
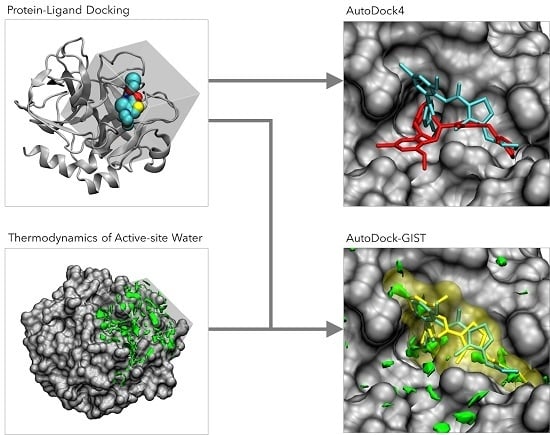
AutoDock-GIST: Incorporating Thermodynamics of Active-Site Water into Scoring Function for Accurate Protein-Ligand Docking

Abstract
:
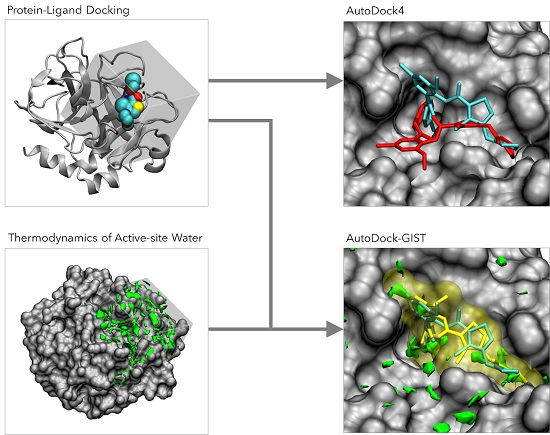
1. Introduction
2. Materials and Methods
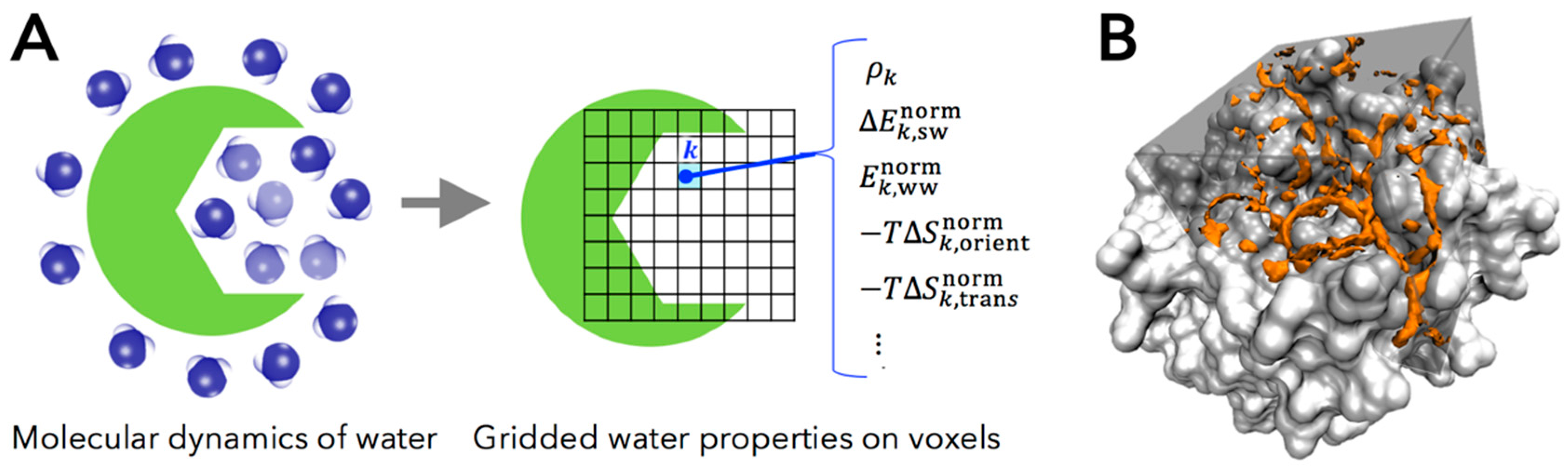
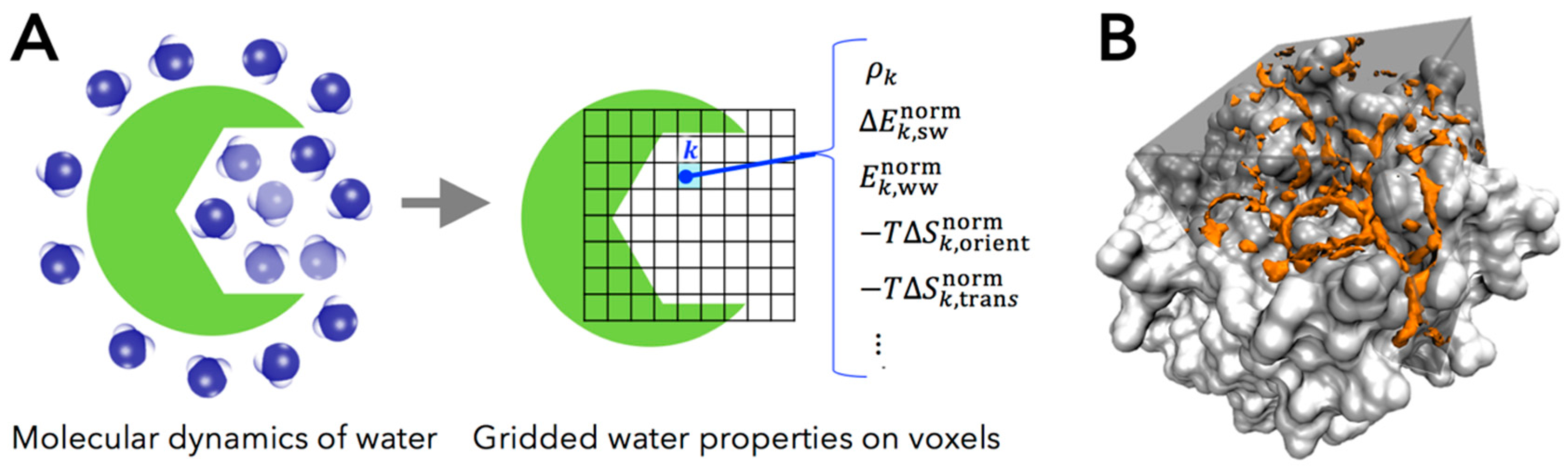
2.1. Grid Inhomogeneous Solvation Theory (GIST)
- , the number density of oxygen atom of water molecule found in a voxel , in units of the density in bulk region (i.e., the number density of bulk water ).
- , the mean energy of solute-water interaction per water molecule in a voxel (kcal/mol/water). This quantity is referenced to bulk water, in the trivial sense that the energetic contribution of solute-water interaction is zero in bulk region.
- , one-half the mean energy of water-water interaction per water molecule in a voxel with all other water molecules (kcal/mol/water). The one-half factor prevents double counting of two water-water interactions and preserves the total energy of neat water being written as the sum of the single water energy [38].
- , first-order orientational entropy per water molecule in a voxel (kcal/mole/water), referenced to bulk water (i.e., the orientational entropy of bulk water is set to be zero).
- , first-order translational entropy per water molecules in a voxel (kcal/mol/water), referenced to bulk water (i.e., the translational entropy of bulk water is set to be zero).
