Synthesis, Crystal Structure, Spectra and Quantum Chemical Study on 1-Phenyl-3-(4-nitrophenyl)-5-(2-thienyl)-2-pyrazoline

Abstract
:1. Introduction

2. Results and Discussion
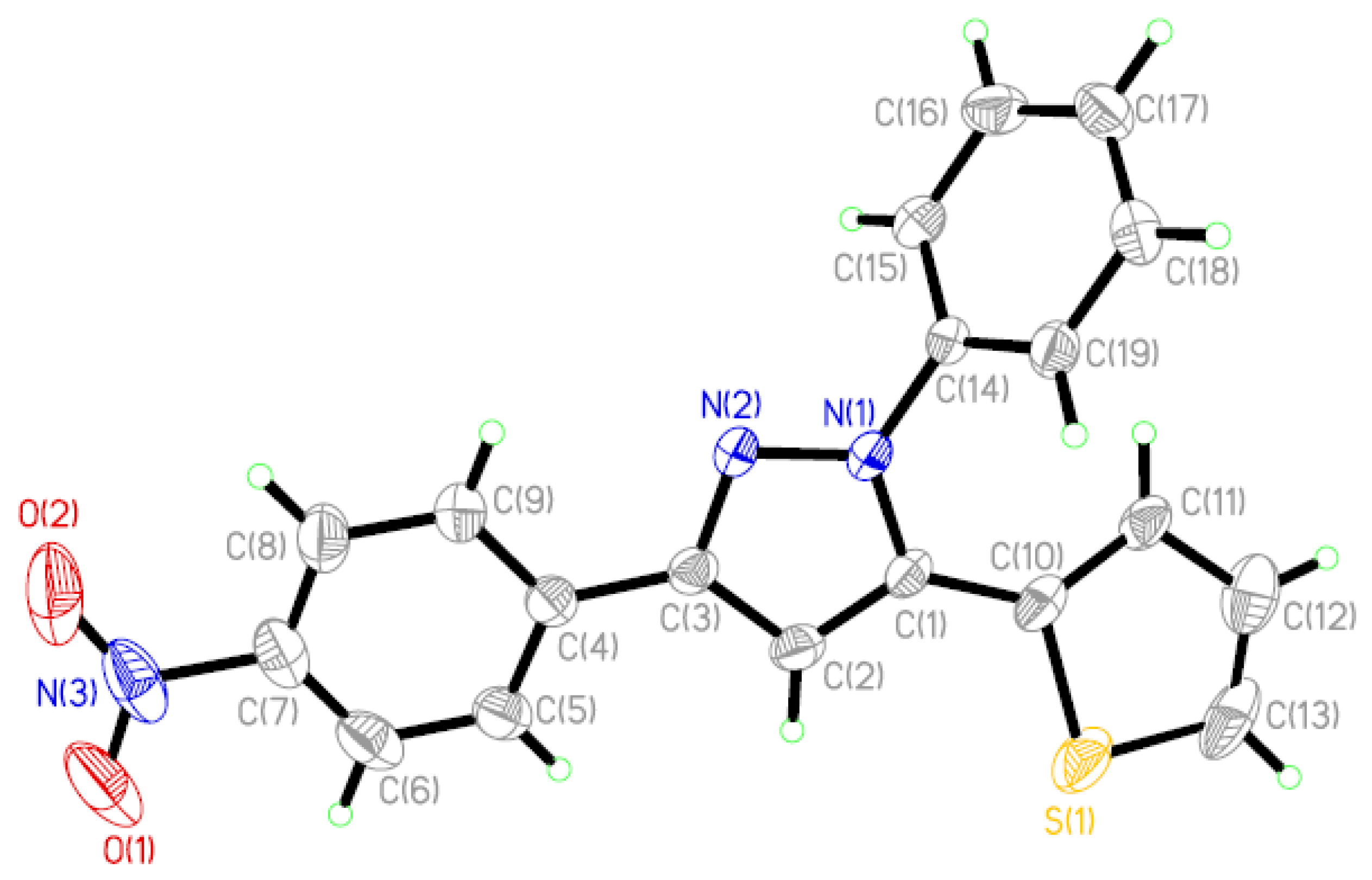
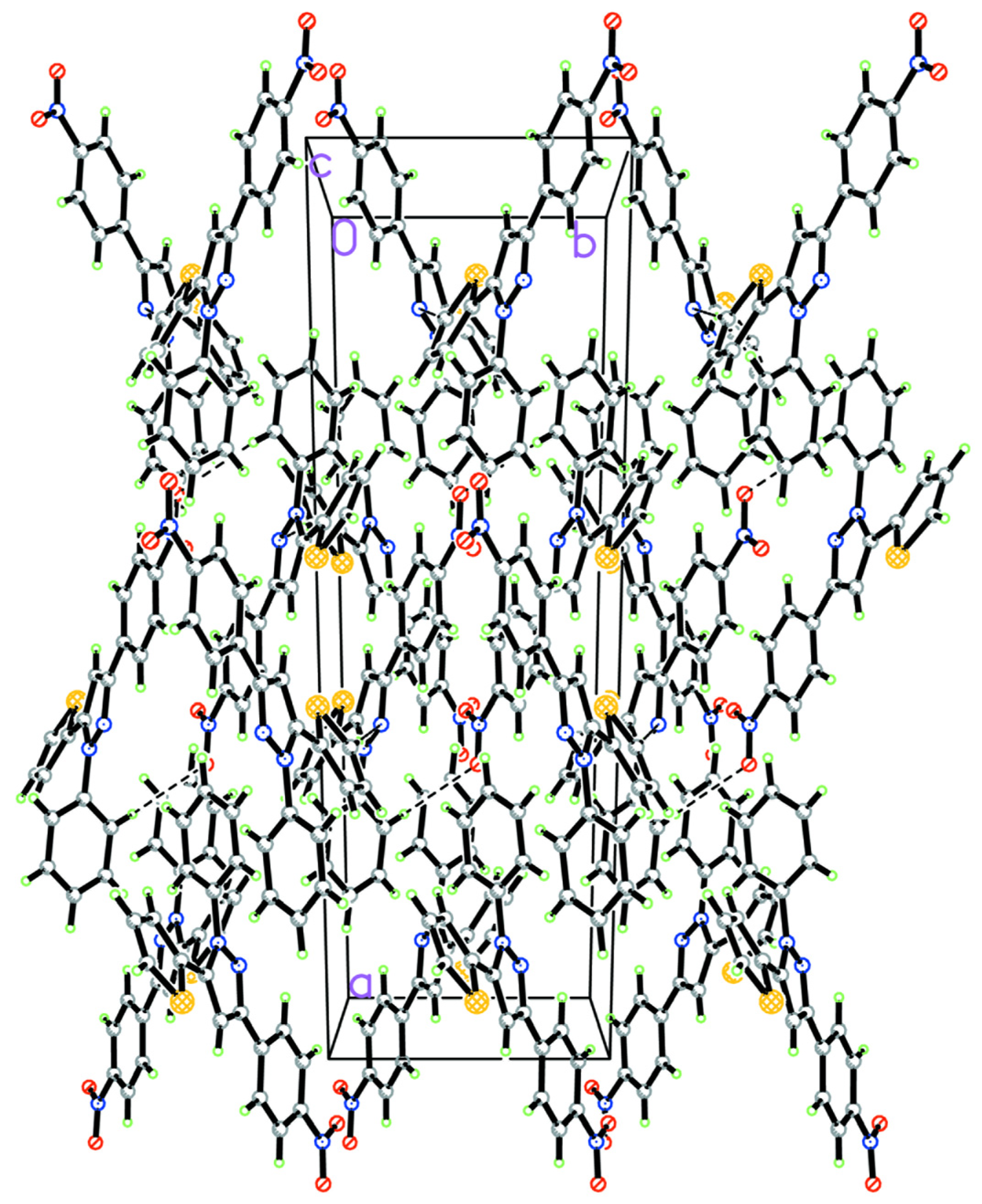
2.1. Description of the Crystal Structure



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bond Lengths (Å) | Experiment | B3LYP/6-311G** | Bond Lengths (Å) | Experiment | B3LYP/6-311G** |
|---|---|---|---|---|---|
| S(1)–C(13) | 1.675(5) | 1.730 | C(1)–C(10) | 1.458(4) | 1.457 |
| S(1)–C(10) | 1.704(3) | 1.750 | C(2)–C(3) | 1.402(4) | 1.415 |
| O(1)–N(3) | 1.201(6) | 1.225 | C(3)–C(4) | 1.466(4) | 1.468 |
| O(2)–N(3) | 1.225(6) | 1.225 | C(4)–C(5) | 1.389(5) | 1.403 |
| N(1)–N(2) | 1.356(3) | 1.348 | C(4)–C(9) | 1.393(5) | 1.405 |
| N(1)–C(1) | 1.365(4) | 1.380 | C(5)–C(6) | 1.381(5) | 1.387 |
| N(1)–C(14) | 1.430(4) | 1.429 | C(10)–C(11) | 1.402(5) | 1.373 |
| N(2)–C(3) | 1.332(4) | 1.334 | C(14)–C(19) | 1.373(4) | 1.394 |
| N(3)–C(7) | 1.480(5) | 1.474 | C(15)–C(16) | 1.381(5) | 1.391 |
| C(1)–C(2) | 1.374(4) | 1.383 | C(17)–C(18) | 1.362(5) | 1.394 |
| Bond Angles (°) | Bond Angles (°) | ||||
| C(13)–S(1)–C(10) | 92.7(2) | 91.6 | C(8)–C(7)–C(6) | 121.6(4) | 121.6 |
| N(2)–N(1)–C(1) | 112.0(2) | 111.9 | C(11)–C(10)–S(1) | 109.6(2) | 110.5 |
| C(3)–N(2)–N(1) | 105.4(2) | 106.0 | C(10)–C(11)–C(12) | 111.4(4) | 112.9 |
| O(1)–N(3)–O(2) | 125.1(5) | 124.6 | C(13)–C(12)–C(11) | 113.4(4) | 113.3 |
| N(1)–C(1)–C(2) | 105.8(2) | 105.8 | C(12)–C(13)–S(1) | 112.9(3) | 111.7 |
| C(1)–C(2)–C(3) | 106.2(3) | 105.7 | C(19)–C(14)–C(15) | 121.0(3) | 120.6 |
