Cancer J. Clin. 2010, 60, 277–300. [Google Scholar] [CrossRef]
- Chang, A. Chemotherapy, chemoresistance and the changing treatment landscape for NSCLC. Lung Cancer 2011, 71, 3–10. [Google Scholar] [CrossRef]
- Tam, I.Y.; Leung, E.L.; Tin, V.P.; Chua, D.T.; Sihoe, A.D.; Cheng, L.C.; Chung, L.P.; Wong, M.P. Double EGFR mutants containing rare EGFR mutant types show reduced in vitro response to gefitinib compared with common activating missense mutations. Mol. Cancer Ther. 2009, 8, 2142–2151. [Google Scholar] [CrossRef]
- Leung, E.L.; Tam, I.Y.; Tin, V.P.; Chua, D.T.; Sihoe, A.D.; Cheng, L.C.; Ho, J.C.; Chung, L.P.; Wong, M.P. SRC promotes survival and invasion of lung cancers with epidermal growth factor receptor abnormalities and is a potential candidate for molecular-targeted therapy. Mol. Cancer Res. 2009, 7, 923–932. [Google Scholar] [CrossRef]
- Wong, D.W.; Leung, E.L.; So, K.K.; Tam, I.Y.; Sihoe, A.D.; Cheng, L.C.; Ho, K.K.; Au, J.S.; Chung, L.P.; Pik Wong, M. The EML4-ALK fusion gene is involved in various histologic types of lung cancers from nonsmokers with wild-type EGFR and KRAS. Cancer 2009, 115, 1723–1733. [Google Scholar] [CrossRef]
- Jun, H.J.; Johnson, H.; Bronson, R.T.; de Feraudy, S.; White, F.; Charest, A. The oncogenic lung cancer fusion kinase CD74-ROS activates a novel invasiveness pathway through E-Syt1 phosphorylation. Cancer Res. 2012, 72, 3764–3774. [Google Scholar] [CrossRef]
- Hirsch, F.R.; Varella-Garcia, M.; Bunn, P.A., Jr.; di Maria, M.V.; Veve, R.; Bremmes, R.M.; Baron, A.E.; Zeng, C.; Franklin, W.A. Epidermal growth factor receptor in non-small-cell lung carcinomas: Correlation between gene copy number and protein expression and impact on prognosis. J. Clin. Oncol. 2003, 21, 3798–3807. [Google Scholar] [CrossRef]
- Selvaggi, G.; Novello, S.; Torri, V.; Leonardo, E.; de Giuli, P.; Borasio, P.; Mossetti, C.; Ardissone, F.; Lausi, P.; Scagliotti, G.V. Epidermal growth factor receptor overexpression correlates with a poor prognosis in completely resected non-small-cell lung cancer. Ann. Oncol. 2004, 15, 28–32. [Google Scholar] [CrossRef]
- Ono, M.; Kuwano, M. Molecular mechanisms of epidermal growth factor receptor (EGFR) activation and response to gefitinib and other EGFR-targeting drugs. Clin. Cancer Res. 2006, 12, 7242–7251. [Google Scholar] [CrossRef]
- Lee, J.Y.; Lee, Y.M.; Chang, G.C.; Yu, S.L.; Hsieh, W.Y.; Chen, J.J.; Chen, H.W.; Yang, P.C. Curcumin induces EGFR degradation in lung adenocarcinoma and modulates p38 activation in intestine: The versatile adjuvant for gefitinib therapy. PLoS One 2011, 6, e23756. [Google Scholar]
- Sequist, L.V.; Waltman, B.A.; Dias-Santagata, D.; Digumarthy, S.; Turke, A.B.; Fidias, P.; Bergethon, K.; Shaw, A.T.; Gettinger, S.; Cosper, A.K.; et al. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci. Transl. Med. 2011, 3. [Google Scholar] [CrossRef]
- Sudo, M.; Chin, T.M.; Mori, S.; Doan, N.B.; Said, J.W.; Akashi, M.; Koeffler, H.P. Inhibiting proliferation of gefitinib-resistant, non-small cell lung cancer. Cancer Chemother. Pharmacol. 2013, 71, 1325–1334. [Google Scholar] [CrossRef]
- Choi, Y.J.; Rho, J.K.; Jeon, B.S.; Choi, S.J.; Park, S.C.; Lee, S.S.; Kim, H.R.; Kim, C.H.; Lee, J.C. Combined inhibition of IGFR enhances the effects of gefitinib in H1650: A lung cancer cell line with EGFR mutation and primary resistance to EGFR-TK inhibitors. Cancer Chemother. Pharmacol. 2010, 66, 381–388. [Google Scholar] [CrossRef]
