Gypmacrophin A, a Rare Pentacyclic Sesterterpenoid, Together with Three Depsides, Functioned as New Chemical Evidence for Gypsoplaca macrophylla (Zahlbr.) Timdal Identification

 ,
,
Abstract
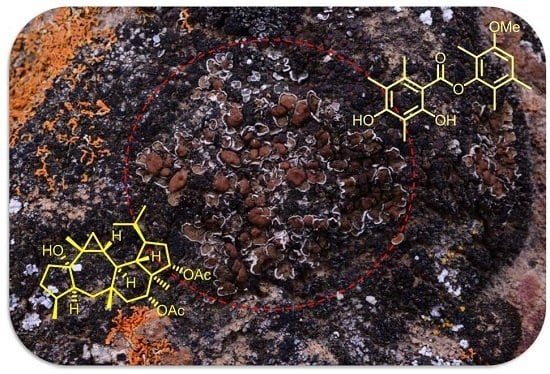
:
1. Introduction
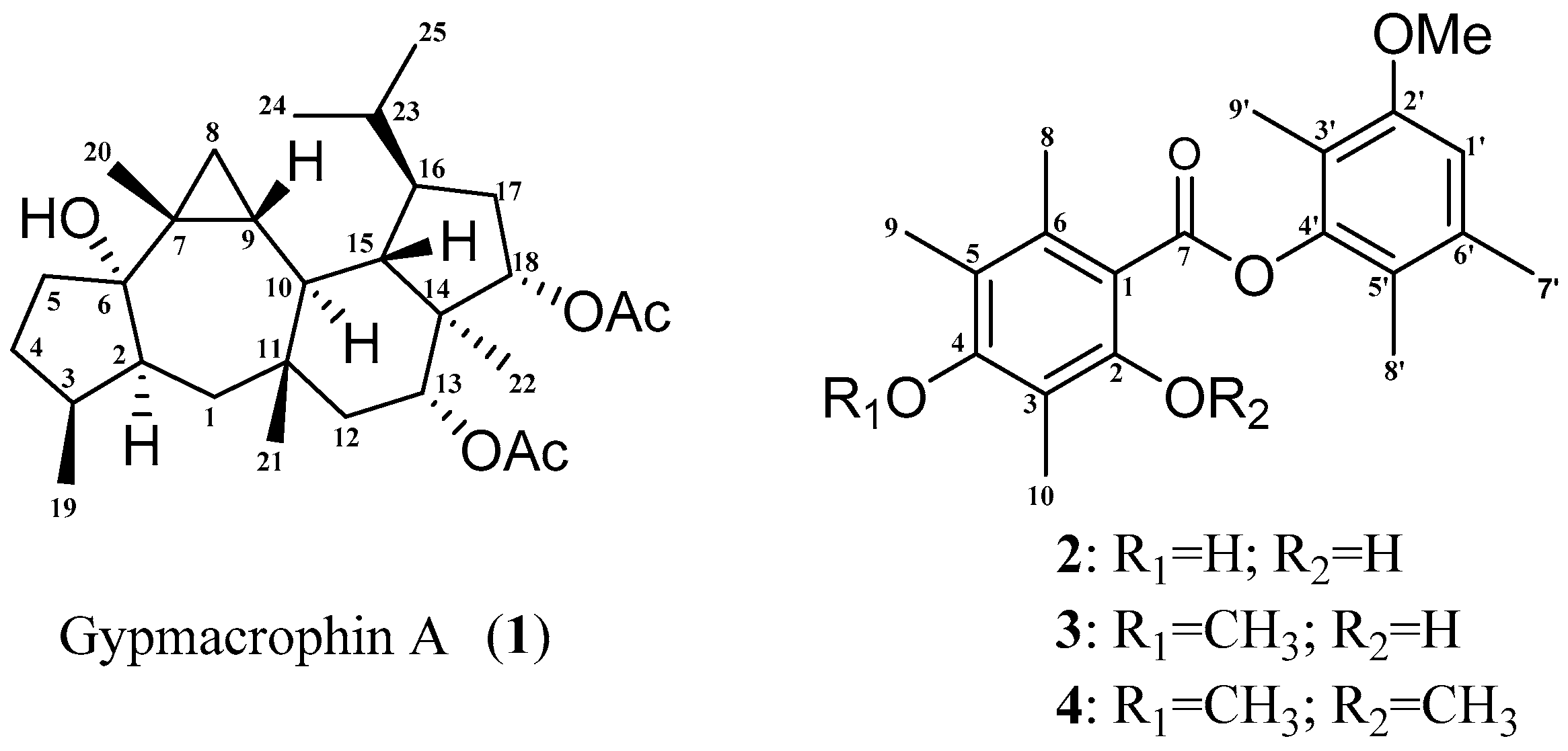
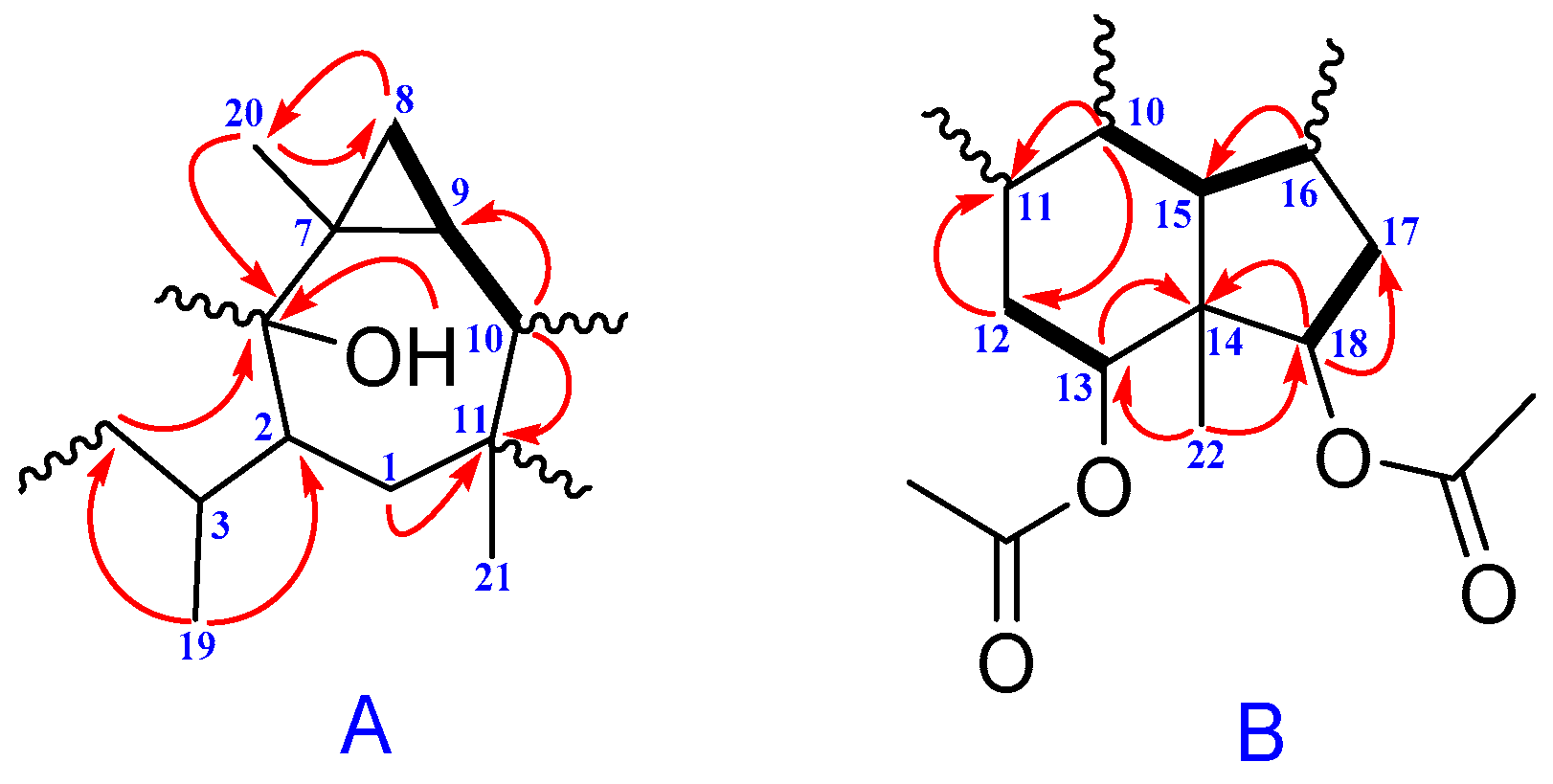
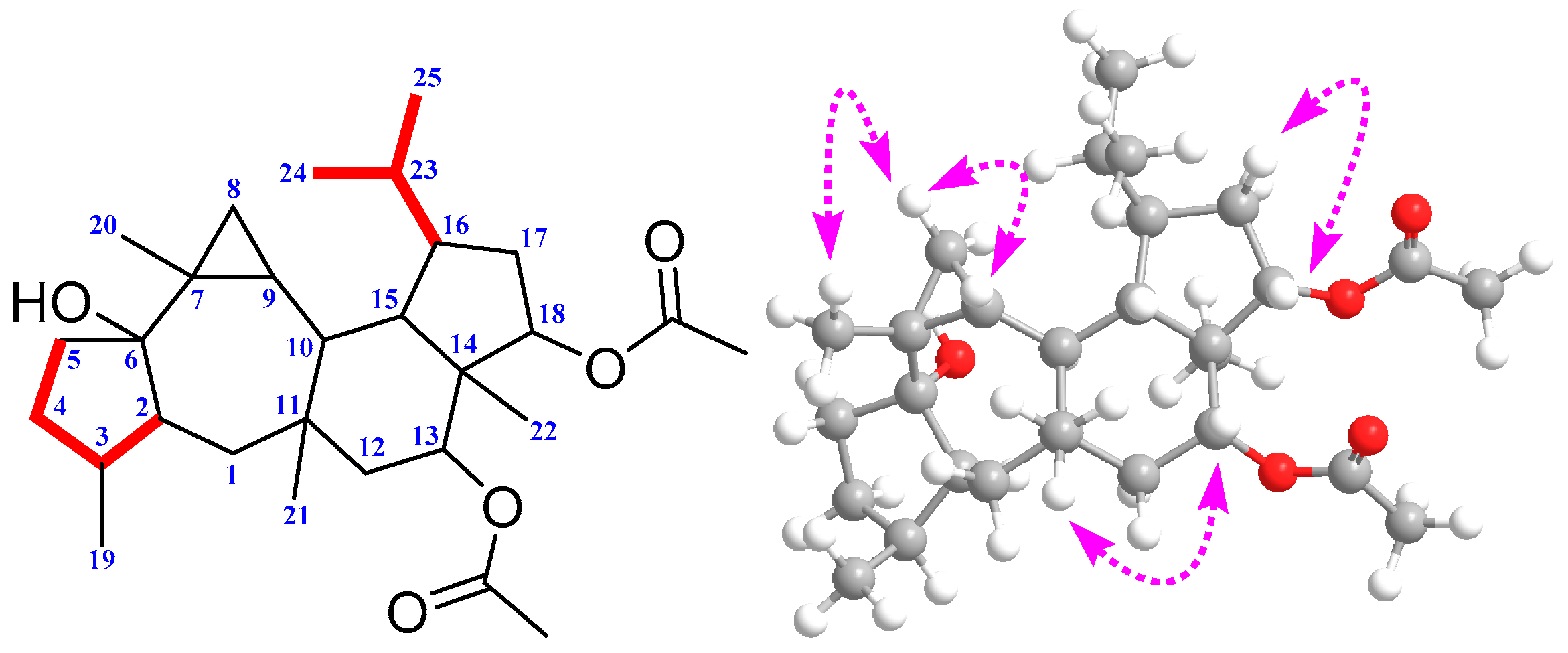
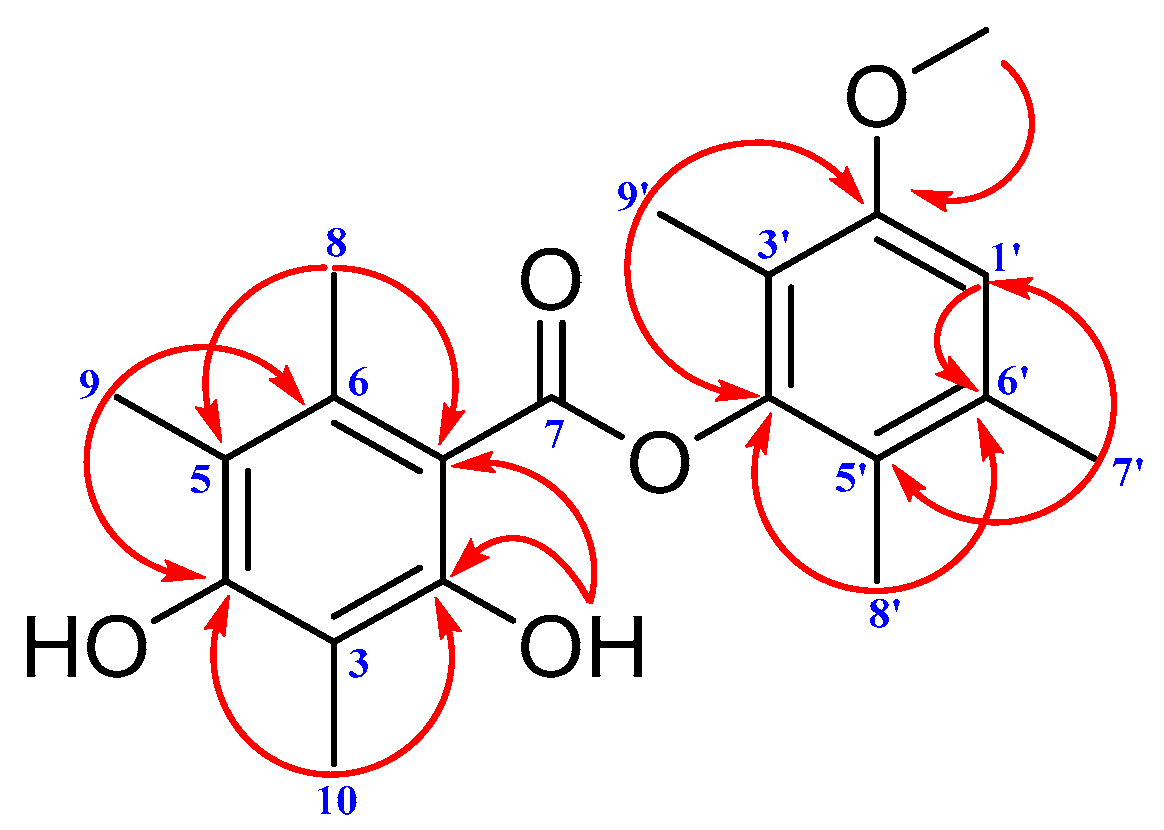
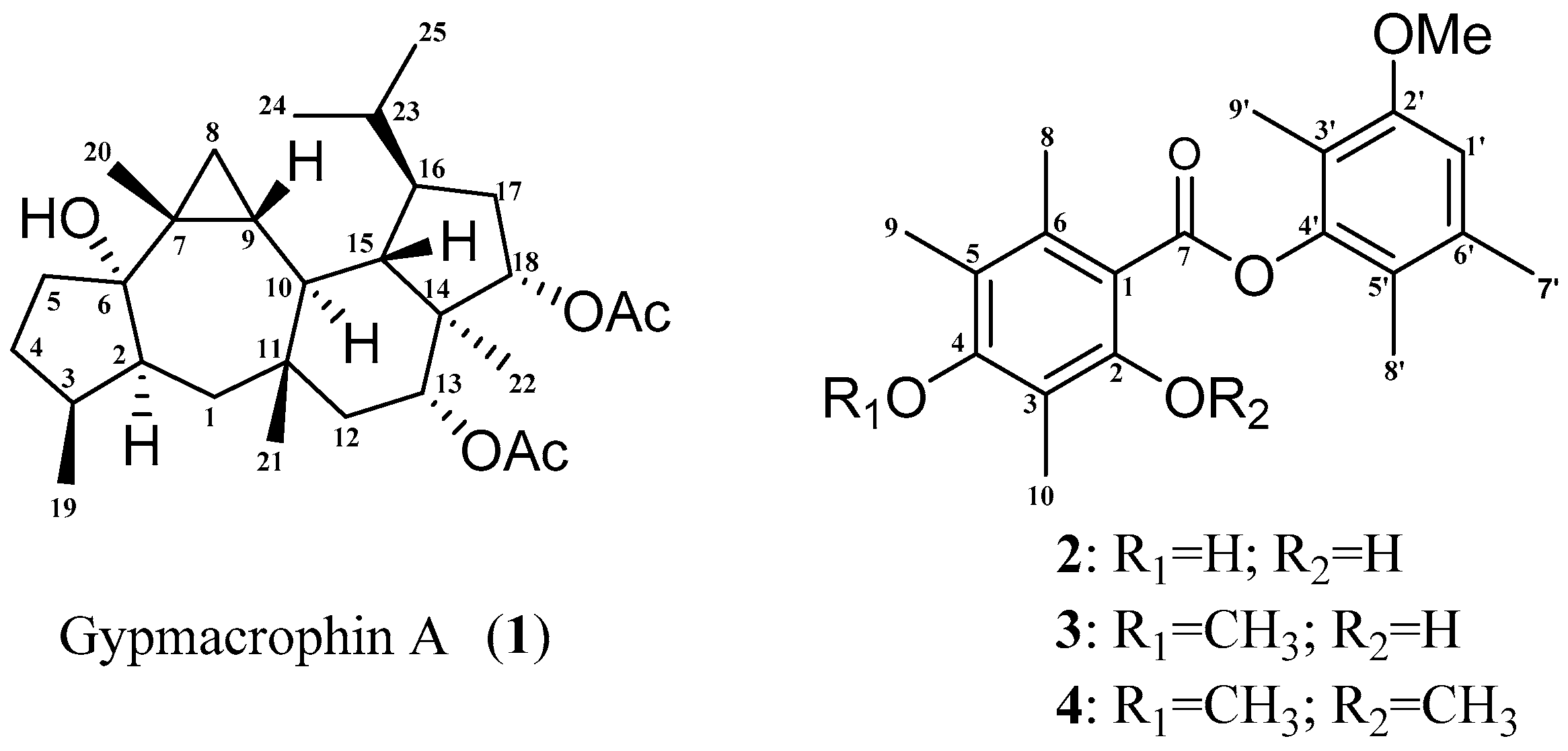
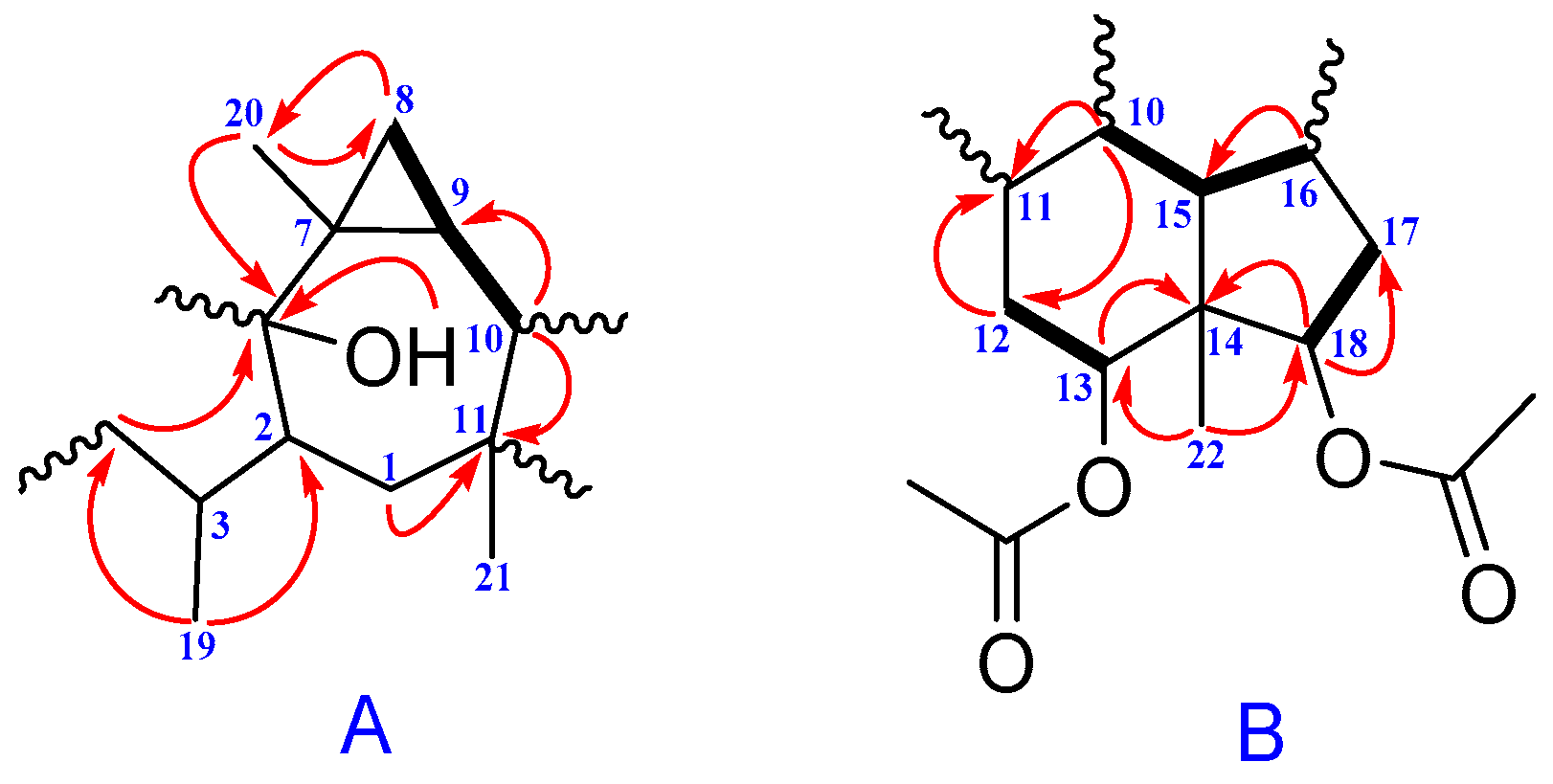
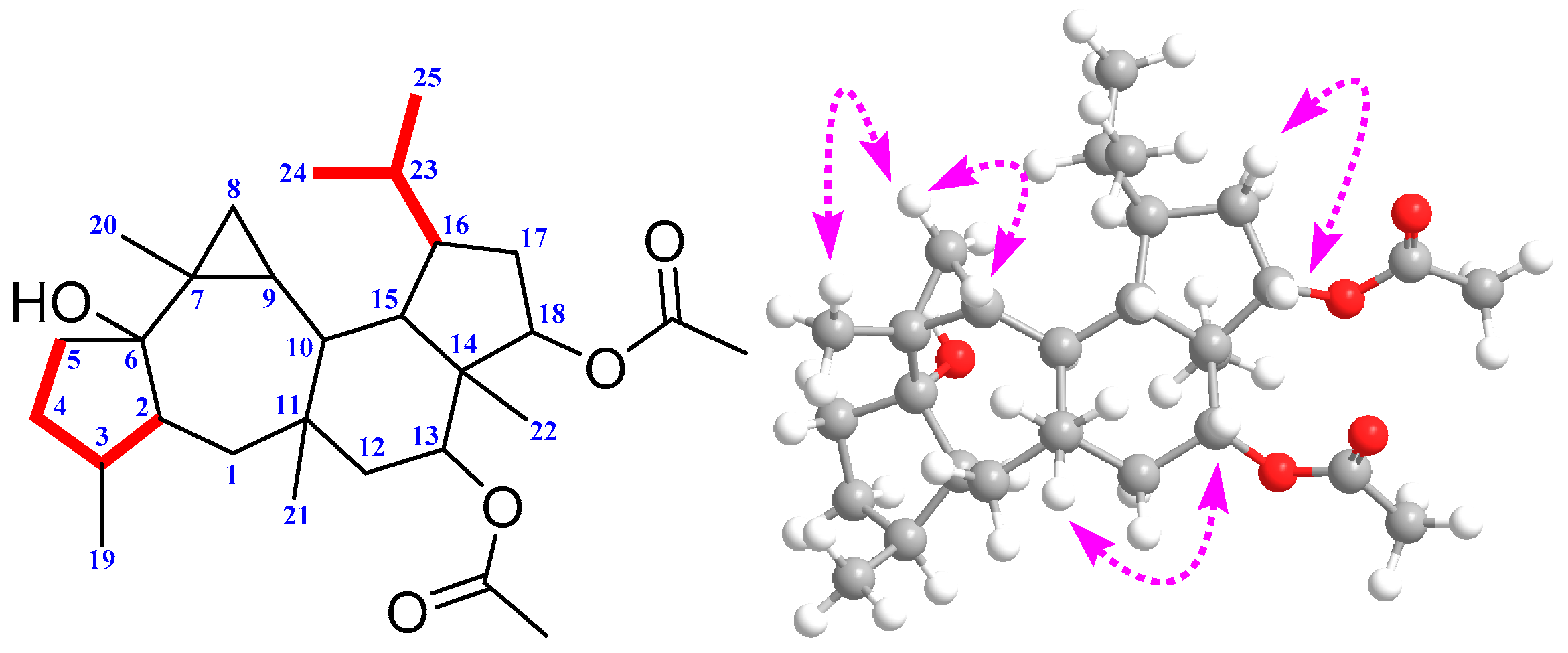
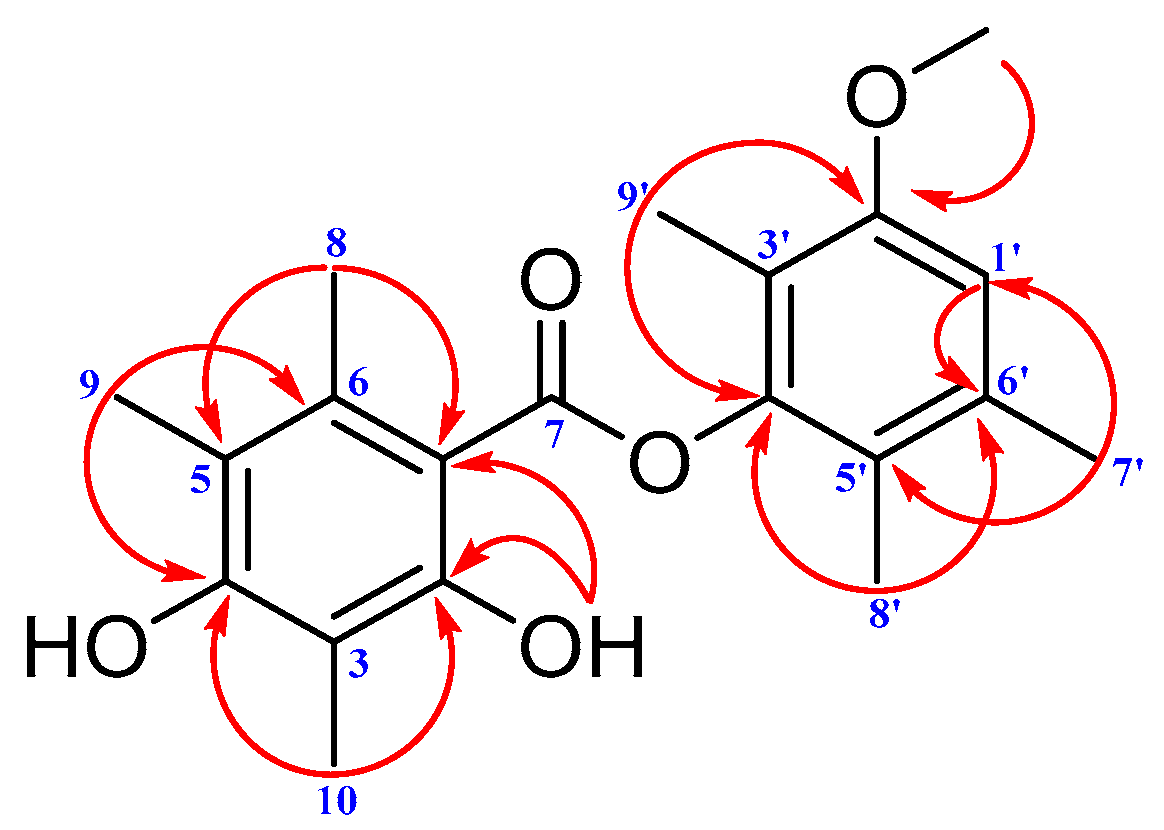
2. Results and Discussion
3. Experimental Section
3.1. General Experimental Procedures
3.2. Collection and Identification of Biological Materials
3.3. Extraction and Isolation
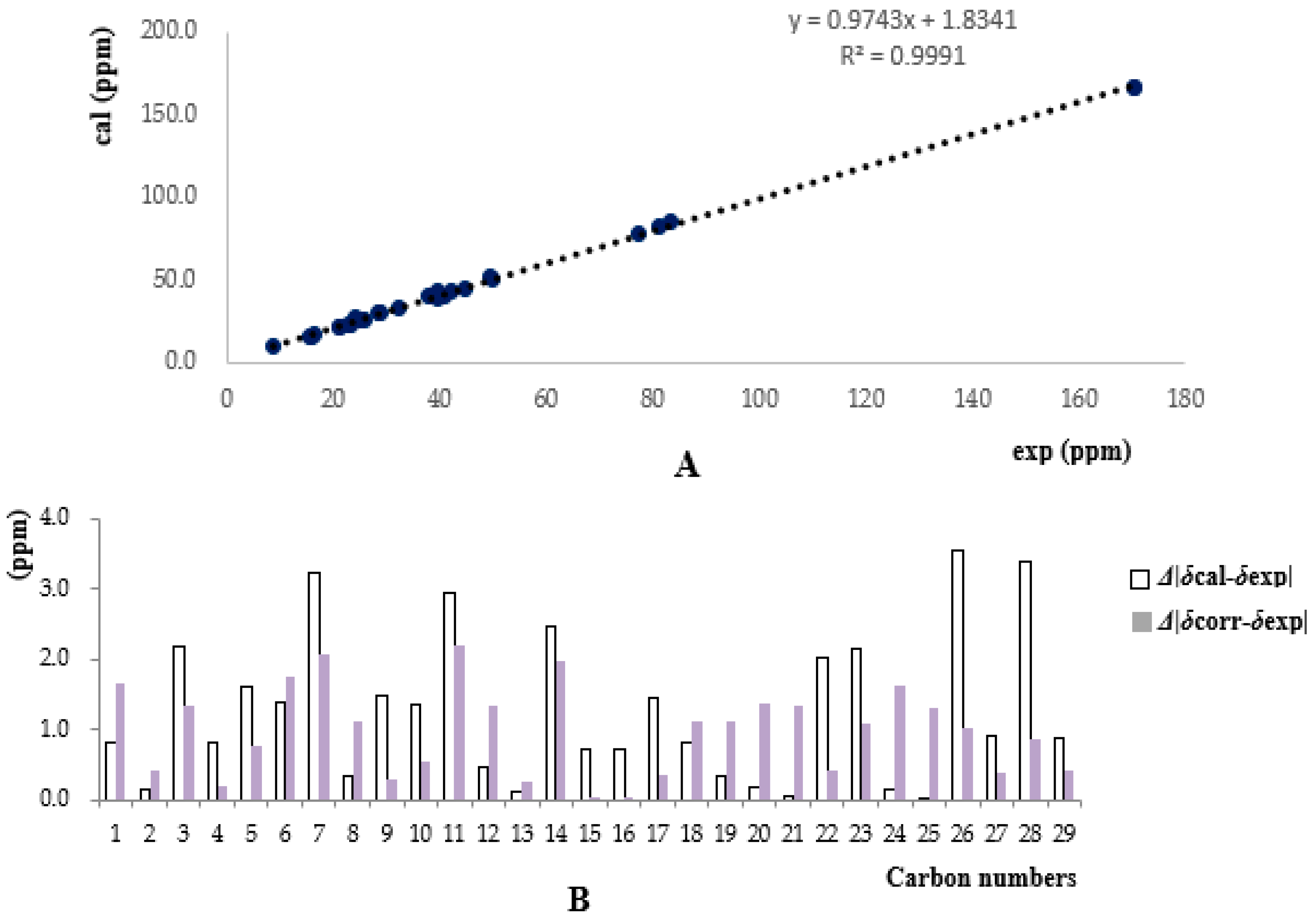
3.4. 13C-NMR and OR Calculations
3.5. Bioactive Assay of Gypmacrophin A
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Tian, W.; Deng, Z.; Hong, K. The biological activities of sesterterpenoid-Type ophiobolins. Mar. Drugs 2017, 15, 229. [Google Scholar] [CrossRef] [PubMed]