| N(2)–C(3)–C(2) | 110.6(3) | 110.6 | C(17)–C(16)–C(15) | 120.5(3) | 120.3 |
| C(5)–C(4)–C(9) | 117.9(3) | 118.7 | C(17)–C(18)–C(19) | 119.9(3) | 120.4 |
| D–H···A | Symmetry | H···A (Å) | D···A (Å) | ∠D–H···A (°) |
|---|---|---|---|---|
| C(13)–H(13)···N(2) | x, 1 − y, −1/2 + z | 2.510(3) | 3.439(3) | 176.98 |
| C(19)–H(19)···O(1) | −x, −y, −z | 2.552(2) | 3.279(1) | 135.30 |
| C(11)–H(11)···Cg(4) | x, y, z | 3.037(1) | 3.743(1) | 134 |
| C(16)–H(16)···Cg(4) | 1/2 − x, 1/2 + y, 1/2 − z | 3.209(2) | 3.879(3) | 131 |
| C(18)–H(18)···Cg(1) | 1/2 − x, 1/2 − y, −z | 2.809(2) | 3.596(2) | 143 |
2.2. Optimized Geometry
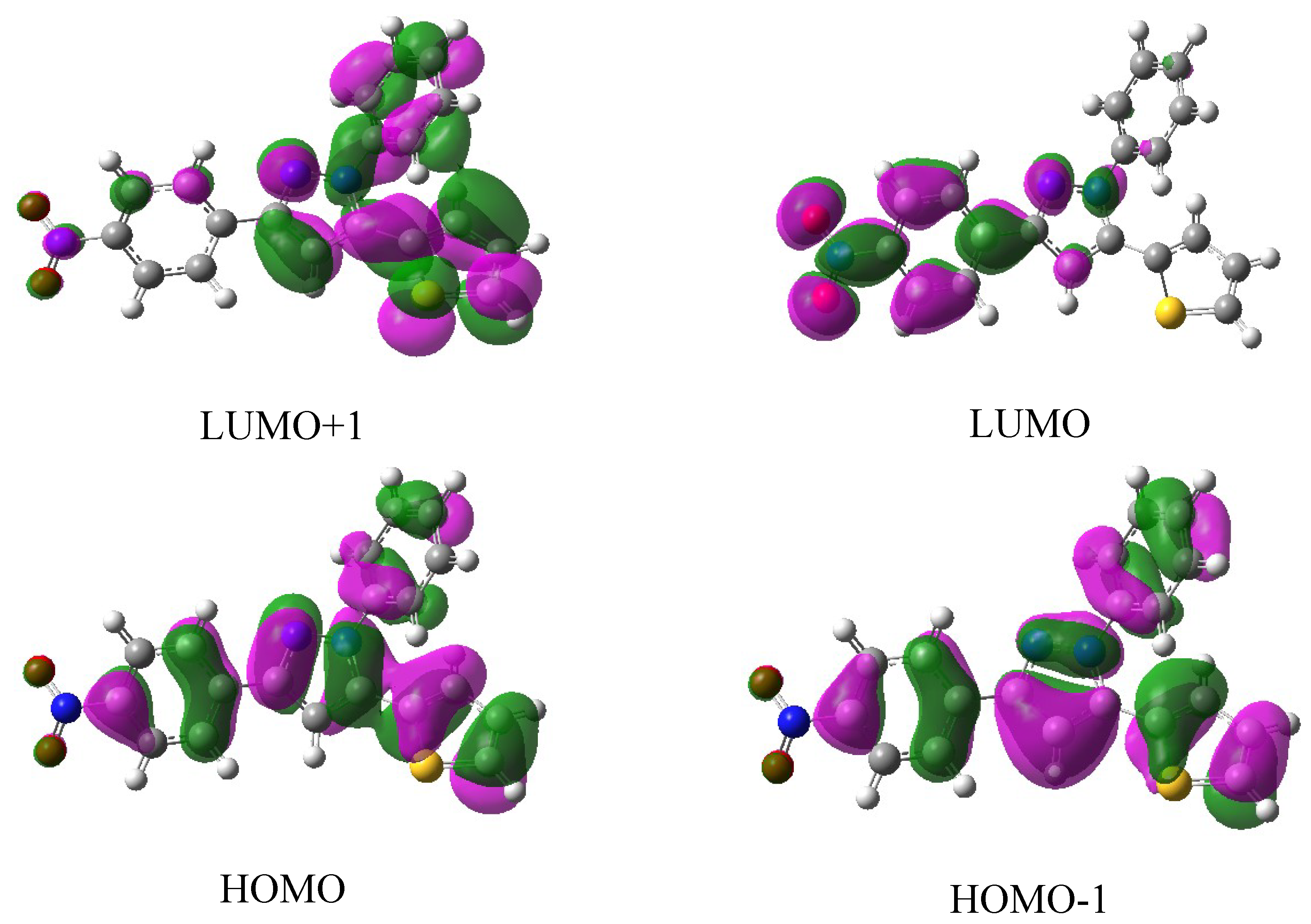
2.3. Atomic Charge Distributions
| Atom | Charges (e) | Atom | Charges (e) | Atom | Charges (e) |
|---|---|---|---|---|---|
| N(1) | −0.16920 | C(4) | −0.03329 | C(11) | −0.22313 |
| N(2) | −0.28191 | C(5) | −0.18105 | C(12) | −0.24353 |
| C(1) | 0.13219 | C(6) | −0.17616 | C(13) | −0.37017 |
| C(2) | −0.27626 | C(7) | 0.05712 | C(14) | 0.14793 |
| C(3) | 0.13913 | C(8) | −0.17632 | C(15) | −0.19406 |
| N(3) | 0.51394 | C(9) | −0.16957 | C(16) | −0.18771 |
| O(1) | −0.38790 | S(1) | 0.41951 | C(17) | −0.19584 |
| O(2) | −0.38977 | C(10) | −0.22371 | C(18) | −0.18649 |
| C(19) | −0.20411 |
2.4. Calculations of Nonlinear Optical Property
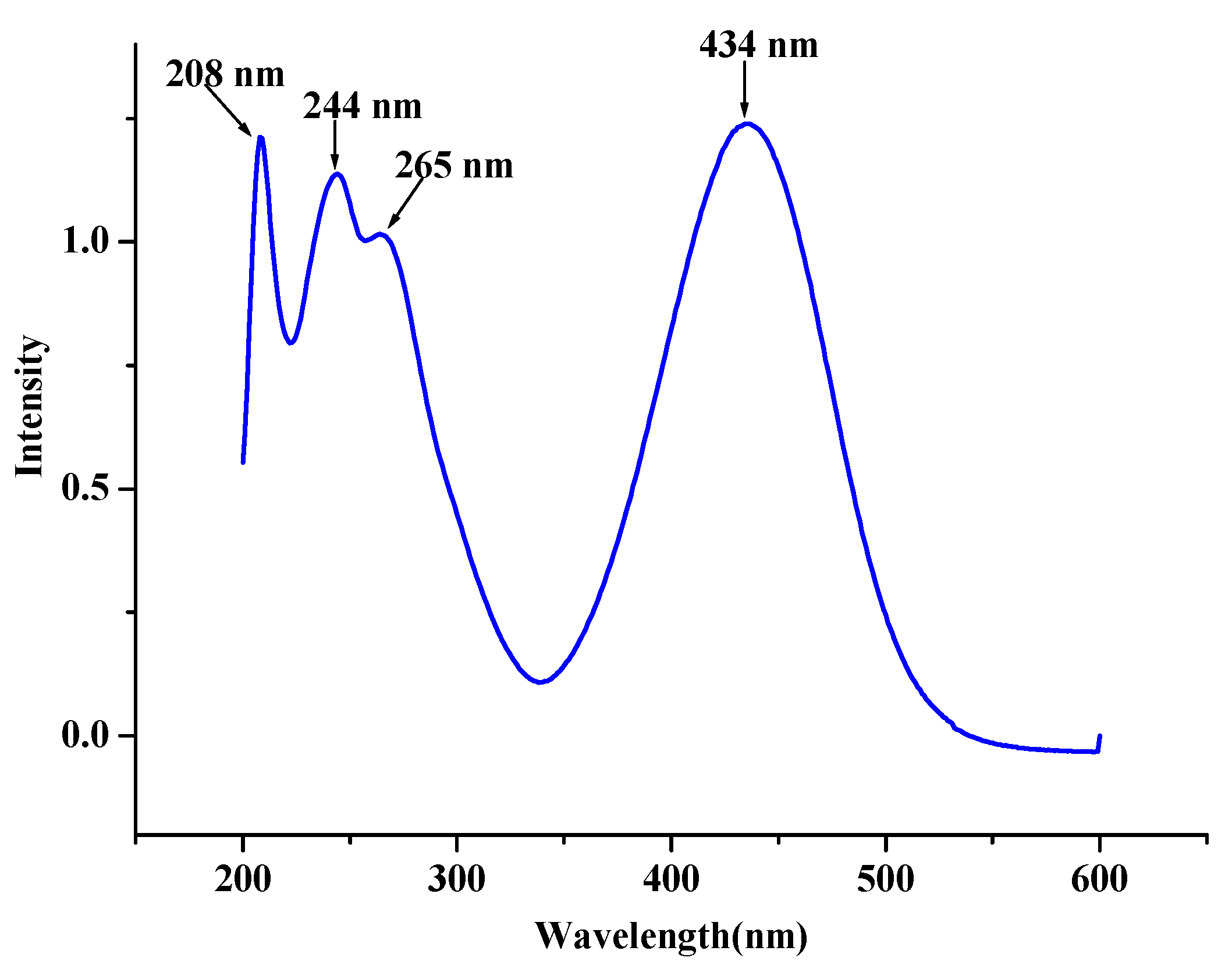
2.5. Electronic Absorption Spectra


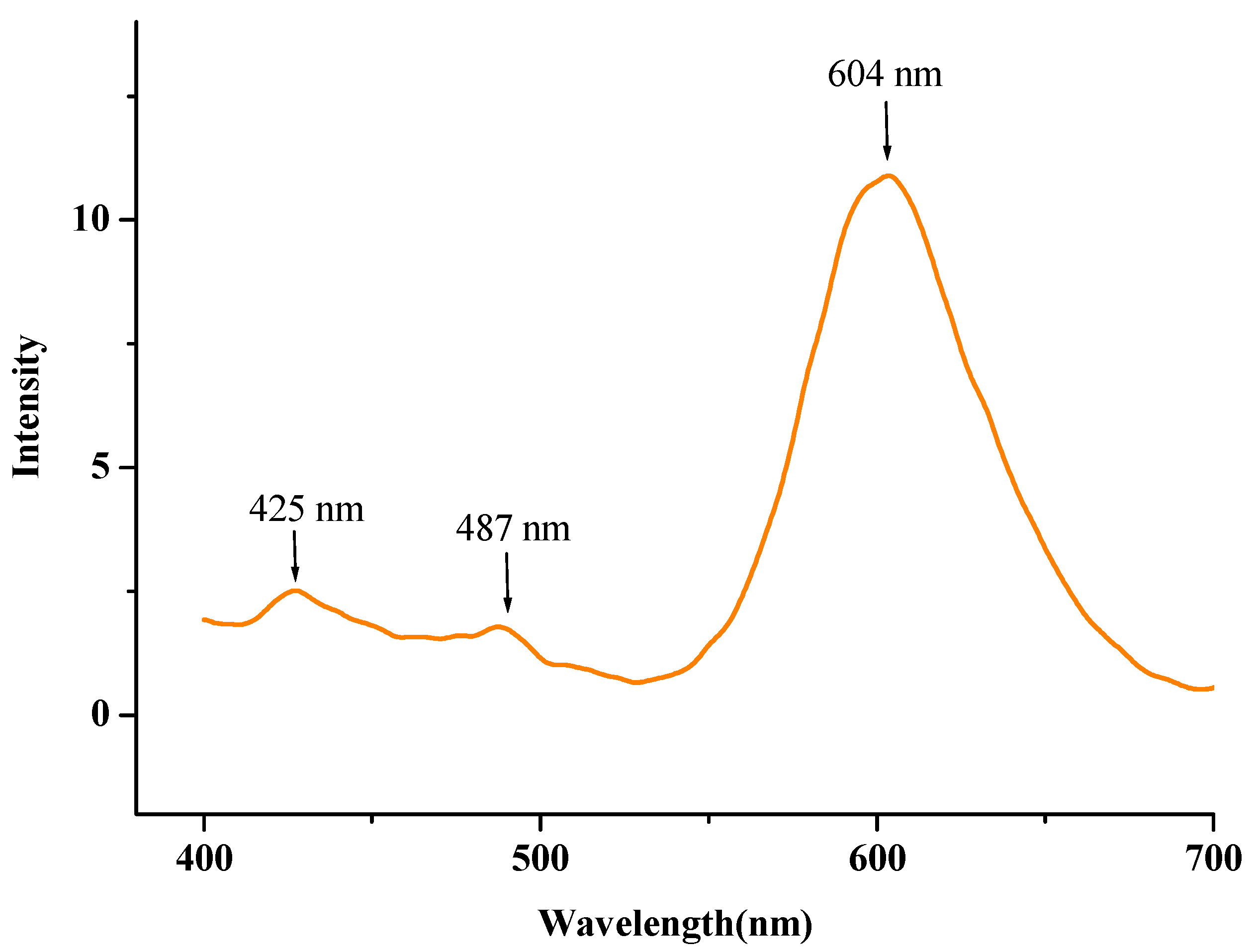
2.6. Fluorescence Spectra

3. Experimental and Theoretical Methods
3.1. Physical Measurements
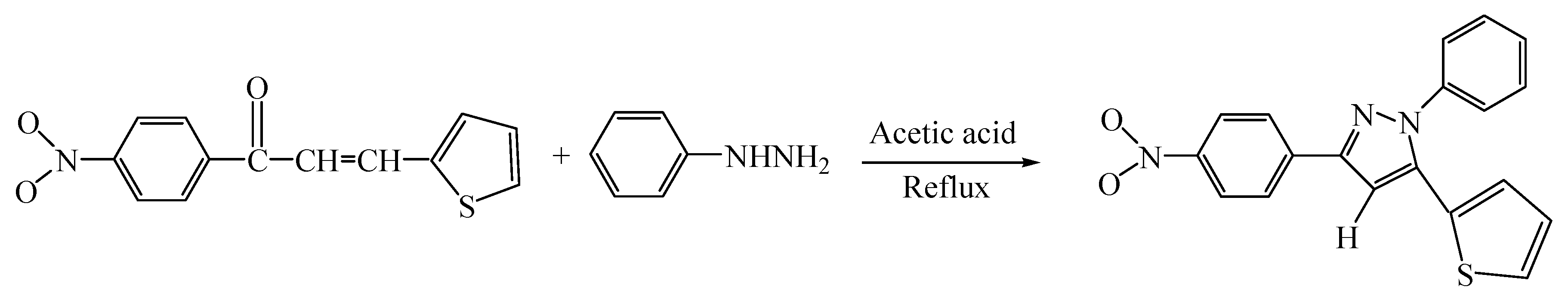
3.2. Synthesis
3.3. Crystallographic Study