- Bivona, T.G.; Hieronymus, H.; Parker, J.; Chang, K.; Taron, M.; Rosell, R.; Moonsamy, P.; Dahlman, K.; Miller, V.A.; Costa, C.; et al. FAS and NF-kappaB signalling modulate dependence of lung cancers on mutant EGFR. Nature 2011, 471, 523–526. [Google Scholar] [CrossRef]
- Kim, H.P.; Han, S.W.; Kim, S.H.; Im, S.A.; Oh, D.Y.; Bang, Y.J.; Kim, T.Y. Combined lapatinib and cetuximab enhance cytotoxicity against gefitinib-resistant lung cancer cells. Mol. Cancer Ther. 2008, 7, 607–615. [Google Scholar] [CrossRef]
- Koivunen, J.P.; Mermel, C.; Zejnullahu, K.; Murphy, C.; Lifshits, E.; Holmes, A.J.; Choi, H.G.; Kim, J.; Chiang, D.; Thomas, R.; et al. EML4-ALK fusion gene and efficacy of an ALK kinase inhibitor in lung cancer. Clin. Cancer Res. 2008, 14, 4275–4283. [Google Scholar] [CrossRef]
- Zou, J.; Chen, Q.; Jin, X.; Tang, S.; Chen, K.; Zhang, T.; Xiao, X. Olaquindox induces apoptosis through the mitochondrial pathway in HepG2 cells. Toxicology 2011, 285, 104–113. [Google Scholar] [CrossRef]
- Liu, T.; Hannafon, B.; Gill, L.; Kelly, W.; Benbrook, D. Flex-Hets differentially induce apoptosis in cancer over normal cells by directly targeting mitochondria. Mol. Cancer Ther. 2007, 6, 1814–1822. [Google Scholar] [CrossRef]
- Gong, K.; Li, W. Shikonin, a Chinese plant-derived naphthoquinone, induces apoptosis in hepatocellular carcinoma cells through reactive oxygen species: A potential new treatment for hepatocellular carcinoma. Free Radic. Biol. Med. 2011, 51, 2259–2271. [Google Scholar] [CrossRef]
- Lakhani, S.A.; Masud, A.; Kuida, K.; Porter, G.A., Jr.; Booth, C.J.; Mehal, W.Z.; Inayat, I.; Flavell, R.A. Caspases 3 and 7: Key mediators of mitochondrial events of apoptosis. Science 2006, 311, 847–851. [Google Scholar] [CrossRef]
- Boatright, K.M.; Salvesen, G.S. Mechanisms of caspase activation. Curr. Opin. Cell. Biol. 2003, 15, 725–731. [Google Scholar] [CrossRef]
- Maddika, S.; Booy, E.P.; Johar, D.; Gibson, S.B.; Ghavami, S.; Los, M. Cancer-specific toxicity of apoptin is independent of death receptors but involves the loss of mitochondrial membrane potential and the release of mitochondrial cell-death mediators by a Nur77-dependent pathway. J. Cell Sci. 2005, 118, 4485–4493. [Google Scholar] [CrossRef]
- Eeva, J.; Nuutinen, U.; Ropponen, A.; Matto, M.; Eray, M.; Pellinen, R.; Wahlfors, J.; Pelkonen, J. The involvement of mitochondria and the caspase-9 activation pathway in rituximab-induced apoptosis in FL cells. Apoptosis 2009, 14, 687–698. [Google Scholar] [CrossRef]
- Chen, G.; Zhang, X.; Zhao, M.; Wang, Y.; Cheng, X.; Wang, D.; Xu, Y.; Du, Z.; Yu, X. Celastrol targets mitochondrial respiratory chain complex I to induce reactive oxygen species-dependent cytotoxicity in tumor cells. BMC Cancer 2011, 11, 170. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, T.; Schwartz, S.J.; Sun, D. New developments in Hsp90 inhibitors as anti-cancer therapeutics: Mechanisms, clinical perspective and more potential. Drug Resist. Updat. 2009, 12, 17–27. [Google Scholar] [CrossRef]
- Nomoto, K.; Tsuta, K.; Takano, T.; Fukui, T.; Yokozawa, K.; Sakamoto, H.; Yoshida, T.; Maeshima, A.M.; Shibata, T.; Furuta, K.; et al. Detection of EGFR mutations in archived cytologic specimens of non-small cell lung cancer using high-resolution melting analysis. Am. J. Clin. Pathol. 2006, 126, 608–615. [Google Scholar] [CrossRef]
- He, D.; Xu, Q.; Yan, M.; Zhang, P.; Zhou, X.; Zhang, Z.; Duan, W.; Zhong, L.; Ye, D.; Chen, W. The NF-kappa B inhibitor, celastrol, could enhance the anti-cancer effect of gambogic acid on oral squamous cell carcinoma. BMC Cancer 2009, 9, 343. [Google Scholar] [CrossRef]