- Evidente, A.; Kornienko, A.; Lefranc, F.; Cimmino, A.; Dasari, R.; Evidente, M.; Mathieu, V.; Kiss, R. Sesterterpenoids with anticancer activity. Curr. Med. Chem. 2015, 22, 3502–3522. [Google Scholar] [CrossRef] [PubMed]
- Baquero, F.; Coque, T.M.; de la Cruz, F. Ecology and evolution as targets: The need for novel eco-evo drugs and strategies to fight antibiotic resistance. Antimicrob. Agents Chemother. 2011, 55, 3649–3660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- García, A.; Bocanegra-García, V.; Palma-Nicolás, J.P.; Rivera, G. Recent advances in antitubercular natural products. Eur. J. Med. Chem. 2012, 49, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Stowe, S.D.; Richards, J.J.; Tucker, A.T.; Thompson, R.; Melander, C.; Cavanagh, J. Anti-Biofilm compounds derived from marine sponges. Mar. Drugs 2011, 9, 2010–2035. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Yang, B.; Lin, X.P.; Zhou, X.F.; Liu, Y. Sesterterpenoids. Nat. Prod. Rep. 2013, 30, 455–473. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Chen, Y.; Chen, S.; Liu, Y.; Lu, Y.; Chen, D.; Lin, Y.; Huang, X.; She, Z. Aspterpenacids A and B, two sesterterpenoids from a mangrove endophytic fungus Aspergillus terreus H010. Org. Lett. 2016, 18, 1406–1409. [Google Scholar] [CrossRef] [PubMed]
