Synthesis of the New Ring System Bispyrido[4',3':4,5]pyrrolo [1,2-a:1',2'-d]pyrazine and Its Deaza Analogue

Abstract
:1. Introduction


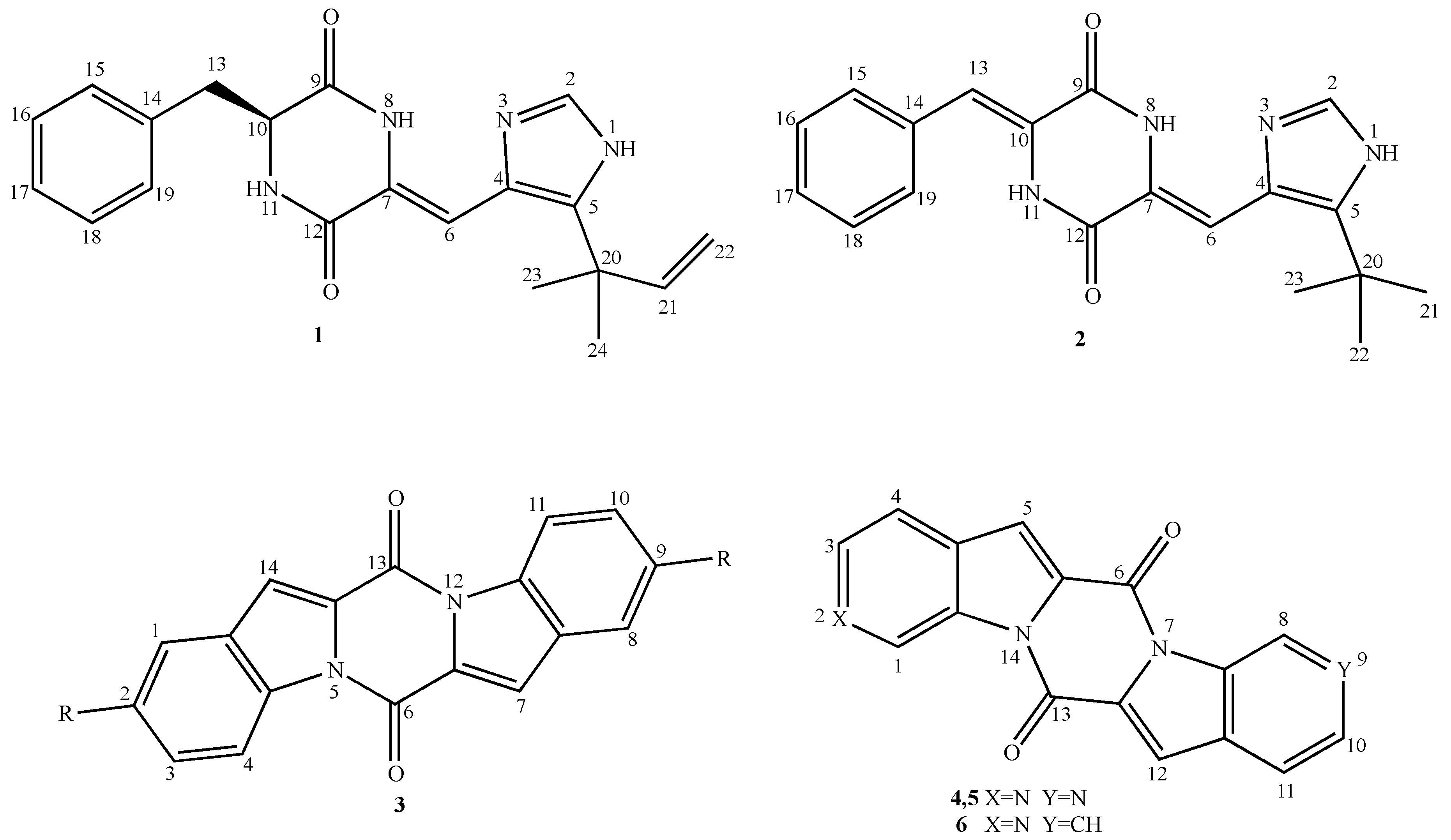{kind=link}
{kind=link}
{kind=link}
| Compound | 3HKD | 1SA0 |
|---|---|---|
| 4a | −8.927 | −6.643 |
| 4b | −9.562 | −6.675 |
| 4c | −9.354 | −6.007 |
| 4d | nd | −6.455 |
| 5a | −9.289 | −6.661 |
| 5b | −9.641 | −6.700 |
| 5c | −9.203 | −4.832 |
| 5d | −9.739 | −6.477 |
| 5e | −9.653 | −6.122 |
| 6a | −9.299 | −6.705 |
| 6b | −9.648 | −6.150 |
| 6c | −9.690 | −6.380 |
| 6d | −9.718 | −6.888 |

2. Results and Discussion

| Compound | R1 | R2 | R3 | R4 | Yields(%) |
|---|---|---|---|---|---|
| 4a | H | H | H | H | 25 |
| 4b | Cl | H | H | Cl | 30 |
| 4c | OCH3 | H | H | OCH3 | 20 |
| 4d | H | OCH3 | OCH3 | H | 28 |
| 5a | H | H | OCH3 | H | 40 |
| 5b | OCH3 | H | OCH3 | H | 55 |
| 5c | H | H | H | OCH3 | 42 |
| 5d | Cl | H | OCH3 | H | 44 |
| 5e | Cl | H | H | OCH3 | 45 |
| 6a | H | H | - | - | 33 |
| 6b | Cl | H | - | - | 65 |
| 6c | OCH3 | H | - | - | 65 |
| 6d | H | OCH3 | - | - | 30 |
3. Experimental Section
3.1. Chemistry
3.1.1. General Procedure for the Preparation of 2-Methoxy-pyridines 7c,d
3.1.2. General Procedure for the Preparation of Ethyl-3-(nitropyridin-4-yl)-2-oxopropanoates 8a,b
3.1.3. General Procedure for the Preparation of Ethyl-3-(nitropyridin-4-yl)-2-oxopropanoates 8c,d
3.1.4. General Procedure for the Preparation of Ethyl 1H-pyrrolo[2,3-c]pyridine-2-carboxylates 9a,b
3.1.5. General Procedure for the Preparation of Ethyl 1H-pyrrolo[2,3-c]pyridine-2-carboxylates 9c,d
3.1.6. General Procedure for the Preparation of 1H-pyrrolo[2,3-c]pyridine-2-carboxylic Acids 10a–d
3.1.7. General Procedure for the Preparation of 6H,13H-Bispyrido[4',3':4,5]pyrrolo[1,2-a:1',2'-d]pyrazine-6,13-diones 4a–d
3.1.8. General Procedure for the Preparation of 6H,13H-bispyrido[4',3':4,5]pyrrolo[1,2-a:1',2'-d]pyrazine-6,13-diones 5a–e
3.1.9. General Procedure for the Preparation of 6H,13H-Pyrido[4'',3'':4',5']pyrrolo[1',2':4,5]pyrazino[1,2-a]indole-6,13-diones 6a–d
3.2. Docking
3.3. Biology
Methodology of the in Vitro Cancer Screen