- Lee, M.S.; Chao, J.; Yen, J.C.; Lin, L.W.; Tsai, F.S.; Hsieh, M.T.; Peng, W.H.; Cheng, H.Y. Schizandrin protects primary rat cortical cell cultures from glutamate-induced apoptosis by inhibiting activation of the MAPK family and the mitochondria dependent pathway. Molecules 2012, 18, 354–72. [Google Scholar] [CrossRef]
- Mao, X.; Yu, C.R.; Li, W.H.; Li, W.X. Induction of apoptosis by shikonin through a ROS/JNK-mediated process in Bcr/Abl-positive chronic myelogenous leukemia (CML) cells. Cell Res. 2008, 18, 879–888. [Google Scholar] [CrossRef]
- Moser, C.; Lang, S.A.; Stoeltzing, O. Heat-shock protein 90 (Hsp90) as a molecular target for therapy of gastrointestinal cancer. Anticancer Res. 2009, 29, 2031–2042. [Google Scholar]
- Basso, A.D.; Solit, D.B.; Chiosis, G.; Giri, B.; Tsichlis, P.; Rosen, N. Akt forms an intracellular complex with heat shock protein 90 (Hsp90) and Cdc37 and is destabilized by inhibitors of Hsp90 function. J. Biol. Chem. 2002, 277, 39858–39866. [Google Scholar]
- Schulte, T.W.; Blagosklonny, M.V.; Ingui, C.; Neckers, L. Disruption of the Raf-1-Hsp90 molecular complex results in destabilization of Raf-1 and loss of Raf-1-Ras association. J. Biol. Chem. 1995, 270, 24585–24588. [Google Scholar]
- Sawai, A.; Chandarlapaty, S.; Greulich, H.; Gonen, M.; Ye, Q.; Arteaga, C.L.; Sellers, W.; Rosen, N.; Solit, D.B. Inhibition of Hsp90 down-regulates mutant epidermal growth factor receptor (EGFR) expression and sensitizes EGFR mutant tumors to paclitaxel. Cancer Res. 2008, 68, 589–596. [Google Scholar] [CrossRef]
- Guo, A.; Villen, J.; Kornhauser, J.; Lee, K.A.; Stokes, M.P.; Rikova, K.; Possemato, A.; Nardone, J.; Innocenti, G.; Wetzel, R.; et al. Signaling networks assembled by oncogenic EGFR and c-Met. Proc. Natl. Acad. Sci. USA 2008, 105, 692–697. [Google Scholar] [CrossRef]
- Liu, Z.; Ma, L.; Wen, Z.S.; Hu, Z.; Wu, F.Q.; Li, W.; Liu, J.; Zhou, G.B. Cancerous inhibitor of PP2A is targeted by natural compound celastrol for degradation in non-small-cell lung cancer. Carcinogenesis 2013. [Google Scholar] [CrossRef]
- Mou, H.; Zheng, Y.; Zhao, P.; Bao, H.; Fang, W.; Xu, N. Celastrol induces apoptosis in non-small-cell lung cancer A549 cells through activation of mitochondria- and Fas/FasL-mediated pathways. Toxicol. In Vitro 2011, 25, 1027–1032. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds are available from the authors.
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Fan, X.-X.; Li, N.; Wu, J.-L.; Zhou, Y.-L.; He, J.-X.; Liu, L.; Leung, E.L.-H. Celastrol Induces Apoptosis in Gefitinib-Resistant Non-Small Cell Lung Cancer Cells via Caspases-Dependent Pathways and Hsp90 Client Protein Degradation. Molecules 2014, 19, 3508-3522. https://doi.org/10.3390/molecules19033508
Fan X-X, Li N, Wu J-L, Zhou Y-L, He J-X, Liu L, Leung EL-H. Celastrol Induces Apoptosis in Gefitinib-Resistant Non-Small Cell Lung Cancer Cells via Caspases-Dependent Pathways and Hsp90 Client Protein Degradation. Molecules. 2014; 19(3):3508-3522. https://doi.org/10.3390/molecules19033508
Chicago/Turabian StyleFan, Xing-Xing, Na Li, Jian-Lin Wu, Yan-Ling Zhou, Jian-Xing He, Liang Liu, and Elaine Lai-Han Leung. 2014. "Celastrol Induces Apoptosis in Gefitinib-Resistant Non-Small Cell Lung Cancer Cells via Caspases-Dependent Pathways and Hsp90 Client Protein Degradation" Molecules 19, no. 3: 3508-3522. https://doi.org/10.3390/molecules19033508