- Hanson, J.R. The sesterterpenoids. Nat. Prod. Rep. 1992, 9, 481–489. [Google Scholar] [CrossRef]
- Okada, M.; Matsuda, Y.; Mitsuhashi, T.; Hoshino, S.; Mori, T.; Nakagawa, K.; Quan, Z.; Qin, B.; Zhang, H.; Hayashi, F.; et al. Genome-based discovery of an unprecedented cyclization mode in fungal sesterterpenoid biosynthesis. J. Am. Chem. Soc. 2016, 138, 10011–10018. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, Y.; Mitsuhashi, T.; Lee, S.; Hoshino, M.; Mori, T.; Okada, M.; Zhang, H.P.; Hayashi, F.; Fujita, M.; Abe, I. Astellifadiene: Structure determination by NMR spectroscopy and crystalline sponge method, and elucidation of its biosynthesis. Angew. Chem. Int. Ed. 2016, 55, 5785–5788. [Google Scholar] [CrossRef] [PubMed]
- Lutzoni, F.; Pagel, M.; Reeb, V. Major fungal lineages are derived from lichen symbiotic ancestors. Nature 2001, 411, 937–940. [Google Scholar] [CrossRef] [PubMed]
- Mittermeier, K.V.; Schmitt, N.; Volk, P.L.; Suárez, P.J.; Beck, A.; Eisenreich, W. Metabolic profiling of alpine and ecuadorian lichens. Molecules 2015, 20, 18047–18065. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.Y.; Wang, X.Y.; Liu, D.; Shi, H.X.; Ye, X.; Yang, M.X.; Wang, L.S. The genus Bulbothrix (Parmeliaceae) in China. Lichenologist 2016, 48, 121–133. [Google Scholar] [CrossRef]