2.2. AutoDock4
2.3. Development of GIST-Based Desolvation Function and Its Incorporation into AutoDock4
2.4. Datasets and Preparation
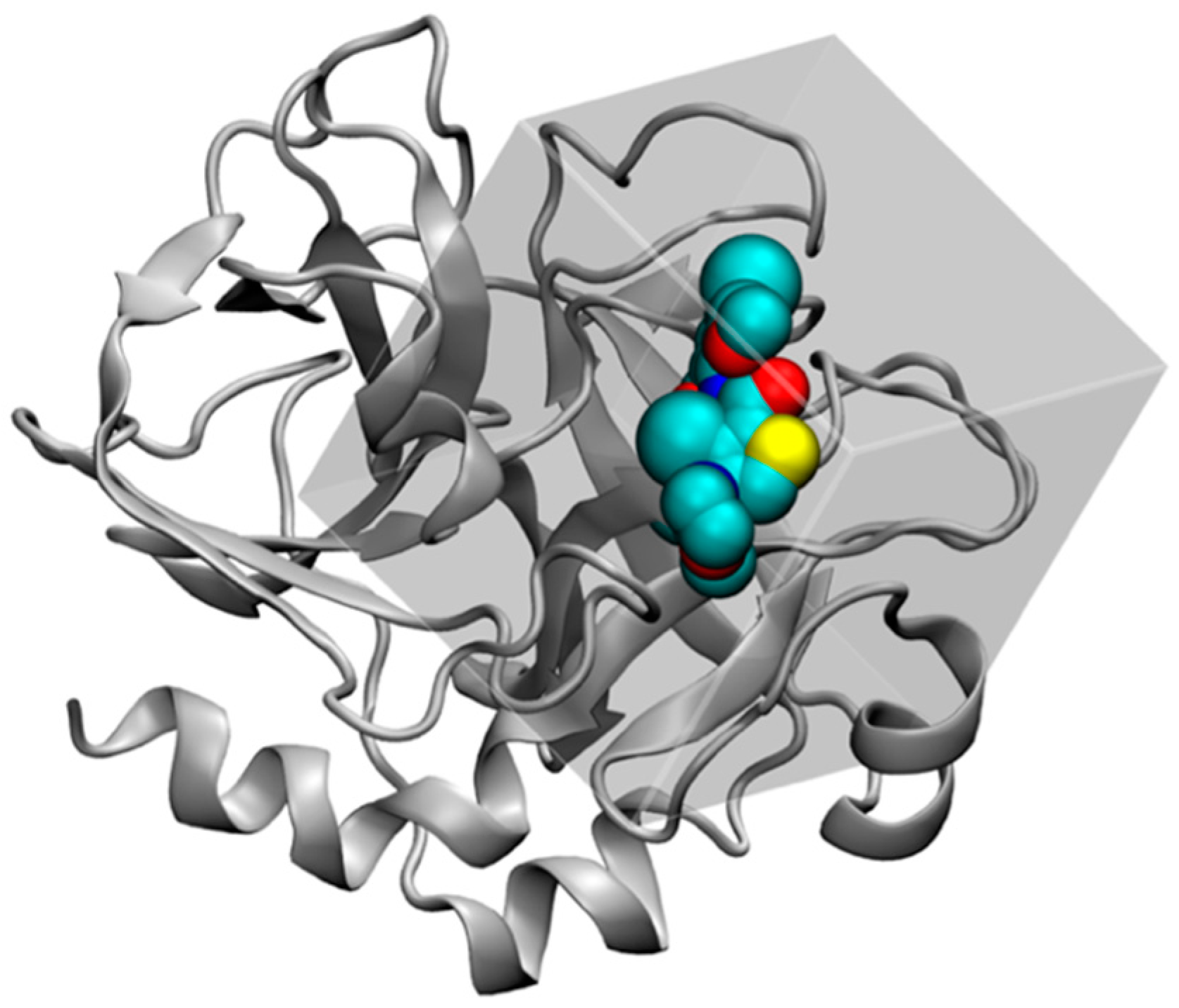
2.4.1. Structure Preparation and MD Simulation for FXa
2.4.2. GIST Calculation and Docking Set-up
2.4.3. Dataset Preparation and Docking Metrics
3. Results and Discussion
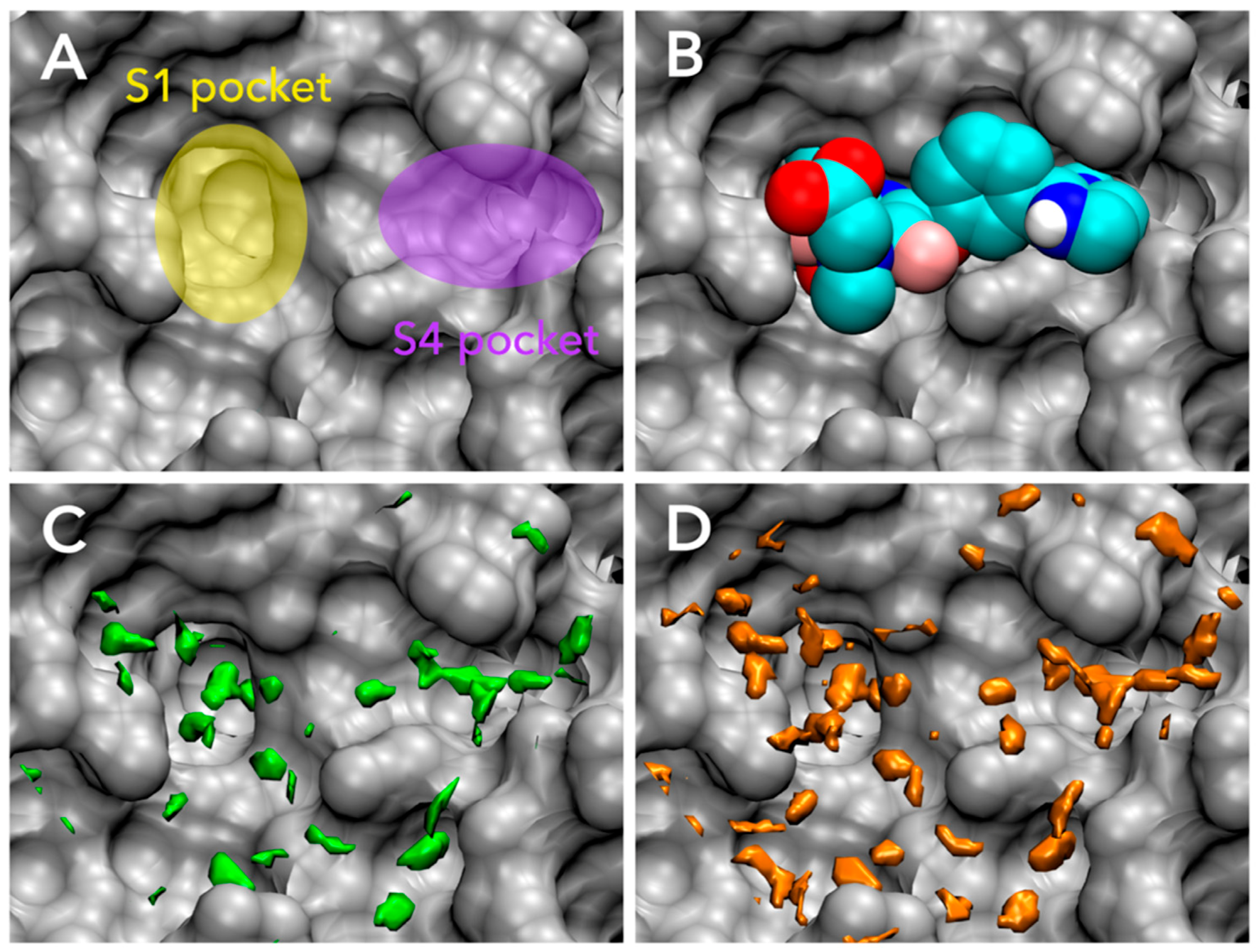
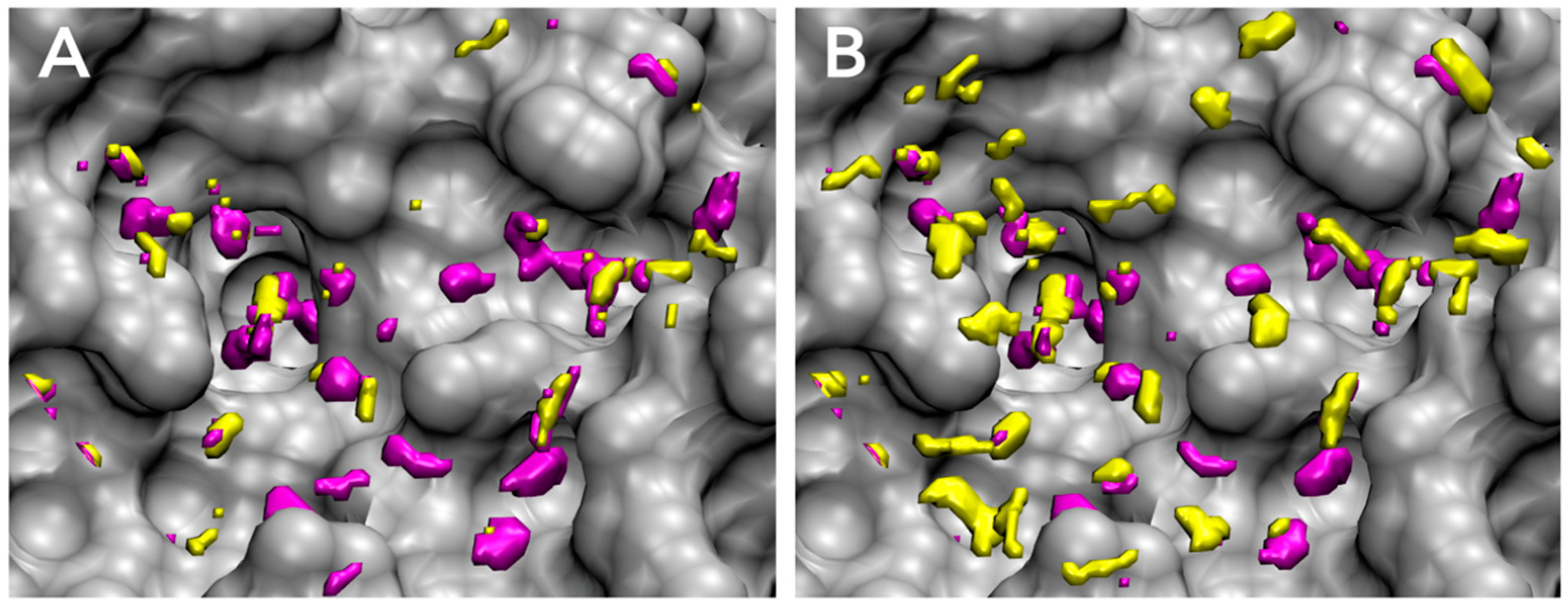
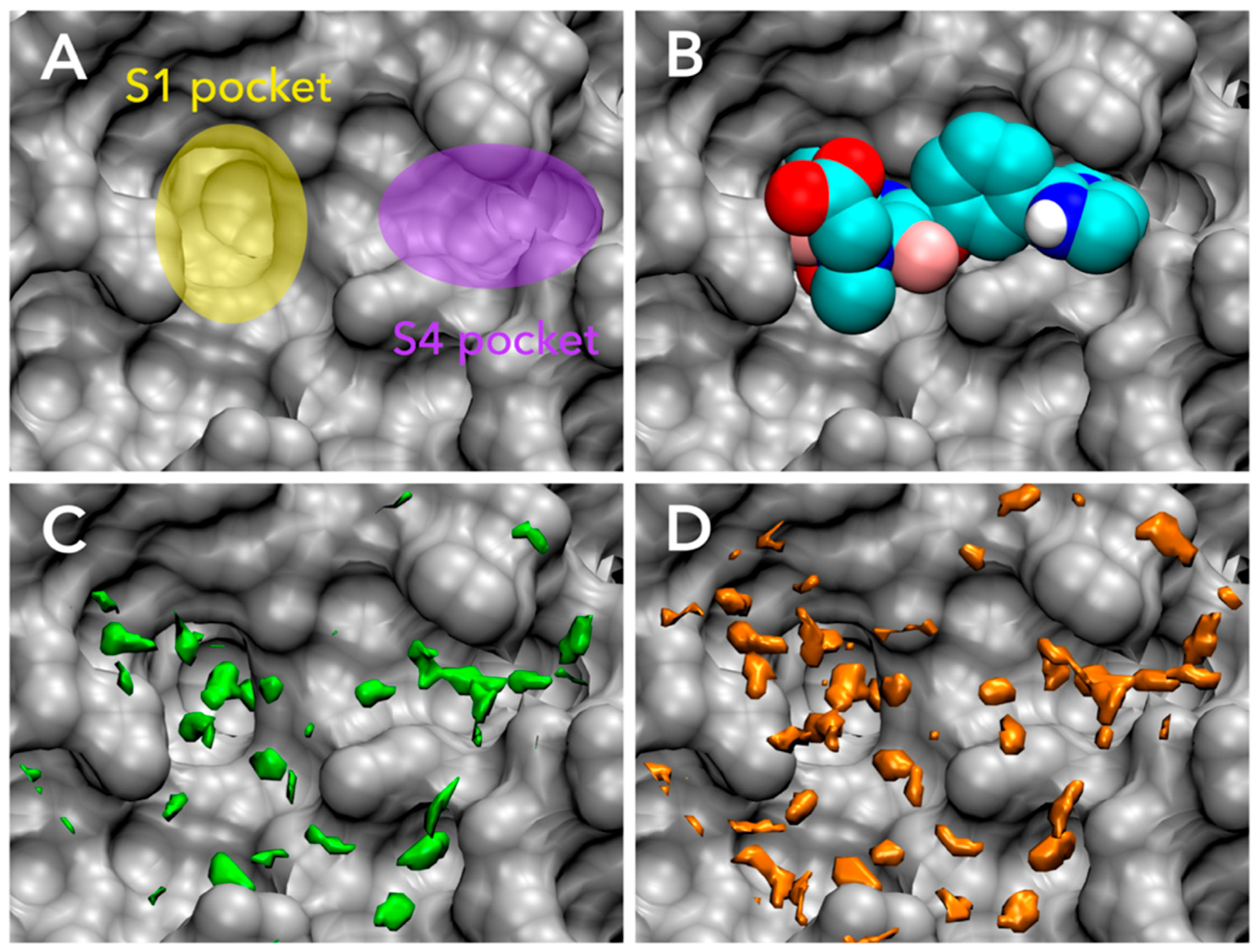
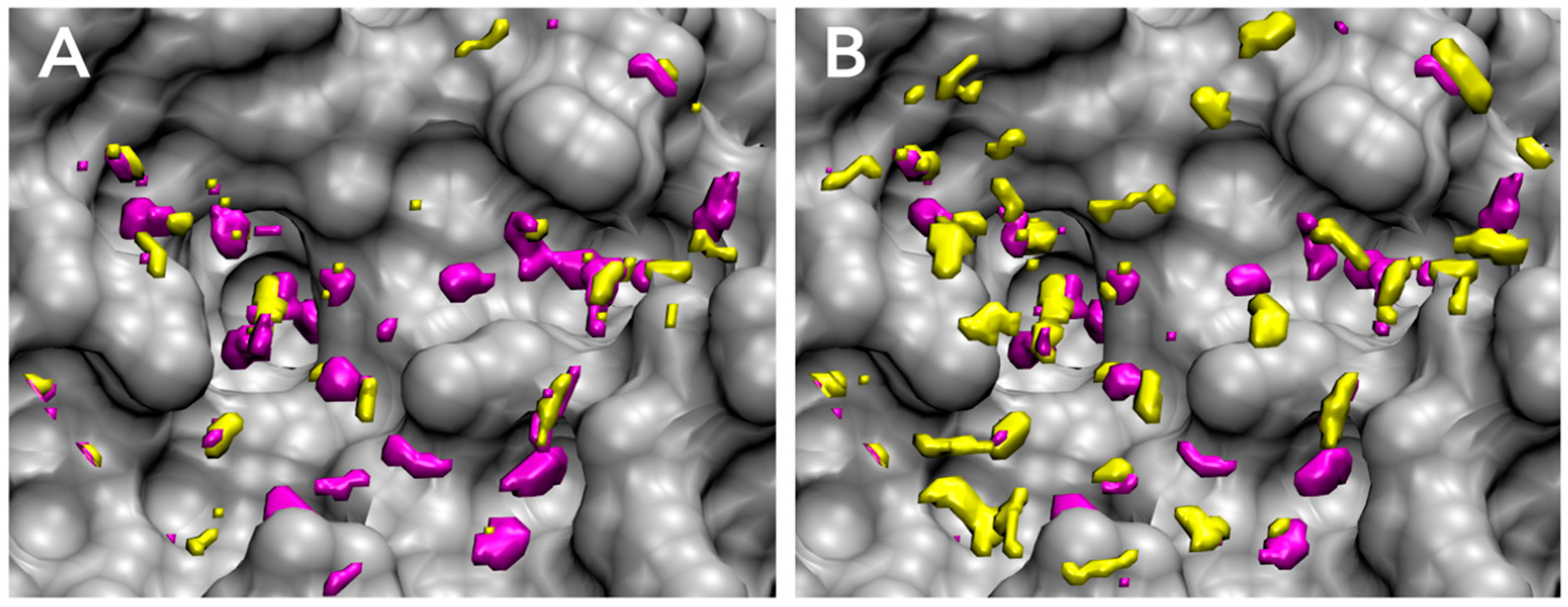
3.1. Parameter Fitting for GIST-Based Desolvation Function
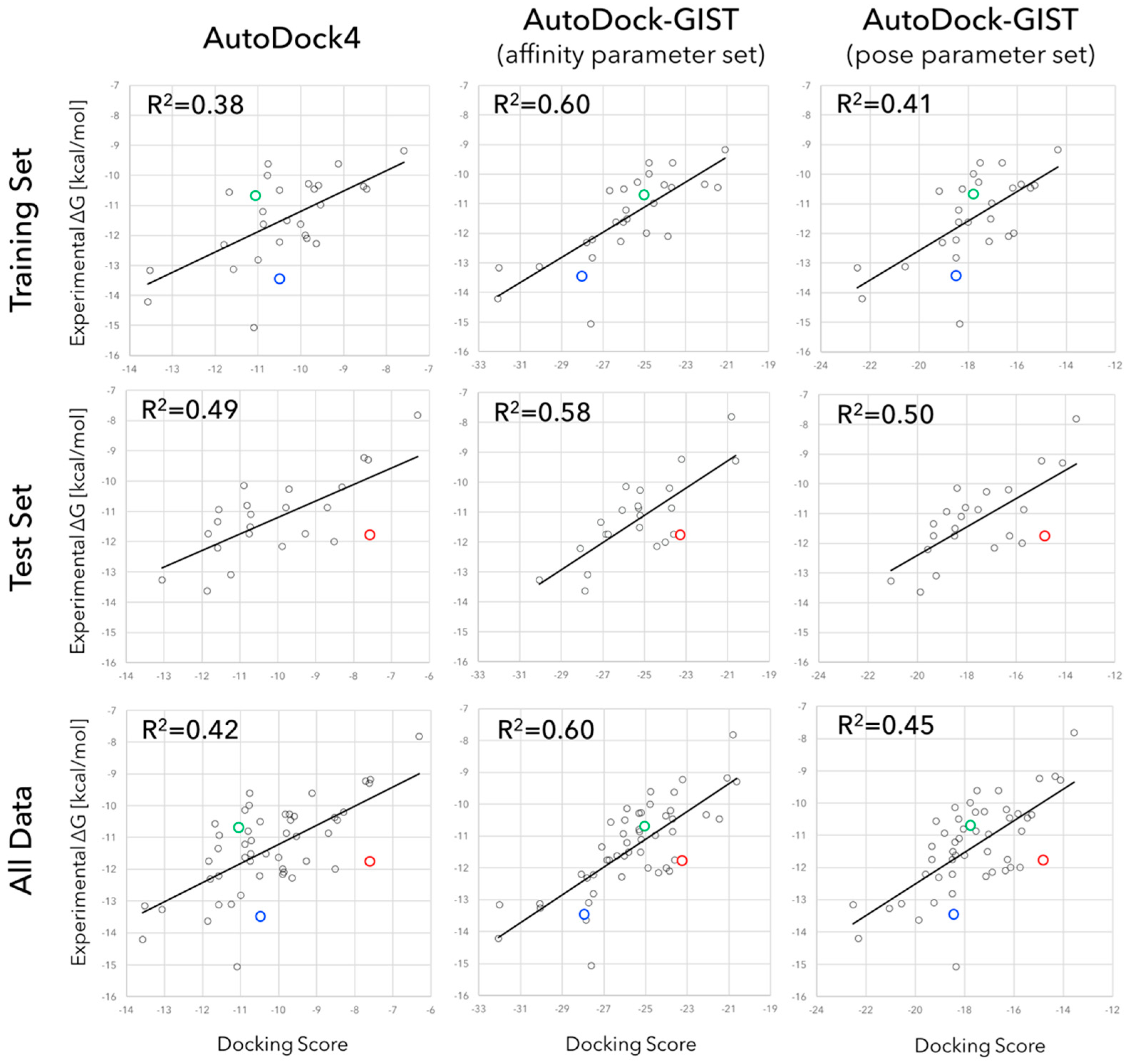
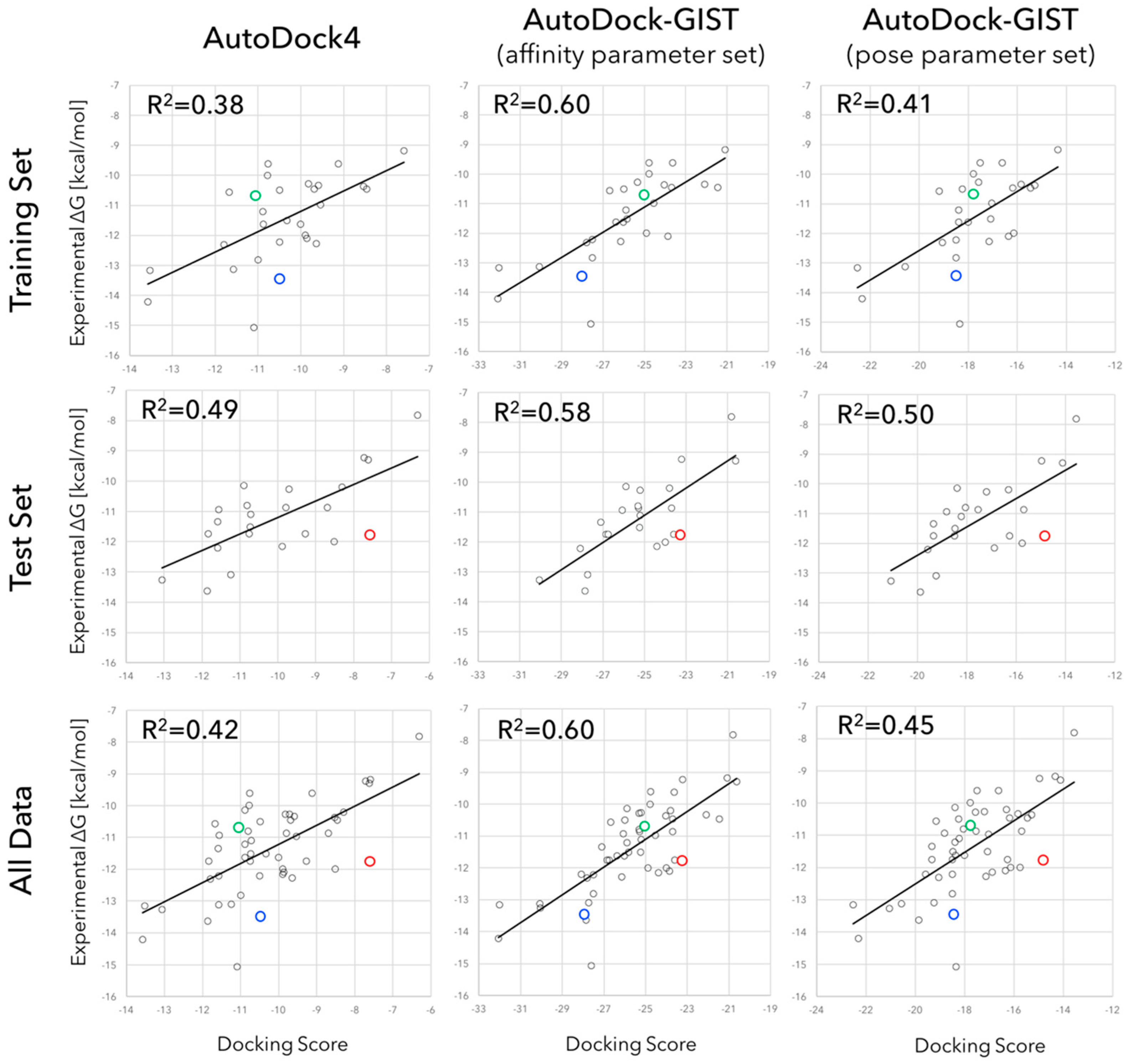
3.2. Accuracy of Binding Affinity Prediction for FXa Ligands
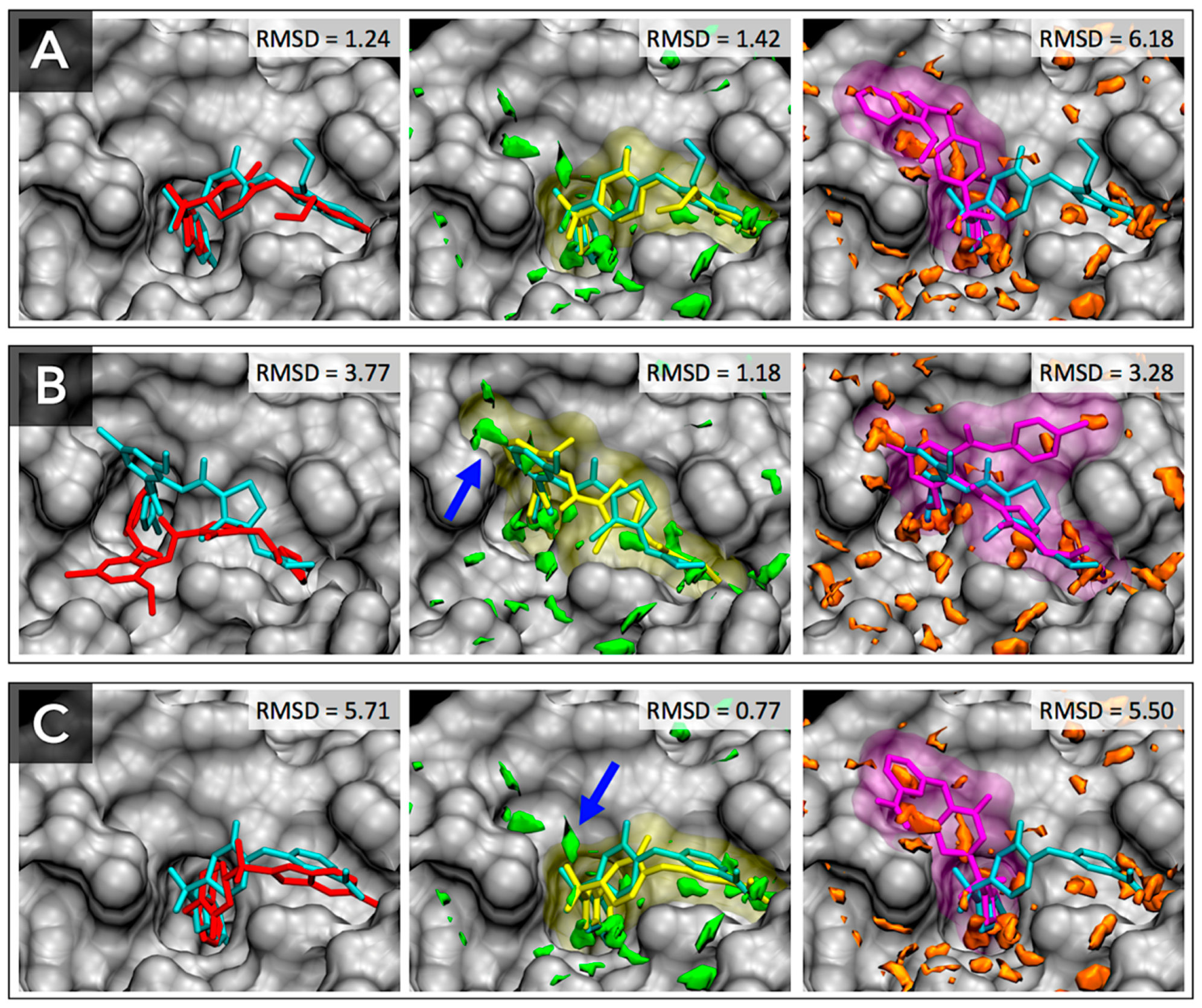
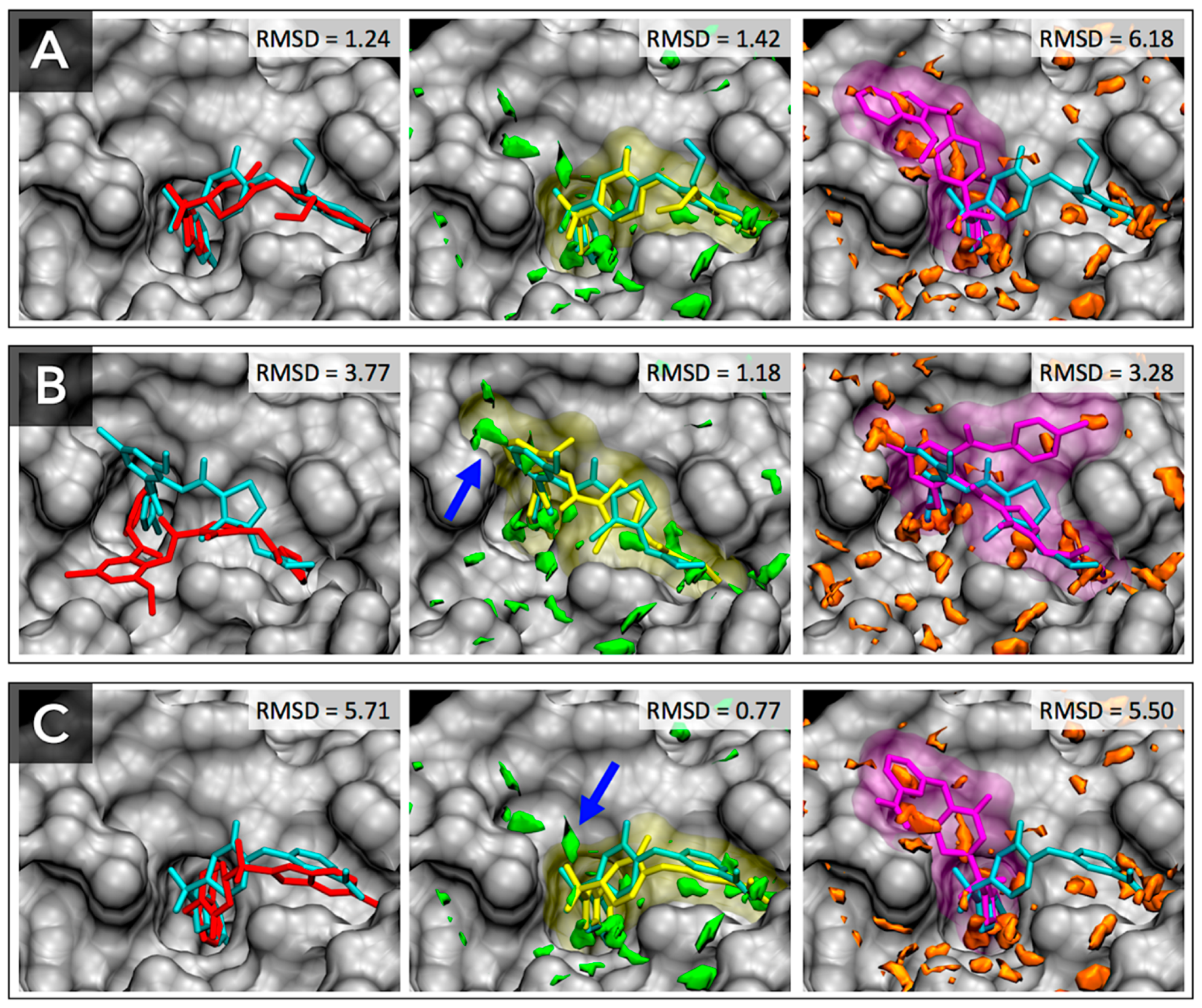
3.3. Docking Success Rate for FXa Ligands
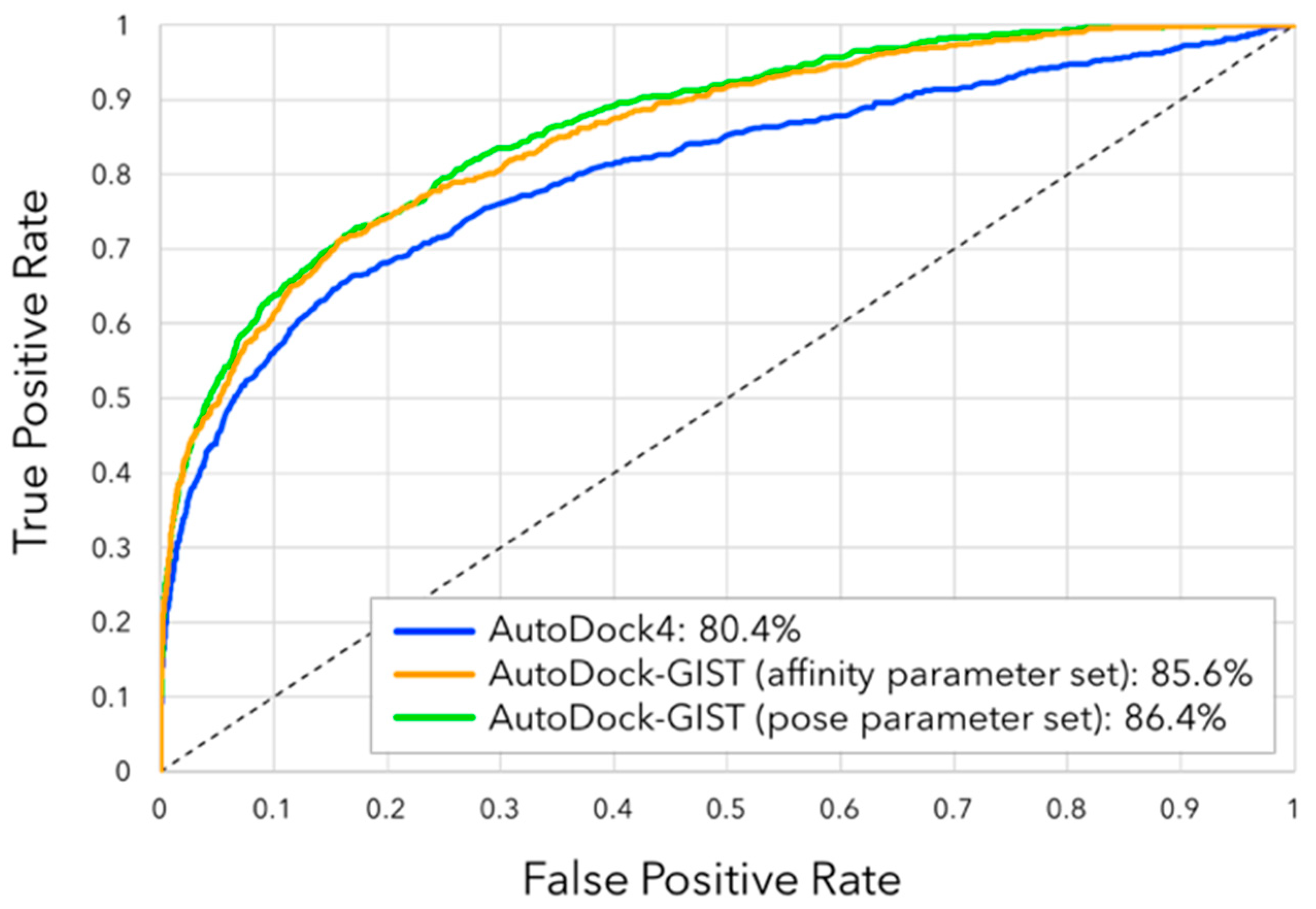
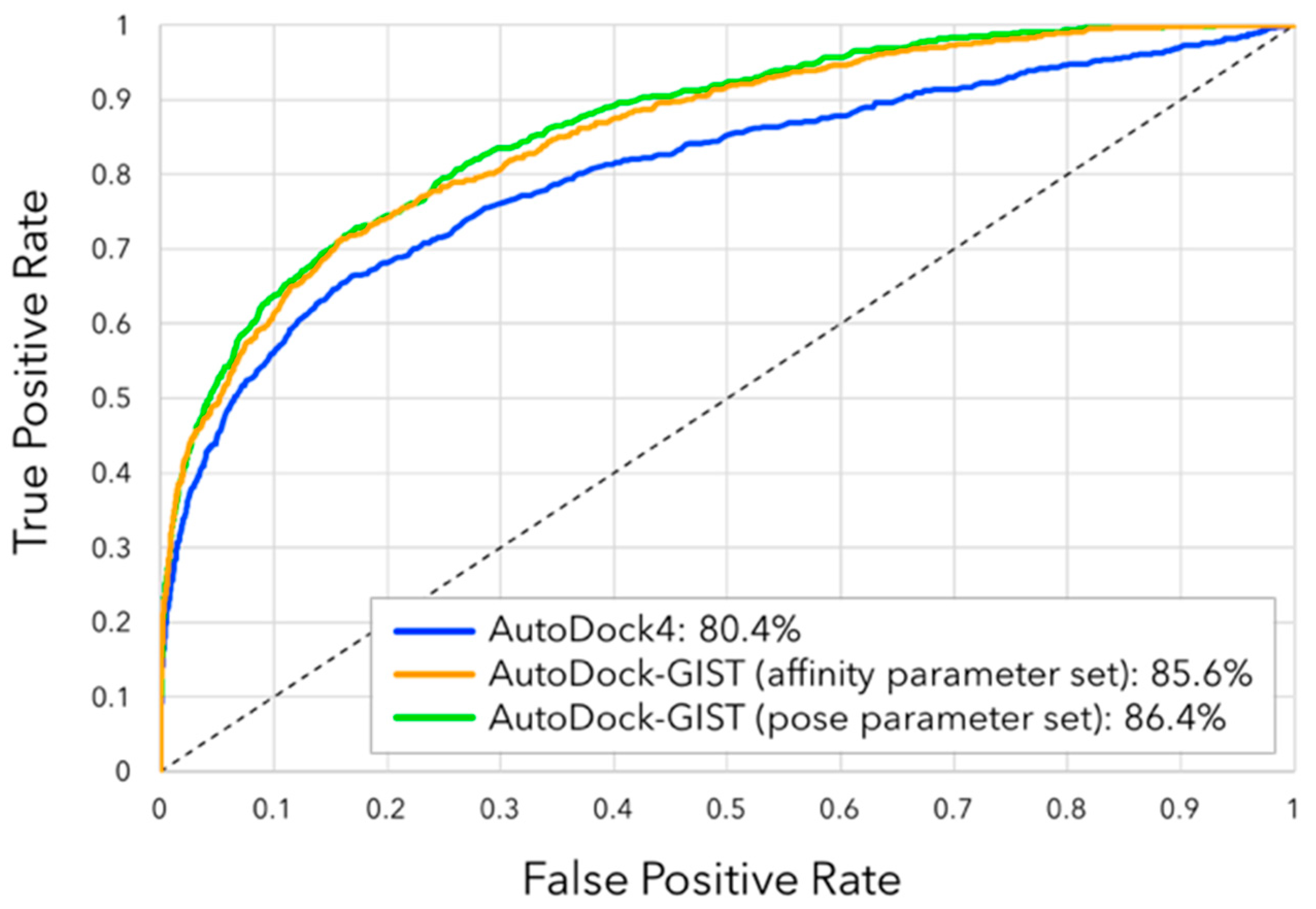
3.4. Virtual Screening Performance of AutoDock-GIST
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| SBDD | Structure-Based Drug Design |
| IST | Inhomogeneous Solvation Theory |
| GIST | Grid Inhomogeneous Solvation Theory |
| HAS | Hydration Site Analysis |
| MD | Molecular Dynamics |
| PDB | Protein Data Bank |
| RMSD | Root Mean Square Deviation |
| FXa | Factor Xa |
| DUD-E | Directory of Useful Decoys-Enhanced |
| ROC | Receiver Operating Characteristic |
| AUC | Area Under the Curve |
| EF | Enrichment Factor |
References
- Ladbury, J.E. Just add water! The effect of water on the specificity of protein-ligand binding sites and its potential application to drug design. Chem. Biol. 1996, 3, 973–980. [Google Scholar] [CrossRef]
- Poornima, C.S.; Dean, P.M. Hydration in drug design. 1. Multiple hydrogen-bonding features of water molecules in mediating protein-ligand interactions. J. Comput. Aided Mol. Des. 1995, 9, 500–512. [Google Scholar] [CrossRef] [PubMed]
- Hummer, G. Molecular binding: Under water’s influence. Nat. Chem. 2010, 2, 906–907. [Google Scholar] [CrossRef] [PubMed]
- Baron, R.; Setny, P.; McCammon, J.A. Water in Cavity−Ligand Recognition. J. Am. Chem. Soc. 2010, 132, 12091–12097. [Google Scholar] [CrossRef] [PubMed]
- Chung, E.; Henriques, D.; Renzoni, D.; Zvelebil, M.; Bradshaw, J.M.; Waksman, G.; Robinson, C.V.; Ladbury, J.E. Mass spectrometric and thermodynamic studies reveal the role of water molecules in complexes formed between SH2 domains and tyrosyl phosphopeptides. Structure 1998, 6, 1141–1151. [Google Scholar] [CrossRef]
- McPhalen, C.A.; James, M.N. Structural comparison of two serine proteinase-protein inhibitor complexes: Eglin-c-subtilisin Carlsberg and CI-2-subtilisin Novo. Biochemistry 1988, 27, 6582–6598. [Google Scholar] [CrossRef] [PubMed]
- Quiocho, F.A.; Wilson, D.K.; Vyas, N.K. Substrate specificity and affinity of a protein modulated by bound water molecules. Nature 1989, 340, 404–407. [Google Scholar] [CrossRef] [PubMed]
- Baron, R.; Setny, P.; Paesani, F. Water Structure, Dynamics, and Spectral Signatures: Changes upon model cavity–ligand recognition. J. Phys. Chem. B 2012, 116, 13774–13780. [Google Scholar] [CrossRef] [PubMed]
