Microsomal Prostaglandin E Synthase-1 Deficiency Exacerbates Pulmonary Fibrosis Induced by Bleomycin in Mice

Abstract
:1. Introduction
2. Results and Discussion
2.1. Results

2.1.2. Bleomycin Treatment Reduced mPGES-1 Expression Levels in mPGES-1+/+ Mice, Inhibited PGE2 Production whereas Increased LTC4 Content in the Lungs from both mPGES-1+/+ and mPGES-1−/− Mice

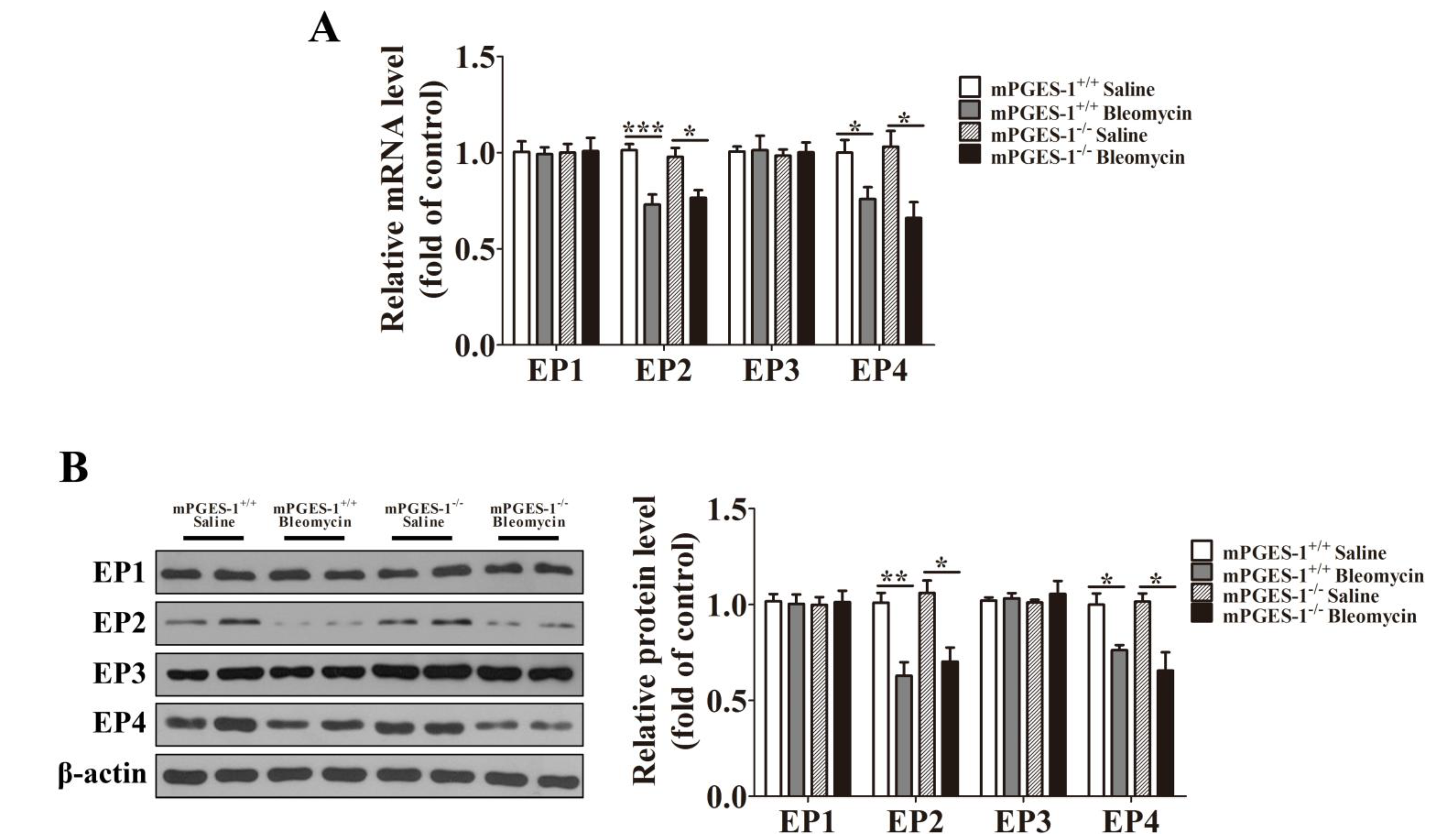
2.1.3. Bleomycin Treatment Reduced the Expression of EP2 and EP4 in mPGES-1−/− and mPGES-1+/+ Mice Lungs

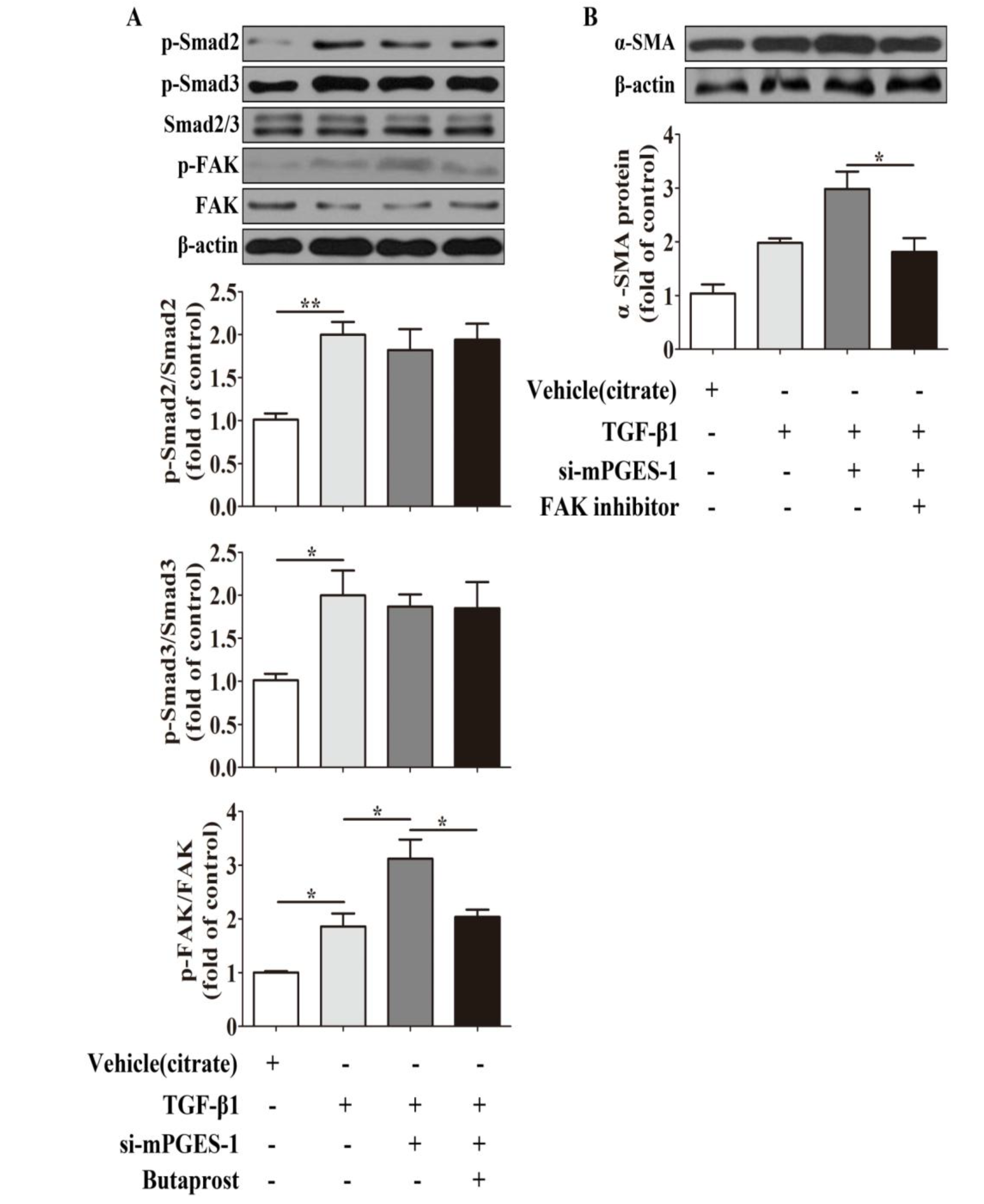
2.1.4. Aggravation of Bleomycin-Induced Lung Fibrosis in mPGES-1−/− Mice Was not Dependent on Smad2/3 Pathway

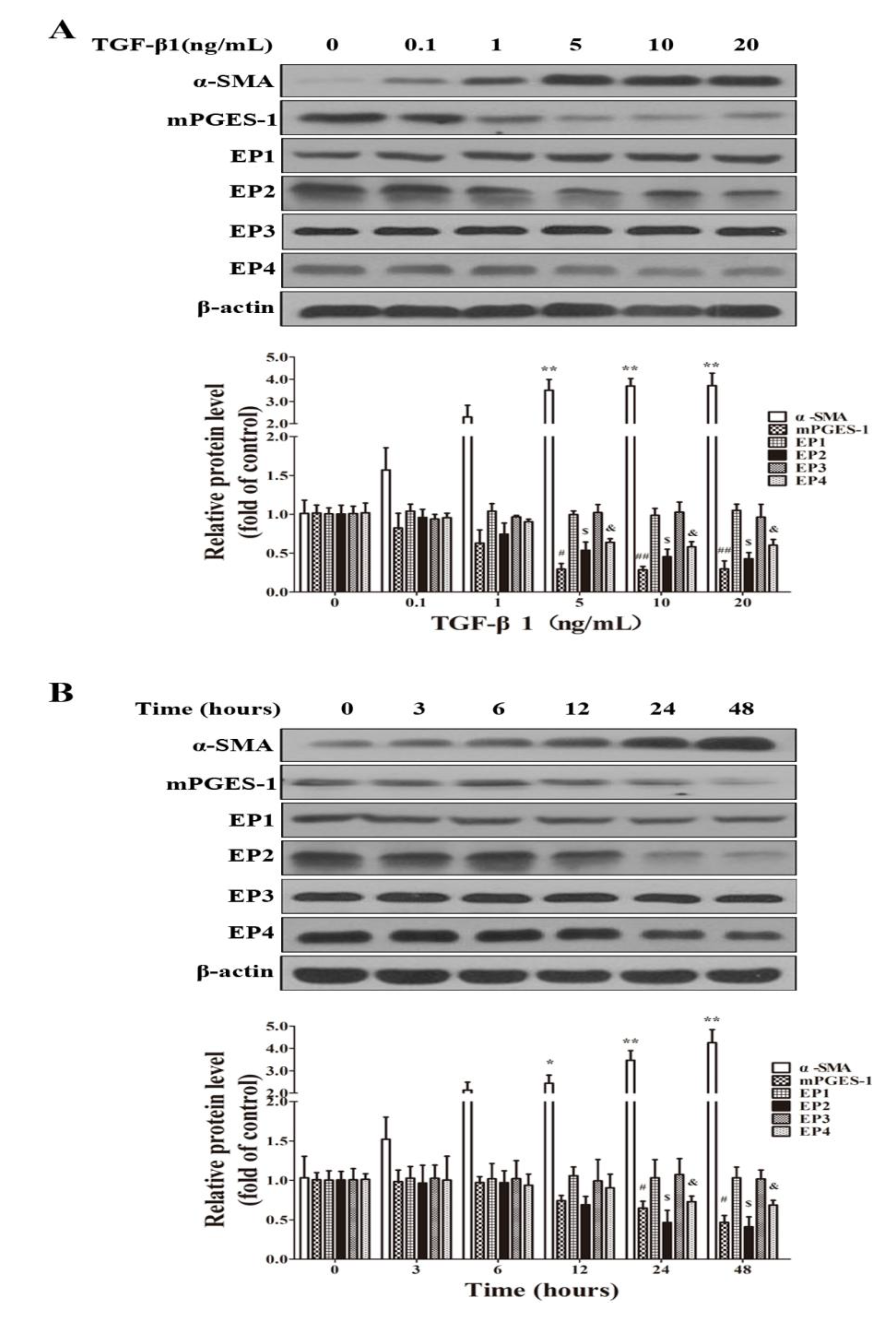
2.1.5. TGF-β1 Treatment Reduced the Expression of mPGES-1, EP2 and EP4 in MRC-5 Cells


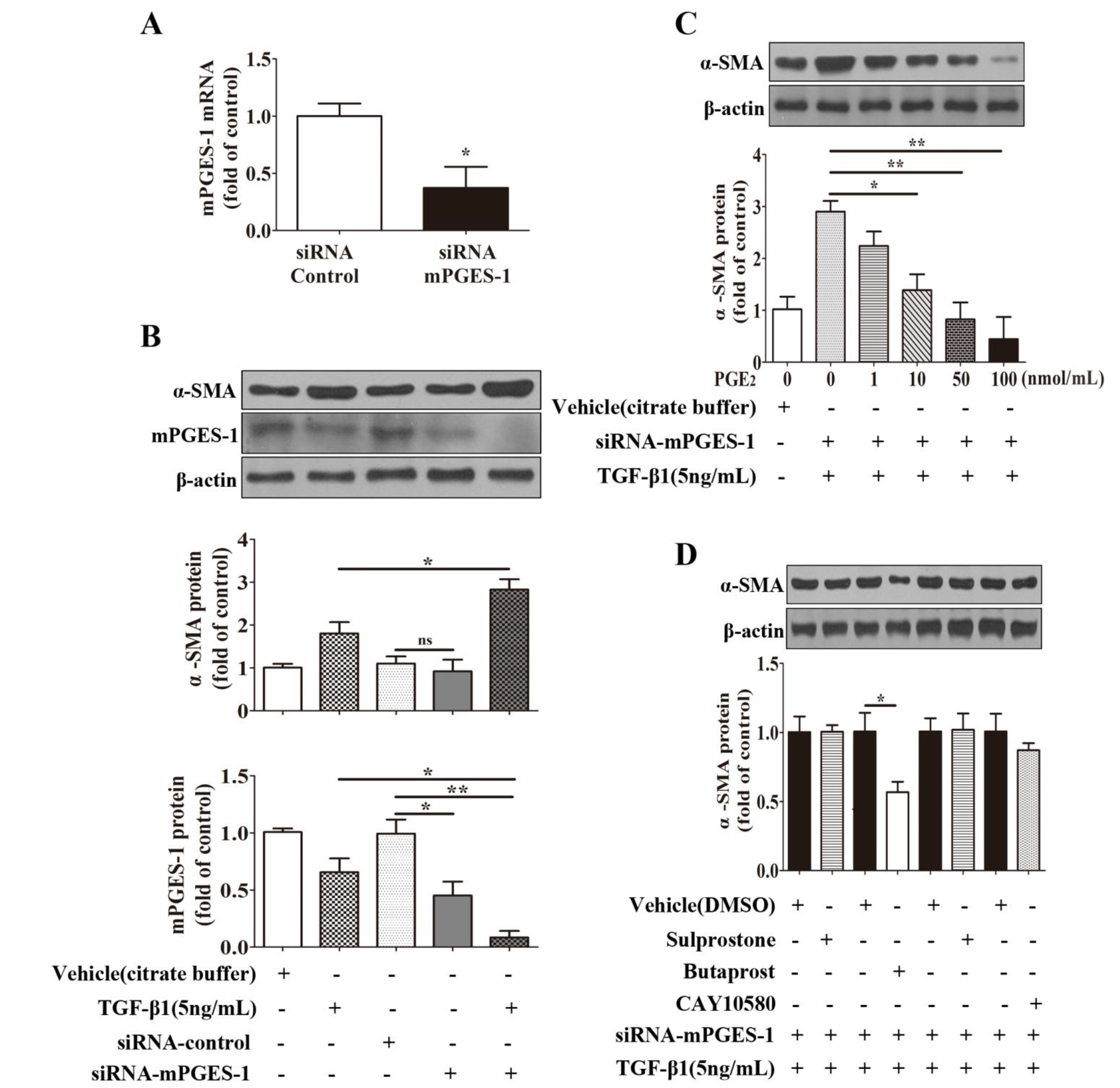
2.1.6. mPGES-1-Derived PGE2 Exerts a Beneficial Effect against α-SMA Expression Induced by TGF-β1 in MRC-5 Cells via EP2 Receptor-Mediated Signaling Transduction
2.1.7. mPGES-1-Derived PGE2 Exerts a Beneficial Effect against α-SMA Expression Induced by TGF-β1 in MRC-5 Cells via Inhibition of FAK Autophosphorylation
2.2. Discussion

3. Experimental Section
3.1. mPGES-1 Knockout Mice
3.2. Animal Groups and Management
3.3. Cell Culture
3.4. Knockdown of mPGES-1 Expression in Human Lung Fibroblasts by RNA Interference
3.5. Lung Histological Examination
3.6. RNA Extraction and Real-Time PCR (RT-PCR)
3.7. Western Blotting