- Wang, X.Y.; Goffinet, B.; Liu, D.; Liang, M.M.; Shi, H.X.; Zhang, Y.Y.; Zhang, J.; Wang, L.S. Taxonomic study of the genus Anzia (Lecanorales, lichenized Ascomycota) from Hengduan Mountains, China. Lichenologist 2015, 47, 99–115. [Google Scholar] [CrossRef]
- Timdal, E. Gypsoplacaceae and Gypsoplaca, a new family and genus of squamiform lichens. Bibl. Lichenol. 1990, 38, 419–427. [Google Scholar]
- Abdulla, A.; Hurnisa, X.; Reyim, M.; Adilijiang, A. A new record of lichen family from Xinjiang. Arid Zone Res. 2015, 32, 509–511. [Google Scholar]
- Zhao, Z.-Z.; Chen, H.-P.; Wu, B.; Zhang, L.; Li, Z.-H.; Feng, T.; Liu, J.-K. Matsutakone and Matsutoic Acid, Two (Nor)steroids with unusual skeletons from the edible mushroom Tricholoma matsutake. J. Org. Chem. 2017, 82, 7974–7979. [Google Scholar] [CrossRef] [PubMed]
- Elix, J.A.; Barclay, C.E.; David, F.; Griffin, F.K.; Hill, A.M.; Mcconnell, D.B.; Wardlaw, J.H. Synthesis of further lichen depsides. Aust. J. Chem. 1993, 46, 301–313. [Google Scholar] [CrossRef]