3.4. Computational Methods
| Empirical Formula | C19H13N3O2S |
|---|---|
| Formula weight | 347.38 |
| Temperature | 293(2) K |
| Wavelength | 0.71073 Å |
| Crystal system, space group | Monoclinic, C2/c |
| Unit cell dimensions | a = 28.023(7) Å |
| b = 7.8005(15) Å β = 122.69(2)° | |
| c = 18.008(7) Å | |
| Volume | 3312.9(17) Å3 |
| Z, Calculated density | 8, 1.393 Mg/m3 |
| Absorption coefficient | 0.213 mm−1 |
| F(000) | 1440 |
| θ range for data collection | 3.26 to 25.00° |
| Limiting indices | −33 ≤ h ≤ 33, −9 ≤ k ≤ 8, −21 ≤ l ≤ 21 |
| Reflections collected/unique | 10,658/2887 [Rint = 0.0476 ] |
| Refinement method | Full-matrix least-squares on F2 |
| Data/restraints/parameters | 2887/0/226 |
| Goodness-of-fit on F2 | 1.120 |
| Final R indices [I > 2σ (I)] | R1 = 0.0707, wR2 = 0.1922 |
| R indices (all data) | R1 = 0.0911, wR2 = 0.2071 |
| Largest diff. peak and hole | 0.402 and −0.404 e. Å−3 |
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Xiao, D.; Xi, L.; Yang, W.; Fu, H.; Shuai, Z.; Fang, Y.; Yao, J. Size-Tunable Emission from 1,3-Diphenyl-5-(2-anthryl)-2-pyrazoline Nanoparticles. J. Am. Chem. Soc. 2003, 125, 6740–6745. [Google Scholar] [CrossRef]
- Ji, S.J.; Shi, H.B. Synthesis and fluorescent property of some novel benzothiazoyl pyrazoline derivatives containing aromatic heterocycle. Dyes Pigm. 2006, 70, 246–250. [Google Scholar] [CrossRef]
- Bian, B.; Ji, S.J.; Shi, H.B. Synthesis and fluorescent property of some novel bischromophore compounds containing pyrazoline and naphthalimide groups. Dyes Pigm. 2008, 76, 348–352. [Google Scholar] [CrossRef]