- Barillari, C.; Taylor, J.; Viner, R.; Essex, J.W. Classification of water molecules in protein binding sites. J. Am. Chem. Soc. 2007, 129, 2577–2587. [Google Scholar] [CrossRef] [PubMed]
- Michel, J.; Tirado-Rives, J.; Jorgensen, W.L. Prediction of the water content in protein binding sites. J. Phys. Chem. B 2009, 113, 13337–13346. [Google Scholar] [CrossRef] [PubMed]
- Raman, E.P.; Mackerell, A.D. Spatial analysis and quantification of the thermodynamic driving forces in protein-ligand binding: Binding site variability. J. Am. Chem. Soc. 2015, 137, 2608–2621. [Google Scholar] [CrossRef] [PubMed]
- Haider, K.; Huggins, D.J. Combining solvent thermodynamic profiles with functionality maps of the Hsp90 binding site to predict the displacement of water molecules. J. Chem. Inf. Model. 2013, 53, 2571–2586. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Lazaridis, T. Thermodynamics of buried water clusters at a protein-ligand binding interface. J. Phys. Chem. B 2006, 110, 1464–1475. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.M.; Xu, S.L.; Wawrzak, Z.; Basarab, G.S.; Jordan, D.B. Structure-based design of potent inhibitors of scytalone dehydratase: Displacement of a water molecule from the active site. Biochemistry 1998, 37, 17735–17744. [Google Scholar] [CrossRef] [PubMed]
- Robinson, D.; Bertrand, T.; Carry, J.-C.; Halley, F.; Karlsson, A.; Mathieu, M.; Minoux, H.; Perrin, M.-A.; Robert, B.; Schio, L.; et al. Differential Water Thermodynamics Determine PI3K-Beta/Delta Selectivity for Solvent-Exposed Ligand Modifications. J. Chem. Inf. Model. 2016, 56, 886–894. [Google Scholar] [CrossRef] [PubMed]
- Ladbury, J.E.; Klebe, G.; Freire, E. Adding calorimetric data to decision making in lead discovery: A hot tip. Nat. Rev. Drug Discov. 2010, 9, 23–27. [Google Scholar] [CrossRef] [PubMed]
- Barandun, L.J.; Ehrmann, F.R.; Zimmerli, D.; Immekus, F.; Giroud, M.; Grunenfelder, C.; Schweizer, W.B.; Bernet, B.; Betz, M.; Heine, A.; et al. Replacement of water molecules in a phosphate binding site by furanoside-appended lin-benzoguanine ligands of tRNA-guanine transglycosylase (TGT). Chem. A Eur. J. 2015, 21, 126–135. [Google Scholar] [CrossRef] [PubMed]
- Biela, A.; Nasief, N.N.; Betz, M.; Heine, A.; Hangauer, D.; Klebe, G. Dissecting the hydrophobic effect on the molecular level: The role of water, enthalpy, and entropy in ligand binding to thermolysin. Angew. Chem. Int. Ed. 2013, 52, 1822–1828. [Google Scholar] [CrossRef] [PubMed]
- Betz, M.; Wulsdorf, T.; Krimmer, S.G.; Klebe, G. Impact of surface water layers on protein-ligand binding: How well are experimental data reproduced by molecular dynamics simulations in a thermolysin test case? J. Chem. Inf. Model. 2016, 56, 223–233. [Google Scholar] [CrossRef] [PubMed]
- Pearlstein, R.A.; Hu, Q.Y.; Zhou, J.; Yowe, D.; Levell, J.; Dale, B.; Kaushik, V.K.; Daniels, D.; Hanrahan, S.; Sherman, W.; et al. New hypotheses about the structure-function of proprotein convertase subtilisin/kexin type 9: Analysis of the epidermal growth factor-like repeat a docking site using watermap. Proteins Struct. Funct. Bioinform. 2010, 78, 2571–2586. [Google Scholar] [CrossRef] [PubMed]