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Hilton, S.; Rossiter, S. Three heterocyclic ring fused (5-6-5). In Comprehensive Heterocyclic Chemistry III; Jones, K., Ed.; Elsevier Ltd.: Oxford, UK, 2008; Volume 12, pp. 747–751. [Google Scholar]
- Kanoh, K.; Kohno, S.; Asari, T.; Harada, T.; Katada, J.; Muramatsu, M.; Kawashima, H.; Sekiya, H.; Uno, I. (−)-Phenylahistin: A new mammalian cell cycle inhibitor produced by Aspergillus ustus. Bioorg. Med. Chem. Lett. 1997, 7, 2847–2852. [Google Scholar] [CrossRef]
- Kanoh, K.; Kohno, S.; Katada, J.; Takahashi, J.; Uno, I. (−)-Phenylahistin arrests cells in mitosis by inhibiting tubulin polymerization. J. Antibiot. 1999, 52, 134–141. [Google Scholar] [CrossRef]
- Kanoh, K.; Kohno, S.; Katada, J.; Hayashi, Y.; Muramatsu, M.; Uno, I. Antitumor activity of phenylahistin in vitro and in vivo. Biosci. Biotechnol. Biochem. 1999, 63, 1130–1133. [Google Scholar] [CrossRef]
- Kanoh, K.; Kohno, S.; Katada, J.; Takahashi, J.; Uno, I.; Hayashi, Y. Synthesis and biological activities of phenylahistin derivatives. Bioorg. Med. Chem. 1999, 7, 1451–1457. [Google Scholar] [CrossRef]
- Yamazaki, Y.; Sumikura, M.; Hidaka, K.; Yasui, H.; Kiso, Y.; Yakushiji, F.; Hayashi, Y. Anti-microtubule “plinabulin” chemical probe KPU-244-B3 labeled both α- and β-tubulin. Bioorg. Med. Chem. 2010, 18, 3169–3174. [Google Scholar] [CrossRef]
- Yamazaki, Y.; Tanaka, K.; Nicholson, B.; Deyanat-Yazdi, G.; Potts, B.; Yoshida, T.; Oda, A.; Kitagawa, T.; Orikasa, S.; Kiso, Y.; et al. Synthesis and structure-activity relationship study of antimicrotubule agents Phenylahistin derivatives with a didehydropiperazine-2,5-dione structure. J. Med. Chem. 2012, 55, 1056–1071. [Google Scholar] [CrossRef]
- Boger, D.L.; Fink, B.E.; Hedrick, M.P. A new class of highly cytotoxic diketopiperazines. Bioorg. Med. Chem. Lett. 2000, 10, 1019–1020. [Google Scholar] [CrossRef]
- Paleni, D. Preparation of Dioxopyrazinodiindoles as Neoplasm Inhibitors. FR 2650590, 8 February 1991. [Google Scholar]
- Paleni, D. Preparation and Formulation of Dioxopyrazinodiindoles as Cell Differentiation Inhibitors. DE 4102921, 6 August 1992. [Google Scholar]
- Barraja, P.; Caracausi, L.; Diana, P.; Carbone, A.; Montalbano, A.; Cirrincione, G.; Brun, P.; Palù, G.; Castagliuolo, I.; Dall’Acqua, F.; et al. Synthesis of pyrrolo[3,2-h]quinolinones with good photochemotherapeutic activity and no DNA damage. Bioorg. Med. Chem. 2010, 18, 4830–4843. [Google Scholar] [CrossRef]