- Zhang, X.H.; Lai, W.Y.; Gao, Z.Q.; Wong, T.C.; Lee, C.S.; Kwong, H.L. Photoluminescence and electroluminescence of pyrazoline monomers and dimers. Chem. Phys. Lett. 2000, 320, 77–80. [Google Scholar] [CrossRef]
- Wei, X.Q.; Yang, G.; Cheng, J.B.; Lu, Z.Y.; Xie, M.G. Synthesis of novel light-emitting calix[4]arene derivatives and their luminescent properties. Opt. Mater. 2007, 29, 936–940. [Google Scholar] [CrossRef]
- Pramanik, S.; Banerjee, P.; Sarkar, A.; Mukherjee, A.; Mahalanabis, K.K.; Bhattacharya, S.C. Spectroscopic investigation of 3-pyrazolyl 2-pyrazoline derivative in homogeneous solvents. Spectrochim. Acta A 2008, 71, 1327–1332. [Google Scholar] [CrossRef]
- Pokladko, M.; Gondek, E.; Sanetra, J.; Nizioł, J.; Danel, A.; Kityk, I.V.; Reshak Ali, H. Spectral emission properties of 4-aryloxy-3-methyl-1-phenyl-1H-pyrazolo[3,4-b]quinolines. Spectrochim. Acta A 2009, 73, 281–285. [Google Scholar] [CrossRef]
- Sun, Y.F.; Cui, Y.P. The synthesis, structure and spectroscopic properties of novel oxazolone-, pyrazolone- and pyrazoline-containing heterocycle chromophores. Dyes Pigm. 2009, 81, 27–34. [Google Scholar] [CrossRef]
- Fu, H.B.; Yao, J.N. Size Effects on the Optical Properties of Organic Nanoparticles. J. Am. Chem. Soc. 2001, 123, 1434–1439. [Google Scholar] [CrossRef]
- Oh, S.W.; Kang, Y.S. The size-dependent optical properties of 1-phenyl-3-naphthyl-5-((dimethyl amino) phenyl)-2-pyrazoline nanoparticles. Colloids Surf. A: Physicochem. Eng. Aspects 2005, 257, 415–418. [Google Scholar]