- Pearlstein, R.A.; Sherman, W.; Abel, R. Contributions of water transfer energy to protein-ligand association and dissociation barriers: Watermap analysis of a series of p38α MAP kinase inhibitors. Proteins Struct. Funct. Bioinform. 2013, 81, 1509–1526. [Google Scholar] [CrossRef] [PubMed]
- Bortolato, A.; Tehan, B.G.; Bodnarchuk, M.S.; Essex, J.W.; Mason, J.S. Water network perturbation in ligand binding: Adenosine A 2A antagonists as a case study. J. Chem. Inf. Model. 2013, 53, 1700–1713. [Google Scholar] [CrossRef] [PubMed]
- Barillari, C.; Duncan, A.L.; Westwood, I.M.; Blagg, J.; van Montfort, R.L.M. Analysis of water patterns in protein kinase binding sites. Proteins Struct. Funct. Bioinform. 2011, 79, 2109–2121. [Google Scholar] [CrossRef] [PubMed]
- Imai, T.; Hiraoka, R.; Kovalenko, A.; Hirata, F. Locating missing water molecules in protein cavities by the three-dimensional reference interaction site model theory of molecular solvation. Proteins Struct. Funct. Bioinform. 2006, 66, 804–813. [Google Scholar] [CrossRef] [PubMed]
- Bayden, A.S.; Moustakas, D.T.; Joseph-McCarthy, D.; Lamb, M.L. Evaluating Free Energies of Binding and Conservation of Crystallographic Waters Using SZMAP. J. Chem. Inf. Model. 2015, 55, 1552–1565. [Google Scholar] [CrossRef] [PubMed]
- Goodford, P.J. A computational procedure for determining energetically favorable binding sites on biologically important macromolecules. J. Med. Chem. 1985, 28, 849–857. [Google Scholar] [CrossRef] [PubMed]
- Goodfellow, J.M.; Vovelle, F. Biomolecular energy calculations using transputer technology. Eur. Biophys. J. 1989, 17, 167–172. [Google Scholar] [CrossRef] [PubMed]
- Pitt, W.R.; Murray-Rust, J.; Goodfellow, J.M. AQUARIUS2: Knowledge-based modeling of solvent sites around proteins. J. Comput. Chem. 1993, 14, 1007–1018. [Google Scholar] [CrossRef]
- Verdonk, M.L.; Cole, J.C.; Taylor, R. SuperStar: A knowledge-based approach for identifying interaction sites in proteins. J. Mol. Biol. 1999, 289, 1093–1108. [Google Scholar] [CrossRef] [PubMed]
- Abel, R.; Young, T.; Farid, R.; Berne, B.J.; Friesner, R.A. Role of the active-site solvent in the thermodynamics of factor Xa ligand binding. J. Am. Chem. Soc. 2008, 130, 2817–2831. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Lazaridis, T. Computing the Thermodynamic Contributions of Interfacial Water. Methods Mol. Biol. 2012, 819, 393–404. [Google Scholar] [PubMed]
- Hu, B.; Lill, M.A. WATsite: Hydration site prediction program with PyMOL interface. J. Comput. Chem. 2014, 35, 1255–1260. [Google Scholar] [CrossRef] [PubMed]
- Czapiewski, D.; Zielkiewicz, J. Structural properties of hydration shell around various conformations of simple polypeptides. J. Phys. Chem. B 2010, 114, 4536–4550. [Google Scholar] [CrossRef] [PubMed]
- Henchman, R.H.; McCammon, J.A. Extracting hydration sites around proteins from explicit water simulations. J. Comput. Chem. 2002, 23, 861–869. [Google Scholar] [CrossRef] [PubMed]
- Lazaridis, T. Inhomogeneous fluid approach to solvation thermodynamics. 1. Theory. J. Phys. Chem. B 1998, 102, 3531–3541. [Google Scholar] [CrossRef]