- Seo, C.; HanYim, J.; Kum Lee, H.; Oh, H. PTP1B inhibitory secondary metabolites from the Antarctic lichen Lecidella carpathica. Mycology 2011, 2, 18–23. [Google Scholar] [CrossRef]
- Huang, X.; Huang, H.; Li, H.; Sun, X.; Huang, H.; Lu, Y.; Lin, Y.; Long, Y.; She, Z. Asperterpenoid A, a new sesterterpenoid as an inhibitor of Mycobacterium tuberculosis protein tyrosine phosphatase B from the culture of Aspergillus sp. 16–5c. Org. Lett. 2013, 15, 721–723. [Google Scholar] [CrossRef] [PubMed]
- Ellman, G.L.; Courtney, K.D.; Andres, V., Jr.; Featherstone, R.M. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem. Pharmacol. 1961, 7, 88–95. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are available from the authors. |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Position | δH-exp a | δC-exp b | δC-cal c |
|---|---|---|---|
| 1β | 1.41 (overlapped, 1H) | 40.9 | 40.1 |
| 1α | 1.13 (d, J = 14.1 Hz, 1H) | -- | -- |
| 2 | 1.99 (overlapped, 1H) | 49.7 | 49.8 |
| 3 | 2.28 (m, 1H) | 37.7 | 39.9 |
| 4β | 1.29 (m, 1H) | 32.4 | 33.2 |
| 4α | 1.94 (m, 1H) | -- | -- |
| 5β | 2.12 (m, 1H) | 38.8 | 40.4 |
| 5α | 1.53 (dd, J = 13.0, 5.7 Hz, 1H) | -- | -- |
| 6 | -- | 83.3 | 84.7 |
| 7 | -- | 24.2 | 27.4 |