- Fu, H.B.; Loo, B.H.; Xiao, D.B.; Xie, R.M.; Ji, X.H.; Yao, J.N.; Zhang, B.W.; Zhang, L.Q. Multiple Emissions from 1,3-Diphenyl-5-pyrenyl-2-pyrazoline Nanoparticles: Evolution from Molecular to Nanoscale to Bulk Materials. Angew. Chem. Int. Ed. 2002, 41, 962–965. [Google Scholar] [CrossRef]
- Zhao, P.S.; Li, Y.F.; Guo, H.M.; Jian, F.F.; Wang, X. Synthesis, Crystal Structure and Density Functional Calculations on 1-Phenyl-3-p-fluorophenyl-5-p-chlorophenyl-2-pyrazoline. Bull. Korean Chem. Soc. 2007, 28, 1539–1544. [Google Scholar] [CrossRef]
- Zhao, P.S.; Li, Y.F.; Guo, H.M.; Wang, X.; Jian, F.F. Synthesis, Characterization and ab initio Calculations on 1-Phenyl-3-p-fluorophenyl-5-p-methoxyphenyl-2-pyrazoline. Pol. J. Chem. 2007, 81, 1735–1742. [Google Scholar]
- Jian, F.F.; Zhao, P.S.; Guo, H.M.; Li, Y.F. Synthesis, characterization, crystal structure and DFT studies on 1-acetyl-3-(2,4-dichloro-5-fluoro-phenyl)-5-phenyl-pyrazoline. Spectrochim. Acta A 2008, 69, 647–653. [Google Scholar] [CrossRef]
- Zhao, P.S.; Wang, H.Y.; Li, R.Q.; Guo, H.M. Synthesis, crystal structure, electronic spectra and density functional studies on 1N-phenyl-3-(3,4-dichlorophenyl)-5-phenyl-2-pyrazoline. Indian J. Chem. A 2008, 47, 986–991. [Google Scholar]
- Zhao, P.S.; Li, R.Q.; Sun, X.J.; Guo, H.M.; Jian, F.F. Comparative study on two 2-pyrazoline derivatives with experimental and theoretical methods. Struct. Chem. 2009, 20, 443–451. [Google Scholar] [CrossRef]
- Zhao, P.S.; Li, R.Q.; Wang, H.Y.; Jian, F.F.; Guo, H.M. Experimental and theoretical comparative studies on two 2-pyrazoline derivatives. Spectrochim. Acta A 2009, 74, 87–93. [Google Scholar] [CrossRef]