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genes | Sense/Antisense | Primer Sequence (5' to 3') | Products(bp) |
|---|---|---|---|
| α-SMA | Sense | GAAGGAATAGCCACGCTCAG | 185 |
| Antisense | TGCTGTCCCTCTATGCCTCT | ||
| FN | Sense | AATGGAAAAGGGGAATGGAC | 134 |
| Antisense | CTCGGTTCTCCTTCTTTGCTC | ||
| mPGES-1 | Sense | CGCGGTGGCTGTCATCA | 205 |
| Antisense | AGGGTTGGGTCCCAGGAAT | ||
| mPGES-1 (H) | Sense | GAAGAAGGCCTTTGCCAAC | 200 |
| Antisense | GGAAGACCAGGAAGTGCATC | ||
| EP1 | Sense | TAACGATGGTCACGCGATGG | 291 |
| Antisense | ATGCAGTAGTGGGCTTAGGG | ||
| EP2 | Sense | ATGCTCCTGCTGCTTATCGT | 126 |
| Antisense | AGGGCCTCTTAGGCTACTGC | ||
| EP3 | Sense | TTGGGCTTGCTGGCTCTG | 109 |
| Antisense | CGTCTCCGTGGTGATTCTGC | ||
| EP4 | Sense | GTTCCGAGACAGCAAAAGC | 203 |
| Antisense | CACCCCGAAGATGAACATCAC | ||
| β-actin | Sense | GAGCCCTTTTAGAGCCTT | 541 |
| Antisense | GAGCCCTTTTAGAGCCTT |
3.8. Measurement of Lung Hydroxyproline Content
3.9. Quantification of PGE2 in Lung
3.10. Statistical Analysis
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Raghu, G.; Collard, H.R.; Egan, J.J.; Martinez, F.J.; Behr, J.; Brown, K.K.; Colby, T.V.; Cordier, J.F.; Flaherty, K.R.; Lasky, J.A.; et al. An official ats/ers/jrs/alat statement: Idiopathic pulmonary fibrosis: Evidence-based guidelines for diagnosis and management. Am. J. Respir. Crit. Care Med. 2011, 183, 788–824. [Google Scholar] [CrossRef]
- Gross, T.J.; Hunninghake, G.W. Idiopathic pulmonary fibrosis. N. Engl. J. Med. 2001, 345, 517–525. [Google Scholar] [CrossRef]