- Barraja, P.; Caracausi, L.; Diana, P.; Montalbano, A.; Carbone, A.; Salvador, A.; Brun, P.; Castagliuolo, I.; Tisi, S.; Dall’Acqua, F.; et al. Pyrrolo[3,2-h]quinazolines as photochemotherapeutic agents. ChemMedChem 2011, 6, 1238–1248. [Google Scholar] [CrossRef]
- Carbone, A.; Parrino, B.; Barraja, P.; Spanò, V.; Cirrincione, G.; Diana, P.; Maier, A.; Kelter, G.; Fiebig, H.-H. Synthesis and antiproliferative activity of 2,5-bis(3'-indolyl)pyrroles, analogues of the marine alkaloid nortopsentin. Mar. Drugs 2013, 11, 643–654. [Google Scholar] [CrossRef]
- Barraja, P.; Caracausi, L.; Diana, P.; Spanò, V.; Montalbano, A.; Carbone, A.; Parrino, B.; Cirrincione, G. Synthesis and antiproliferative activity of the ring system [1,2]oxazolo[4,5-g]indole. ChemMedChem 2012, 7, 1901–1904. [Google Scholar] [CrossRef]
- Diana, P.; Carbone, A.; Barraja, P.; Montalbano, A.; Parrino, B.; Lopergolo, A.; Pennati, M.; Zaffaroni, N; Cirrincione, G. Synthesis and antitumor activity of 3-(2-phenyl-1,3-thiazol-4-yl)-1H-indoles and 3-(2-phenyl-1,3-thiazol-4-yl)-1H-7-azaindoles. ChemMedChem 2011, 6, 1300–1309. [Google Scholar] [CrossRef]
- Carbone, A.; Pennati, M.; Parrino, B.; Lopergolo, A.; Barraja, P.; Montalbano, A.; Spanò, V.; Sbarra, S.; Doldi, V.; de Cesare, M.; et al. Novel 1H-pyrrolo[2,3-b]pyridine derivatives nortopsentin analogues: Synthesis and antitumor activity in peritoneal mesothelioma experimental models. J. Med. Chem. 2013, 56, 7060–7072. [Google Scholar] [CrossRef]
- Carbone, A.; Pennati, M.; Barraja, P.; Montalbano, A.; Parrino, B.; Spanò, V.; Lopergolo, A.; Sbarra, S.; Doldi, V.; Zaffaroni, N.; et al. Synthesis and antiproliferative activity of substituted 3-[2-(1H-indol-3-yl)-1,3-thiazol-4-yl]-1H-pyrrolo[3,2-b]pyridines, marine alkaloid nortopsentin analogues. Curr. Med. Chem. 2014, 21, 1654–1666. [Google Scholar] [CrossRef]
- Barraja, P.; Diana, P.; Montalbano, A.; Dattolo, G.; Cirrincione, G.; Viola, G.; Vedaldi, D.; Dall’Acqua, F. Pyrrolo[2,3-h]quinolinones: A new ring system with potent photoantiproliferative activity. Bioorg. Med. Chem. 2006, 14, 8712–8728. [Google Scholar] [CrossRef]