- Zhao, P.S.; Zhou, S.S.; Guo, Z.Y.; Zhu, Y. Crystal structure, spectra properties and comparative studies on a 2-pyrazoline derivative. Spectrochim. Acta A 2012, 94, 65–71. [Google Scholar] [CrossRef]
- Jin, Z.N.; Wu, J.S.; Wang, C.F.; Dai, G.L.; Liu, S.Y.; Lu, J.M.; Jiang, H.J. Novel fluorescent 1,8-naphthalimide derivatives containing thiophene and pyrazole moieties: Synthesis by direct C–H arylation and evaluation of photophysical and electrochemical properties. Spectrochim. Acta A: Mol. Biomol. Spectr 2014, 117, 527–534. [Google Scholar] [CrossRef]
- Willy, B.; Müller, T.J.J. Regioselective Three-Component Synthesis of Highly Fluorescent 1,3,5-Trisubstituted Pyrazoles. Eur. J. Org. Chem. 2008, 24, 4157–4168. [Google Scholar] [CrossRef]
- Yang, Y.W.; Kuang, C.X.; Jin, H.; Yang, Q.; Zhang, Z.K. Efficient synthesis of 1,3-diaryl-4-halo-1H-pyrazoles from 3-arylsydnones and 2-aryl-1,1-dihalo-1-alkenes. Beilstein. J. Org. Chem. 2011, 7, 1656–1662. [Google Scholar] [CrossRef]
- Raghava, B.; Prasad, T.N.M.; Lakshminarayana, B.N.; Sridhar, M.A.; Prasad, J.S.; Rangappa, K.S. Synthesis and crystal structure of 3-(2,5-dimethylphenyl)-1-(4-methoxy phenyl)-5-(thiophen-2-yl)-1H-pyrazole. X-ray Struct. Anal. Online 2012, 28, 51–52. [Google Scholar] [CrossRef]
- Koch, W.; Holthausen, M.C. A Chemistry Guide to Density Functional Theory; Wiley-VCH: Weinheim, Germany, 2000. [Google Scholar]
- Parr, R.R.; Yang, R.G. Density Functional Theory of Atoms and Molecules; Oxford University Press: New York, NY, USA, 1989; and references therein. [Google Scholar]
- Guo, H.M.; Jian, F.F.; Zhao, P.S.; Zhang, Y.C.; Li, Y.F. 5-(2-Chlorophenyl)-3-(4-chlorophenyl)-1-phenyl-2-pyrazoline. Acta. Cryst. 2007, E63, o215–o216. [Google Scholar]