- Gabbiani, G. The myofibroblast: A key cell for wound healing and fibrocontractive diseases. Prog. Clin. Biol. Res. 1981, 54, 183–194. [Google Scholar]
- Hagiwara, S.I.; Ishii, Y.; Kitamura, S. Aerosolized administration of n-acetylcysteine attenuates lung fibrosis induced by bleomycin in mice. Am. J. Respir. Crit. Care Med. 2000, 162, 225–231. [Google Scholar] [CrossRef]
- Gribbin, J.; Hubbard, R.B.; Le Jeune, I.; Smith, C.J.; West, J.; Tata, L.J. Incidence and mortality of idiopathic pulmonary fibrosis and sarcoidosis in the uk. Thorax 2006, 61, 980–985. [Google Scholar] [CrossRef]
- Smith, W.L.; Dewitt, D.L. Prostaglandin endoperoxide h synthases-1 and -2. Adv. Immunol. 1996, 62, 167–215. [Google Scholar] [CrossRef]
- Jakobsson, P.J.; Thoren, S.; Morgenstern, R.; Samuelsson, B. Identification of human prostaglandin e synthase: A microsomal, glutathione-dependent, inducible enzyme, constituting a potential novel drug target. Proc. Natl. Acad. Sci. USA 1999, 96, 7220–7225. [Google Scholar]
- Tanioka, T.; Nakatani, Y.; Semmyo, N.; Murakami, M.; Kudo, I. Molecular identification of cytosolic prostaglandin e2 synthase that is functionally coupled with cyclooxygenase-1 in immediate prostaglandin e2 biosynthesis. J. Biol. Chem. 2000, 275, 32775–32782. [Google Scholar]
- Murakami, M.; Nakashima, K.; Kamei, D.; Masuda, S.; Ishikawa, Y.; Ishii, T.; Ohmiya, Y.; Watanabe, K.; Kudo, I. Cellular prostaglandin e2 production by membrane-bound prostaglandin e synthase-2 via both cyclooxygenases-1 and -2. J. Biol. Chem. 2003, 278, 37937–37947. [Google Scholar] [CrossRef]