| 8β | 0.24 (dd, J = 8.5, 4.1 Hz, 1H) | 16.4 | 16.7 |
| 8α | 1.02 (overlapped, 1H) | -- | -- |
| 9 | 0.54 (ddd, J = 11.4, 8.5, 5.5 Hz, 1H) | 24.8 | 26.3 |
| 10 | 1.76 (t, J = 11.4 Hz, 1H) | 38.8 | 40.2 |
| 11 | -- | 39.7 | 42.6 |
| 12β | 1.18 (dd, J = 12.7, 4.9 Hz, 1H) | 39.4 | 38.9 |
| 12α | 1.60 (overlapped, 1H) | -- | -- |
| 13 | 4.91 (dd, J = 11.7, 4.9 Hz, 1H) | 77.3 | 77.4 |
| 14 | -- | 49.4 | 51.9 |
| 15 | 1.60 (m, 1H) | 44.6 | 45.3 |
| 16 | 2.02 (overlapped, 1H) | 42.2 | 42.9 |
| 17β | 1.92 (overlapped, 1H) | 28.7 | 30.1 |
| 17α | 1.42 (m, 1H) | -- | -- |
| 18 | 4.65 (t, J = 9.0 Hz, 1H) | 81.1 | 81.9 |
| 19 | 0.83 (d, J = 7.4, 3H) | 15.9 | 16.2 |
| 20 | 1.07 (s, 3H) | 25.8 | 25.6 |
| 21 | 1.03 (s, 3H) | 22.8 | 22.8 |
| 22 | 0.92 (s, 3H) | 8.7 | 10.7 |
| 23 | 2.31 (m, 1H) | 28.4 | 30.6 |
| 24 | 0.85 (d, J = 7.3, 3H) | 23.2 | 23.2 |
| 25 | 0.86 (d, J = 7.3, 3H) | 15.5 | 15.4 |
| AcO-13 | -- | 170.4 | 166.9 |
| -- | 1.93 (s, 3H) | 21.2 | 22.1 |
| AcO-18 | -- | 170.2 | 166.8 |
| -- | 1.90 (s, 3H) | 20.9 | 21.8 |
| HO-6 | 2.96 (s, 1H) | -- | -- |
| Position | δH (2) a | δC (2) b | δH (3) a | δC (3) b | δH (4) a | δC (4) b |
|---|---|---|---|---|---|---|