- Steiner, T. C–H–O Hydrogen Bonding in Crystals. Cryst. Rev. 1996, 6, 1–51. [Google Scholar] [CrossRef]
- Jeffrey, G.A.; Maluszynska, H.; Mitra, J. Hydrogen bonding in nucleosides and nucleotides. Int. J. Biol. Macromol. 1985, 7, 336–348. [Google Scholar] [CrossRef]
- Hunter, R.H.; Haueisen, R.H.; Irving, A. The First Water-Dependent Liquid Clathrate: X-Ray Evidence in the Solid for a C–H···π (Heteroarene) π···H–C Interaction. Angew. Chem. Int. Ed. Engl. 1994, 33, 566–568. [Google Scholar] [CrossRef]
- Joaquín, B.; Koen, C.; Raquel, G.; Stephan, H.; André, P.; José, L.S. Versatile optical materials: Fluorescence, non-linear optical and mesogenic properties of selected 2-pyrazoline derivatives. J. Mater. Chem. 1998, 8, 1725–1730. [Google Scholar] [CrossRef]
- You, X.Z. Molecular-based Materials -Opto-electronic Functional Compounds; Science and Technology Publishing Company: Shanghai, China, 2001; pp. 1–164. [Google Scholar]
- Sheldrick, G.M. SHELXTL; v5 Reference Manual; Siemens Analytical X-Ray Systems: Madison, WI, USA, 1997. [Google Scholar]
- Wilson, A.J. International Table for X-Ray Crystallography; Kluwer Academic: Dordrecht, The Netherlands, 1992; Volume C, Tables 4.2.6.8 and 6.1.1.4; pp. 219–222, 500–502, respectively. [Google Scholar]
- Peng, C.; Ayala, P.Y.; Schlegel, H.B.; Frisch, M.J. Using redundant internal coordinates to optimize equilibrium geometries and transition states. J. Comput. Chem. 1996, 17, 49–56. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Montgomery, J.A.; Vreven, T., Jr.; Kudin, K.N.; Burant, J.C.; et al. Gaussian 03W; Gaussian, Inc.: Wallingford, CT, USA, 2004. [Google Scholar]