- Kojima, F.; Naraba, H.; Sasaki, Y.; Okamoto, R.; Koshino, T.; Kawai, S. Coexpression of microsomal prostaglandin e synthase with cyclooxygenase-2 in human rheumatoid synovial cells. J. Rheumatol. 2002, 29, 1836–1842. [Google Scholar]
- Stichtenoth, D.O.; Thoren, S.; Bian, H.; Peters-Golden, M.; Jakobsson, P.J.; Crofford, L.J. Microsomal prostaglandin e synthase is regulated by proinflammatory cytokines and glucocorticoids in primary rheumatoid synovial cells. J. Immunol. 2001, 167, 469–474. [Google Scholar]
- Kamei, D.; Yamakawa, K.; Takegoshi, Y.; Mikami-Nakanishi, M.; Nakatani, Y.; Oh-Ishi, S.; Yasui, H.; Azuma, Y.; Hirasawa, N.; Ohuchi, K.; et al. Reduced pain hypersensitivity and inflammation in mice lacking microsomal prostaglandin e synthase-1. J. Biol. Chem. 2004, 279, 33684–33695. [Google Scholar] [CrossRef]
- Trebino, C.E.; Stock, J.L.; Gibbons, C.P.; Naiman, B.M.; Wachtmann, T.S.; Umland, J.P.; Pandher, K.; Lapointe, J.M.; Saha, S.; Roach, M.L.; et al. Impaired inflammatory and pain responses in mice lacking an inducible prostaglandin e synthase. Proc. Natl. Acad. Sci. USA 2003, 100, 9044–9049. [Google Scholar] [CrossRef]
- Saha, S.; Engstrom, L.; Mackerlova, L.; Jakobsson, P.J.; Blomqvist, A. Impaired febrile responses to immune challenge in mice deficient in microsomal prostaglandin e synthase-1. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2005, 288, R1100–R1107. [Google Scholar] [CrossRef]