| 1 | -- | 105.1 | -- | 123.1 | -- | 121.5 |
| 2 | -- | 161.8 | -- | 162.3 | -- | 159.8 |
| 3 | -- | 109.3 | -- | 109.8 | -- | 117.1 |
| 4 | -- | 159.8 | -- | 161.0 | -- | 155.4 |
| 5 | -- | 117.3 | -- | 117.2 | -- | 125.7 |
| 6 | -- | 138.4 | -- | 138.6 | -- | 136. 0 |
| 7 | -- | 171.5 | -- | 171.0 | -- | 167.1 |
| 8 | 2.66 (s, 3H) | 19.5 | 2.64 (s, 3H) | 19.3 | 2.14 (s, 3H) | 17.3 |
| 9 | 2.23 (s, 3H) | 12.4 | 2.23 (s, 3H) | 12.6 | 2.17 (s, 3H) | 12.5 |
| 10 | 2.16 (s, 3H) | 8.7 | 2.16 (s, 3H) | 9.3 | 2.31 (s, 3H) | 9.8 |
| 1′ | 6.79 (s, 1H) | 110.8 | 6.80 (s, 1H) | 111.0 | 6.77 (s, 1H) | 110.7 |
| 2′ | -- | 156.8 | -- | 156.6 | -- | 156.8 |
| 3′ | -- | 116.6 | -- | 116.5 | -- | 122.9 |
| 4′ | -- | 149.2 | -- | 149.2 | -- | 149.6 |
| 5′ | -- | 136.1 | -- | 136.2 | -- | 127.1 |
| 6′ | -- | 121.0 | -- | 121.0 | -- | 133.9 |
| 7′ | 2.31 (s, 3H) | 20.2 | 2.31 (s, 3H) | 20.2 | 2.23 (s, 3H) | 20.3 |
| 8′ | 2.02 (s, 3H) | 12.6 | 2.02 (s, 3H) | 12.7 | 2.27 (s, 3H) | 12.7 |
| 9′ | 1.99 (s, 3H) | 9.7 | 1.99 (s, 3H) | 9.8 | 2.34 (s, 3H) | 9.9 |
| CH3O-2′ | 3.85 (s, 3H) | 56.0 | 3.80 (s, 3H) | 56.1 | 3.74 (s, 3H) | 56.0 |
| R2O-2 | 11.56 (s, 1H) | 11.02 (s, 1H) | 3.84 (s, 3H) | 62.1 | ||