- Barraja, P.; Spanò, V.; Diana, P.; Carbone, A.; Cirrincione, G.; Vedaldi, D.; Salvador, A.; Viola, G.; Dall’Acqua, F. Pyrano[2,3-e]isoindol-2-ones, a new angelicin heteroanalogues. Bioorg. Med. Chem. Lett. 2009, 19, 1711–1714. [Google Scholar] [CrossRef]
- Barraja, P.; Spanò, V.; Diana, P.; Carbone, A.; Cirrincione, G. Synthesis of the new ring system 6,8-dihydro-5H-pyrrolo[3,4-h]quinazoline. Tetrahedron Lett. 2009, 50, 5389–5391. [Google Scholar]
- Barraja, P.; Diana, P.; Montalbano, A.; Carbone, A.; Viola, G.; Basso, G.; Salvador, A.; Vedaldi, D.; Dall’Acqua, F.; Cirrincione, G. Pyrrolo[3,4-h]quinolinones a new class of photochemotherapeutic agents. Bioorg. Med. Chem. 2011, 19, 2326–2341. [Google Scholar] [CrossRef]
- Spanò, V.; Montalbano, A.; Carbone, A.; Parrino, B.; Diana, P.; Cirrincione, G.; Castagliuolo, I.; Brun, P.; Issinger, O.-G.; Tisi, S.; et al. Synthesis of a new class of pyrrolo[3,4-h]quinazolines with antimitotic activity. Eur. J. Med. Chem. 2014, 74, 340–357. [Google Scholar] [CrossRef]
- Barraja, P.; Spanò, V.; Giallombardo, D.; Diana, P.; Montalbano, A.; Carbone, A.; Parrino, B.; Cirrincione, G. Synthesis of [1,2]oxazolo[5,4-e]indazoles as antitumour agents. Tetrahedron 2013, 69, 6474–6477. [Google Scholar] [CrossRef]
- Ravelli, R.B.G.; Gigant, B.; Curmi, P.A.; Jourdain, I.; Lachkar, S.; Sobel, A.; Knossow, M. Insight into tubulin regulation from a complex with colchicine and a stathmin-like domain. Nature 2004, 428, 198–202. [Google Scholar]
- Dorléans, A.; Gigant, B.; Ravelli, R.B.G.; Mailliet, P.; Mikol, V.; Knossow, M. Variations in the colchicine-binding domain provide insight into the structural switch of tubulin. Proc. Natl. Acad. Sci. USA 2009, 106, 13775–13779. [Google Scholar]
- Arai, T. Inhibition of microtubule assembly in vitro by TN-16, a synthetic antitumor drug. FEBS Lett. 1983, 155, 273–276. [Google Scholar] [CrossRef]
- Barbier, P.; Dorléans, A.; Devred, F; Sanz, L.; Allegro, D.; Alfonso, C.; Knossow, M.; Peyrot, V.; Andreu, J.M. Stathmin and interfacial microtubule inhibitors recognize a naturally curved conformation of tubulin dimmers. J. Biol. Chem. 2010, 285, 31672–31681. [Google Scholar] [CrossRef]
- Glide, version 5.9; Schrödinger, LLC: New York, NY, USA, 2013.