- Kamei, D.; Murakami, M.; Nakatani, Y.; Ishikawa, Y.; Ishii, T.; Kudo, I. Potential role of microsomal prostaglandin e synthase-1 in tumorigenesis. J. Biol. Chem. 2003, 278, 19396–19405. [Google Scholar]
- Lazarus, M.; Eguchi, N.; Matsumoto, S.; Nagata, N.; Yano, T.; Killian, G.J.; Urade, Y. Species-specific expression of microsomal prostaglandin e synthase-1 and cyclooxygenases in male monkey reproductive organs. Prostaglandins Leukot. Essent. Fatty Acids 2004, 71, 233–240. [Google Scholar] [CrossRef]
- McCann, M.R.; Monemdjou, R.; Ghassemi-Kakroodi, P.; Fahmi, H.; Perez, G.; Liu, S.; Shi-Wen, X.; Parapuram, S.K.; Kojima, F.; Denton, C.P.; et al. Mpges-1 null mice are resistant to bleomycin-induced skin fibrosis. Arthritis Res. Ther. 2011, 13, R6. [Google Scholar] [CrossRef]
- Wilborn, J.; Crofford, L.J.; Burdick, M.D.; Kunkel, S.L.; Strieter, R.M.; Peters-Golden, M. Cultured lung fibroblasts isolated from patients with idiopathic pulmonary fibrosis have a diminished capacity to synthesize prostaglandin e2 and to express cyclooxygenase-2. J. Clin. Investig. 1995, 95, 1861–1868. [Google Scholar] [CrossRef]
- Umezawa, H.; Maeda, K.; Takeuchi, T.; Okami, Y. New antibiotics, bleomycin a and b. J. Antibiot. (Tokyo) 1966, 19, 200–209. [Google Scholar]
- Mouratis, M.A.; Aidinis, V. Modeling pulmonary fibrosis with bleomycin. Curr. Opin. Pulm. Med. 2011, 17, 355–361. [Google Scholar] [CrossRef]