- Dewar, M.J.S.; Thiel, W. Ground states of molecules. 38. The MNDO method. Approximations and parameters. J. Am. Chem. Soc. 1977, 99, 4899–4907. [Google Scholar] [CrossRef]
- Stewart, J.J.P. Optimization of parameters for semiempirical methods I. Method. J. Comput. Chem. 1989, 10, 209–220. [Google Scholar] [CrossRef]
- Stewart, J.J.P. QCPE Program 455 (1983); Version 6.0; Indiana University: Bloomington, IN, USA, 1990. [Google Scholar]
- Sample Availability: Sample of the title compound is available from authors.
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Guo, H.-M.; Zhao, P.-S.; Wu, Q.; Li, Y.-F. Synthesis, Crystal Structure, Spectra and Quantum Chemical Study on 1-Phenyl-3-(4-nitrophenyl)-5-(2-thienyl)-2-pyrazoline. Molecules 2014, 19, 5313-5324. https://doi.org/10.3390/molecules19045313
Guo H-M, Zhao P-S, Wu Q, Li Y-F. Synthesis, Crystal Structure, Spectra and Quantum Chemical Study on 1-Phenyl-3-(4-nitrophenyl)-5-(2-thienyl)-2-pyrazoline. Molecules. 2014; 19(4):5313-5324. https://doi.org/10.3390/molecules19045313
Chicago/Turabian StyleGuo, Huan-Mei, Pu-Su Zhao, Qian Wu, and Yu-Feng Li. 2014. "Synthesis, Crystal Structure, Spectra and Quantum Chemical Study on 1-Phenyl-3-(4-nitrophenyl)-5-(2-thienyl)-2-pyrazoline" Molecules 19, no. 4: 5313-5324. https://doi.org/10.3390/molecules19045313
APA StyleGuo, H.-M., Zhao, P.-S., Wu, Q., & Li, Y.-F. (2014). Synthesis, Crystal Structure, Spectra and Quantum Chemical Study on 1-Phenyl-3-(4-nitrophenyl)-5-(2-thienyl)-2-pyrazoline. Molecules, 19(4), 5313-5324. https://doi.org/10.3390/molecules19045313