- Lazaridis, T. Inhomogeneous fluid approach to solvation thermodynamics. 2. Applications to Simple Fluids. J. Phys. Chem. B 1998, 102, 3542–3550. [Google Scholar] [CrossRef]
- Nguyen, C.N.; Young, T.K.; Gilson, M.K. Grid inhomogeneous solvation theory: Hydration structure and thermodynamics of the miniature receptor Cucurbit [7] uril. J. Chem. Phys. 2012, 137, 149901. [Google Scholar] [CrossRef]
- Nguyen, C.N.; Cruz, A.; Gilson, M.K.; Kurtzman, T. Thermodynamics of water in an enzyme active site: Grid-based hydration analysis of coagulation factor xa. J. Chem. Theory Comput. 2014, 10, 2769–2780. [Google Scholar] [CrossRef] [PubMed]
- Kitchen, D.B.; Decornez, H.; Furr, J.R.; Bajorath, J. Docking and scoring in virtual screening for drug discovery: Methods and applications. Nat. Rev. Drug Discov. 2004, 3, 935–949. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.-Y.; Zhang, H.-X.; Mezei, M.; Cui, M. Molecular docking: A powerful approach for structure-based drug discovery. Curr. Comput. Aided Drug Des. 2011, 7, 146–157. [Google Scholar] [CrossRef] [PubMed]
- Yuriev, E.; Agostino, M.; Ramsland, P.A. Challenges and advances in computational docking: 2009 in review. J. Mol. Recognit. 2011, 24, 149–164. [Google Scholar] [CrossRef] [PubMed]
- Okimoto, N.; Futatsugi, N.; Fuji, H.; Suenaga, A.; Morimoto, G.; Yanai, R.; Ohno, Y.; Narumi, T.; Taiji, M. High-performance drug discovery: Computational screening by combining docking and molecular dynamics simulations. PLoS Comput. Biol. 2009, 5, e1000528. [Google Scholar] [CrossRef] [PubMed]
- Graaf, D.C.; Pospisil, P.; Pos, W.; Folkers, G.; Vermeulen, N.P.E. Binding mode prediction of cytochrome P450 and thymidine kinase protein-ligand complexes by consideration of water and rescoring in automated docking. J. Med. Chem. 2005, 48, 2308–2318. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Zou, X. Inclusion of solvation and entropy in the knowledge-based scoring function for protein—Ligand interactions. J. Chem. Inf. Model. 2010, 50, 262–273. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Zhang, K.Y.J. Investigation on the effect of key water molecules on docking performance in CSARdock exercise. J. Chem. Inf. Model. 2013, 53, 1880–1892. [Google Scholar] [CrossRef] [PubMed]
- Huang, N.; Shoichet, B.K. Exploiting ordered waters in molecular docking. J. Med. Chem. 2008, 51, 4862–4865. [Google Scholar] [CrossRef] [PubMed]
- Verdonk, M.L.; Chessari, G.; Cole, J.C.; Hartshorn, M.J.; Murray, C.W.; Nissink, J.W.M.; Taylor, R.D.; Taylor, R. Modeling water molecules in protein-ligand docking using GOLD. J. Med. Chem. 2005, 48, 6504–6515. [Google Scholar] [CrossRef] [PubMed]
- Forli, S.; Olson, A.J. A force field with discrete displaceable waters and desolvation entropy for hydrated ligand docking. J. Med. Chem. 2012, 55, 623–638. [Google Scholar] [CrossRef] [PubMed]
- Huey, R.; Morris, G.M.; Olson, A.J.; Goodsell, D.S. A semiempirical free energy force field with charge-based desolvation. J. Comput. Chem. 2007, 28, 1145–1152. [Google Scholar] [CrossRef] [PubMed]