| R1O-4 | 8.18 (s, 1H) | 3.74 (s, 3H) | 60.3 | 3.81 (s, 3H) | 60.3 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, Y.-F.; Shi, H.-X.; Hu, K.; Tang, J.-W.; Li, X.-R.; Du, X.; Sun, H.-D.; Wang, L.-S.; Pu, J.-X. Gypmacrophin A, a Rare Pentacyclic Sesterterpenoid, Together with Three Depsides, Functioned as New Chemical Evidence for Gypsoplaca macrophylla (Zahlbr.) Timdal Identification. Molecules 2017, 22, 1675. https://doi.org/10.3390/molecules22101675
Zhou Y-F, Shi H-X, Hu K, Tang J-W, Li X-R, Du X, Sun H-D, Wang L-S, Pu J-X. Gypmacrophin A, a Rare Pentacyclic Sesterterpenoid, Together with Three Depsides, Functioned as New Chemical Evidence for Gypsoplaca macrophylla (Zahlbr.) Timdal Identification. Molecules. 2017; 22(10):1675. https://doi.org/10.3390/molecules22101675
Chicago/Turabian StyleZhou, Yuan-Fei, Hai-Xia Shi, Kun Hu, Jian-Wei Tang, Xing-Ren Li, Xue Du, Han-Dong Sun, Li-Song Wang, and Jian-Xin Pu. 2017. "Gypmacrophin A, a Rare Pentacyclic Sesterterpenoid, Together with Three Depsides, Functioned as New Chemical Evidence for Gypsoplaca macrophylla (Zahlbr.) Timdal Identification" Molecules 22, no. 10: 1675. https://doi.org/10.3390/molecules22101675