- Fang, L.P.; Lin, Q.; Tang, C.S.; Liu, X.M. Hydrogen sulfide suppresses migration, proliferation and myofibroblast transdifferentiation of human lung fibroblasts. Pulm. Pharmacol. Ther. 2009, 22, 554–561. [Google Scholar] [CrossRef]
- Thomas, P.E.; Peters-Golden, M.; White, E.S.; Thannickal, V.J.; Moore, B.B. PGE(2) inhibition of TGF-beta 1-induced myofibroblast differentiation is Smad-independent but involves cell shape and adhesion-dependent signaling. Am. J. Physiol. Lung Cell. Mol. Physiol. 2007, 293, L417–L428. [Google Scholar] [CrossRef]
- Selman, M.; Pardo, A. Role of epithelial cells in idiopathic pulmonary fibrosis: From innocent targets to serial killers. Proc. Am. Thorac. Soc. 2006, 3, 364–372. [Google Scholar] [CrossRef]
- Lama, V.; Moore, B.B.; Christensen, P.; Toews, G.B.; Peters-Golden, M. Prostaglandin e2 synthesis and suppression of fibroblast proliferation by alveolar epithelial cells is cyclooxygenase-2-dependent. Am. J. Respir. Cell Mol. Biol. 2002, 27, 752–758. [Google Scholar]
- Huang, S.K.; Wettlaufer, S.H.; Chung, J.; Peters-Golden, M. Prostaglandin e2 inhibits specific lung fibroblast functions via selective actions of pka and epac-1. Am. J. Respir. Cell Mol. Biol. 2008, 39, 482–489. [Google Scholar]
- Elias, J.A.; Rossman, M.D.; Zurier, R.B.; Daniele, R.P. Human alveolar macrophage inhibition of lung fibroblast growth. A prostaglandin-dependent process. Am. Rev. Respir. Dis. 1985, 131, 94–99. [Google Scholar]