- Mysinger, M.M.; Shoichet, B.K. Rapid context-dependent ligand desolvation in molecular docking. J. Chem. Inf. Model. 2010, 50, 1561–1573. [Google Scholar] [CrossRef] [PubMed]
- Shoichet, B.K.; Leach, A.R.; Kuntz, I.D. Ligand solvation in molecular docking. Proteins Struct. Funct. Genet. 1999, 34, 4–16. [Google Scholar] [CrossRef]
- Alvarez-Garcia, D.; Barril, X. Molecular simulations with solvent competition quantify water displaceability and provide accurate interaction maps of protein binding sites. J. Med. Chem. 2014, 57, 8530–8539. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed]
- Goodsell, D.S.; Olson, A.J. Automated docking of substrates to proteins by simulated annealing. Proteins Struct. Funct. Genet. 1990, 8, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Mysinger, M.M.; Carchia, M.; Irwin, J.J.; Shoichet, B.K. Directory of useful decoys, enhanced (DUD-E): Better ligands and decoys for better benchmarking. J. Med. Chem. 2012, 55, 6582–6594. [Google Scholar] [CrossRef] [PubMed]
- Young, T.; Hua, L.; Huang, X.; Abel, R.; Friesner, R.; Berne, B.J. Dewetting transitions in protein cavities. Proteins Struct. Funct. Bioinform. 2010, 78, 1856–1869. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VDM: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Uehara, S.; Fujimoto, K.J.; Tanaka, S. Protein-ligand docking using fitness learning-based artificial bee colony with proximity stimuli. Phys. Chem. Chem. Phys. 2015, 17, 16412–16417. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.-M.; Liu, B.-F.; Huang, H.-L.; Hwang, S.-F.; Ho, S.-Y. SODOCK: Swarm optimization for highly flexible protein-ligand docking. J. Comput. Chem. 2007, 28, 612–623. [Google Scholar] [CrossRef] [PubMed]
- Santos-Martins, D.; Forli, S.; Ramos, M.J.; Olson, A.J. AutoDock4Zn: An improved AutoDock force field for small-molecule docking to zinc metalloproteins. J. Chem. Inf. Model. 2014, 54, 2371–2379. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Goodsell, D.S.; Halliday, R.S.; Huey, R.; Hart, W.E.; Belew, R.K.; Olson, A.J. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 1998, 19, 1639–1662. [Google Scholar] [CrossRef]
- Weiner, S.J.; Kollman, P.A.; Case, D.A.; Singh, U.C.; Ghio, C.; Alagona, G.; Profeta, S.; Weinerl, P. A new force field for molecular mechanical simulation of nucleic acids and proteins. J. Am. Chem. Soc. 1984, 106, 765–784. [Google Scholar] [CrossRef]
- Huey, R.; Goodsell, D.; Morris, G.; Olson, A. Grid-based hydrogen bond potentials with improved directionality. Lett. Drug Des. Discov. 2004, 1, 178–183. [Google Scholar] [CrossRef]
- Mehler, E.L.; Solmajer, T. Electrostatic effects in proteins: Comparison of dielectric and charge models. Protein Eng. Des. Sel. 1991, 4, 903–910. [Google Scholar] [CrossRef]
- Stouten, P.F.W.; Frömmel, C.; Nakamura, H.; Sander, C. An Effective Solvation Term Based on Atomic Occupancies for Use in Protein Simulations. Mol. Simul. 1993, 10, 97–120. [Google Scholar] [CrossRef]
- Sgobba, M.; Caporuscio, F.; Anighoro, A.; Portioli, C.; Rastelli, G. Application of a post-docking procedure based on MM-PBSA and MM-GBSA on single and multiple protein conformations. Eur. J. Med. Chem. 2012, 58, 431–440. [Google Scholar] [CrossRef] [PubMed]