- Bradley, S.E.; Krulle, T.M.; Murray, P.J.; Procter, M.J.; Rowley, R.J.; Sambrook, S.C.P.; Thomas, G.H. Preparation of Pyrrolopyridine-2-Carboxylic Acid Amide as Inhibitors of Glycogen Phosphorylase. WO 2004104001, 2 December 2004. [Google Scholar]
- Casara, P.; le Diguarher, T.; Durand, D.; Geneste, O.; Hickman, J. New Tricyclic Derivatives, Their Preparation as Pro-Apoptotic and Antitumor Agents and Their Pharmaceutical Compositions Containing Them. WO 2010007248, 21 January 2010. [Google Scholar]
- Evans, G.B.; Furneaux, R.H.; Hutchison, T.L.; Kezar, H.S.; Morris, P.E., Jr.; Schramm, V.L.; Tyler, P.C. Addition of lithiated 9-deazapurine derivatives to a carbohydrate cyclic imine: Convergent synthesis of the aza-C-nucleoside immucillins. J. Org. Chem. 2001, 66, 5723–5730. [Google Scholar] [CrossRef]
- Monks, A.; Scudiero, D.; Skehan, P.; Shoemaker, R.; Paull, K.; Vistica, D.; Hose, C.; Langley, J.; Cronise, P.; Vaigro-Wolff, A.; et al. Feasibility of a high-flux anticancer drug screen using a diverse panel of cultured human tumor cell lines. J. Natl. Cancer Inst. 1991, 83, 757–766. [Google Scholar] [CrossRef]
- Frydman, B.; Despuy, M.E.; Rapoport, H. Pyrroles from azaindoles. A synthesis of porphobilinogen. J. Am. Chem. Soc. 1965, 3530–3531. [Google Scholar]
- Dubois, L.; Evanno, Y.; Malanda, A. Preparation Of 1-(Arylalkyl)-1H-Pyrrolopyridine-2-Carboxamide Derivatives as VR1 Type Capsaicin Receptor Antagonists. WO 2007010138, 25 January 2007. [Google Scholar]
- Frydman, B.; Reil, S.J.; Boned, J.; Rapoport, H. Synthesis of substituted 4- and 6-azaindoles. J. Org. Chem. 1968, 33, 3762–3766. [Google Scholar] [CrossRef]
- Qiao, G.G.; Meutermans, W.; Wong, M.W.; Traeubel, M.; Wentrup, C. (Cyanovinyl)ketenes from azafulvenones. An apparent retro-Wolff rearrangement. J. Am. Chem. Soc. 1996, 118, 3852–3861. [Google Scholar]
- Protein Data Bank. Available online: http://www.rcsb.org/pdb (accessed on 28 August 2014).
- NCI-60 DTP Human tumor cell line screen. Available online: http://dtp.nci.nih.gov/branches/btb/ivclsp.html (accessed on 20 July 2012).
- Sample Availability: Not available.
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Parrino, B.; Spanò, V.; Carbone, A.; Barraja, P.; Diana, P.; Cirrincione, G.; Montalbano, A. Synthesis of the New Ring System Bispyrido[4',3':4,5]pyrrolo [1,2-a:1',2'-d]pyrazine and Its Deaza Analogue. Molecules 2014, 19, 13342-13357. https://doi.org/10.3390/molecules190913342
Parrino B, Spanò V, Carbone A, Barraja P, Diana P, Cirrincione G, Montalbano A. Synthesis of the New Ring System Bispyrido[4',3':4,5]pyrrolo [1,2-a:1',2'-d]pyrazine and Its Deaza Analogue. Molecules. 2014; 19(9):13342-13357. https://doi.org/10.3390/molecules190913342
Chicago/Turabian StyleParrino, Barbara, Virginia Spanò, Anna Carbone, Paola Barraja, Patrizia Diana, Girolamo Cirrincione, and Alessandra Montalbano. 2014. "Synthesis of the New Ring System Bispyrido[4',3':4,5]pyrrolo [1,2-a:1',2'-d]pyrazine and Its Deaza Analogue" Molecules 19, no. 9: 13342-13357. https://doi.org/10.3390/molecules190913342