- Narumiya, S.; Sugimoto, Y.; Ushikubi, F. Prostanoid receptors: Structures, properties, and functions. Physiol. Rev. 1999, 79, 1193–1226. [Google Scholar]
- Moore, B.B.; Ballinger, M.N.; White, E.S.; Green, M.E.; Herrygers, A.B.; Wilke, C.A.; Toews, G.B.; Peters-Golden, M. Bleomycin-induced e prostanoid receptor changes alter fibroblast responses to prostaglandin e2. J. Immunol. 2005, 174, 5644–5649. [Google Scholar]
- Huang, S.; Wettlaufer, S.H.; Hogaboam, C.; Aronoff, D.M.; Peters-Golden, M. Prostaglandin e(2) inhibits collagen expression and proliferation in patient-derived normal lung fibroblasts via e prostanoid 2 receptor and camp signaling. Am. J. Physiol. Lung Cell. Mol. Physiol. 2007, 292, L405–L413. [Google Scholar]
- Huang, S.K.; Wettlaufer, S.H.; Hogaboam, C.M.; Flaherty, K.R.; Martinez, F.J.; Myers, J.L.; Colby, T.V.; Travis, W.D.; Toews, G.B.; Peters-Golden, M. Variable prostaglandin e2 resistance in fibroblasts from patients with usual interstitial pneumonia. Am. J. Respir. Crit. Care Med. 2008, 177, 66–74. [Google Scholar] [CrossRef]
- Kolodsick, J.E.; Peters-Golden, M.; Larios, J.; Toews, G.B.; Thannickal, V.J.; Moore, B.B. Prostaglandin e2 inhibits fibroblast to myofibroblast transition via e. Prostanoid receptor 2 signaling and cyclic adenosine monophosphate elevation. Am. J. Respir. Cell Mol. Biol. 2003, 29, 537–544. [Google Scholar] [CrossRef]
- Parsons, J.T.; Martin, K.H.; Slack, J.K.; Taylor, J.M.; Weed, S.A. Focal adhesion kinase: A regulator of focal adhesion dynamics and cell movement. Oncogene 2000, 19, 5606–5613. [Google Scholar] [CrossRef]