- Thompson, D.C.; Humblet, C.; Joseph-McCarthy, D. Investigation of MM-PBSA rescoring of docking poses. J. Chem. Inf. Model. 2008, 48, 1081–1091. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Zhao, L.; Peng, S.; Huang, N. Incorporating replacement free energy of binding-site waters in molecular docking. Proteins Struct. Funct. Bioinform. 2014, 82, 1765–1776. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Adler, M.; Davey, D.D.; Phillips, G.B.; Kim, S.H.; Jancarik, J.; Rumennik, G.; Light, D.R.; Whitlow, M. Preparation, characterization, and the crystal structure of the inhibitor ZK-807834 (CI-1031) complexed with factor Xa. Biochemistry 2000, 39, 12534–12542. [Google Scholar] [CrossRef] [PubMed]
- Word, J.M.; Lovell, S.C.; Richardson, J.S.; Richardson, D.C. Asparagine and glutamine: Using hydrogen atom contacts in the choice of side-chain amide orientation. J. Mol. Biol. 1999, 285, 1735–1747. [Google Scholar] [CrossRef] [PubMed]
- Case, D.A.; Babin, V.; Berryman, J.T.; Betz, R.M.; Cai, Q.; Cerutti, D.S.; Cheatham, T.E., III; Darden, T.A.; Duke, R.E.; Gohlke, H.; et al. AMBER 14; University of California: San Francisco, CA, USA, 2014. [Google Scholar]
- Hornak, V.; Abel, R.; Okur, A.; Strockbine, B.; Roitberg, A.; Simmerling, C. Comparison of multiple Amber force fields and development of improved protein backbone parameters. Proteins Struct. Funct. Bioinform. 2006, 65, 712–725. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Salomon-Ferrer, R.; Götz, A.W.; Poole, D.; Le Grand, S.; Walker, R.C. Routine microsecond molecular dynamics simulations with AMBER on GPUs. 2. Explicit solvent particle mesh ewald. J. Chem. Theory Comput. 2013, 9, 3878–3888. [Google Scholar] [CrossRef] [PubMed]
- Ryckaert, J.P.; Ciccotti, G.; Berendsen, H.J.C. Numerical integration of the Cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef]
- Roe, D.R.; Cheatham, T.E., III. PTRAJ and CPPTRAJ: Software for processing and analysis of molecular synamics trajectory data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef] [PubMed]
- Ramsey, S.; Nguyen, C.; Salomon-Ferrer, R.; Walker, R.C.; Gilson, M.K.; Kurtzman, T. Solvation thermodynamic mapping of molecular surfaces in AmberTools: GIST. J. Comput. Chem. 2016, 37, 2029–2037. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Li, Y.; Han, L.; Li, J.; Liu, J.; Zhao, Z.; Nie, W.; Liu, Y.; Wang, R. PDB-wide collection of binding data: Current status of the PDBbind database. Bioinformatics 2014, 31, 405–412. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Huang, N.; Cho, S.; MacKerell, A.D. Consideration of molecular weight during compound selection in virtual target-based database screening. J. Chem. Inf. Comput. Sci. 2003, 43, 267–272. [Google Scholar] [CrossRef] [PubMed]
- Kim, R.; Skolnick, J. Assessment of programs for ligand binding affinity prediction. J. Comput. Chem. 2008, 29, 1316–1331. [Google Scholar] [CrossRef] [PubMed]
- Molecular Operating Environment (MOE), 2013.08; Chemical Computing Group Inc.: Montreal, QC, Canada, 2016.