- Mitra, S.K.; Hanson, D.A.; Schlaepfer, D.D. Focal adhesion kinase: In command and control of cell motility. Nat. Rev. Mol. Cell Biol. 2005, 6, 56–68. [Google Scholar] [CrossRef]
- Ashcroft, T.; Simpson, J.M.; Timbrell, V. Simple method of estimating severity of pulmonary fibrosis on a numerical scale. J. Clin. Pathol. 1988, 41, 467–470. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(t)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds are not available from the authors.
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Wei, B.; Cai, L.; Sun, D.; Wang, Y.; Wang, C.; Chai, X.; Xie, F.; Su, M.; Ding, F.; Liu, J.; et al. Microsomal Prostaglandin E Synthase-1 Deficiency Exacerbates Pulmonary Fibrosis Induced by Bleomycin in Mice. Molecules 2014, 19, 4967-4985. https://doi.org/10.3390/molecules19044967
Wei B, Cai L, Sun D, Wang Y, Wang C, Chai X, Xie F, Su M, Ding F, Liu J, et al. Microsomal Prostaglandin E Synthase-1 Deficiency Exacerbates Pulmonary Fibrosis Induced by Bleomycin in Mice. Molecules. 2014; 19(4):4967-4985. https://doi.org/10.3390/molecules19044967
Chicago/Turabian StyleWei, Bo, Linhong Cai, Dan Sun, Yanhua Wang, Cairui Wang, Xiaoyu Chai, Feng Xie, Ming Su, Fangrui Ding, Jie Liu, and et al. 2014. "Microsomal Prostaglandin E Synthase-1 Deficiency Exacerbates Pulmonary Fibrosis Induced by Bleomycin in Mice" Molecules 19, no. 4: 4967-4985. https://doi.org/10.3390/molecules19044967