- Triballeau, N.; Acher, F.; Brabet, I.; Pin, J.P.; Bertrand, H.O. Virtual screening workflow development guided by the “receiver operating characteristic” curve approach. Application to high-throughput docking on metabotropic glutamate receptor subtype 4. J. Med. Chem. 2005, 48, 2534–2547. [Google Scholar] [CrossRef] [PubMed]
- Bender, A.; Glen, R.C. A discussion of measures of enrichment in virtual screening: Comparing the information content of descriptors with increasing levels of sophistication. J. Chem. Inf. Model. 2005, 45, 1369–1375. [Google Scholar] [CrossRef] [PubMed]
- Perzborn, E.; Roehrig, S.; Straub, A.; Kubitza, D.; Misselwitz, F. The discovery and development of rivaroxaban, an oral, direct factor Xa inhibitor. Nat. Rev. Drug Discov. 2011, 10, 61–75. [Google Scholar] [CrossRef] [PubMed]
- Rai, R.; Sprengeler, P.; Elrod, K.; Young, W. Perspectives on Factor Xa Inhibition. Curr. Med. Chem. 2001, 8, 101–119. [Google Scholar] [CrossRef] [PubMed]
- Lumry, R.; Rajender, S. Enthalpy-entropy compensation phenomena in water solutions of proteins and small molecules: A ubiquitous properly of water. Biopolymers 1970, 9, 1125–1227. [Google Scholar] [CrossRef] [PubMed]
- Meloun, M.; Ferenčíková, Z. Enthalpy–entropy compensation for some drugs dissociation in aqueous solutions. Fluid Phase Equilib. 2012, 328, 31–41. [Google Scholar] [CrossRef]
- Ahmad, M.; Helms, V.; Lengauer, T.; Kalinina, O.V. Enthalpy-entropy compensation upon molecular conformational changes. J. Chem. Theory Comput. 2015, 11, 1410–1418. [Google Scholar] [CrossRef] [PubMed]
- Plewczynski, D.; Łaźniewski, M.; Augustyniak, R.; Ginalski, K. Can we trust docking results? Evaluation of seven commonly used programs on PDBbind database. J. Comput. Chem. 2011, 32, 742–755. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Lai, L.; Wang, S. Further development and validation of empirical scoring functions for structure-based binding affinity prediction. J. Comput. Aided Mol. Des. 2002, 16, 11–26. [Google Scholar] [CrossRef] [PubMed]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra precision glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef] [PubMed]
- Kadirvelraj, R.; Foley, B.L.; Dyekjær, J.D.; Woods, R.J. Involvement of water in carbohydrate−protein binding: concanavalin a revisited. J. Am. Chem. Soc. 2008, 130, 16933–16942. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Lazaridis, T. Thermodynamic contributions of the ordered water molecule in HIV-1 protease. J. Am. Chem. Soc. 2003, 125, 6636–6637. [Google Scholar] [CrossRef] [PubMed]
- Michel, J.; Tirado-Rives, J.; Jorgensen, W.L. Energetics of displacing water molecules from protein binding sites: Consequences for ligand optimization. J. Am. Chem. Soc. 2009, 131, 15403–15411. [Google Scholar] [CrossRef] [PubMed]
- Baldwin, E.T.; Bhat, T.N.; Gulnik, S.; Liu, B.; Topol, I.A.; Kiso, Y.; Mimoto, T.; Mitsuya, H.; Erickson, J.W. Structure of HIV-1 protease with KNI-272, a tight-binding transition-state analog containing allophenylnorstatine. Structure 1995, 3, 581–590. [Google Scholar] [CrossRef]
- Blum, A.P.; Lester, H.A.; Dougherty, D.A. Nicotinic pharmacophore: The pyridine N of nicotine and carbonyl of acetylcholine hydrogen bond across a subunit interface to a backbone NH. Proc. Natl. Acad. Sci. USA 2010, 107, 13206–13211. [Google Scholar] [CrossRef] [PubMed]
- Adachi, M.; Ohhara, T.; Kurihara, K.; Tamada, T.; Honjo, E.; Okazaki, N.; Arai, S.; Shoyama, Y.; Kimura, K.; Matsumura, H.; et al. Structure of HIV-1 protease in complex with potent inhibitor KNI-272 determined by high-resolution X-ray and neutron crystallography. Proc. Natl. Acad. Sci. USA 2009, 106, 4641–4646. [Google Scholar] [CrossRef] [PubMed]
- Murphy, R.B.; Repasky, M.P.; Greenwood, J.R.; Tubert-Brohman, I.; Jerome, S.; Annabhimoju, R.; Boyles, N.A.; Schmitz, C.D.; Abel, R.; Farid, R.; et al. WScore: A flexible and accurate treatment of explicit water molecules in ligand–receptor docking. J. Med. Chem. 2016, 59, 4364–4384. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Not available.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter Set | (kcal/mol/Water) | (kcal/mol) | |
|---|---|---|---|
| Affinity parameter set | 4.8 | 1.0 | −0.50 |
| Pose parameter set | 4.3 | 1.9 | −0.25 |
| Data Set | AutoDock4 | AutoDock-GIST (Pose Parameter Set) | AutoDock-GIST (Affinity Parameter Set) |
|---|---|---|---|
| Training set | 75.0% | 89.3% | 71.4% |
| Test set | 82.6% | 95.7% | 90.4% |
| All data | 78.4% | 92.1% | 80.4% |
| Metrics | AutoDock4 | AutoDock-GIST (Affinity Parameter Set) | AutoDock-GIST (Pose Parameter Set) |
|---|---|---|---|
| EF(0.1%) | 25.47 | 26.75 | 26.75 |
| EF(0.5%) | 23.22 | 25.23 | 24.73 |
| EF(1%) | 21.45 | 22.84 | 23.34 |
| EF(2%) | 16.65 | 17.92 | 19.11 |
| EF(5%) | 10.30 | 10.57 | 12.26 |
| EF(10%) | 6.44 | 6.61 | 7.59 |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Uehara, S.; Tanaka, S. AutoDock-GIST: Incorporating Thermodynamics of Active-Site Water into Scoring Function for Accurate Protein-Ligand Docking. Molecules 2016, 21, 1604. https://doi.org/10.3390/molecules21111604
Uehara S, Tanaka S. AutoDock-GIST: Incorporating Thermodynamics of Active-Site Water into Scoring Function for Accurate Protein-Ligand Docking. Molecules. 2016; 21(11):1604. https://doi.org/10.3390/molecules21111604
Chicago/Turabian StyleUehara, Shota, and Shigenori Tanaka. 2016. "AutoDock-GIST: Incorporating Thermodynamics of Active-Site Water into Scoring Function for Accurate Protein-Ligand Docking" Molecules 21, no. 11: 1604. https://doi.org/10.3390/molecules21111604
APA StyleUehara, S., & Tanaka, S. (2016). AutoDock-GIST: Incorporating Thermodynamics of Active-Site Water into Scoring Function for Accurate Protein-Ligand Docking. Molecules, 21(11), 1604. https://doi.org/10.3390/molecules21111604