The Antitumor Constituents from Hedyotis Diffusa Willd
by
Changfu Wang
1, 
Xuegang Zhou
2,
Youzhi Wang
3,
Donghua Wei
2,
Chengjie Deng
2,
Xiaoyun Xu
2,
Ping Xin
2,* and
Shiqin Sun
2,* 1
College of TCM, Guangdong Pharmaceutical University, No. 280 Outside Loop East Road of Higher Education Mega Center, Panyu District, Guangzhou 510006, China
2
College of Pharmacy, Harbin Medical University-Daqing, Daqing 163319, China
3
Department of Medicinal Chemistry, School of Pharmacy, China Pharmaceutical University, Nanjing 210009, China
*
Authors to whom correspondence should be addressed.
Molecules 2017, 22(12), 2101; https://doi.org/10.3390/molecules22122101
Submission received: 4 November 2017
/
Revised: 18 November 2017
/
Accepted: 27 November 2017
/
Published: 30 November 2017
Abstract
:As a TCM, Hedyotis diffusa Willd. has been using to treat malignant tumors, and many studies also showed that the extracts from Hedyotis diffusa Willd. possessed evident antitumor activities. Therefore, we carried out chemical study on Hedyotis diffusa Willd. and investigated the cytotoxicity of the obtained compounds on a panel of eight tumor cell lines. As a result, four new compounds were isolated from Hedyotis diffusa Willd., including three iridoid glycosides of Shecaoiridoidside A–C (1–3) and a cerebroside of shecaocerenoside A (4). Also, six known iridoid compounds (5–10) were also obtained. The cytotoxicity of all compounds against human tumor cell lines of HL-60, HeLa, HCT15, A459, HepG2, PC-3, CNE-2, and BCG-823 were also evaluated in vitro. New compound 3 exhibited evident cytotoxicity to all tumor cell lines except the Hela, and the IC50 values are from 9.6 µM to 62.2 µM, while new compound 4 showed moderate cytotoxicity to all the cell lines, and the IC50 values are from 33.6 µM to 89.3 µM. By contrast, new compound 1 and known compound 9 showed moderate cytotoxicity to HCT15, A459, and HepG2 selectively. Known compound 7 also exhibited moderate cytotoxicity to HCT15 and A459 selectively.
1. Introduction
As an annual herb, Genus of Hedyotis has been widely distributed in subtropical area of the world [1]. 62 species of Hedyotis are distributed in China, among which more than 20 species are used as medicines, ingcluding Hedyotis diffusa Willd. (H. diffusa Willd.). H. diffusa Willd. has been recorded in Chinese pharmacopoeia (2015 edt) and possesses the efficacies of diuresis to reduce edema, clearing away the heat evil and detoxifying, and promoting blood circulation to arrest pain [2]. Clinically, H. diffusa Willd. has been using to treat urinary tract infection, tonsillitis, appendicitis, pharyngitis, hepatitis, dysentery, diarrhea, and snake bites [1,2,3]. But more importantly, H. diffusa Willd. also showed significant effective on malignant tumors of breast, gastric, colon, rectal, and ovarian [2,4,5]. The components of iridoids, triterpenes, flavonoids, lignans, anthraquinones, alkaloids, cerebrosides, coumarins, and sterols were discovered during the chemical studies of H. diffusa Willd. [2,6,7,8]. There are some differences of the chemical constituents if the H. diffusa Willd. grown in different parts of China. The contents of anthraquinones and iridoids in H. diffusa Willd. from Guangdong province were higher than the H. diffusa Willd. from the provinces of zhejiang, Jiangxi, Hubei, and Fujian. These include 2,7-dihydroxy-3-methyl anthraquinone, 2-hydroxy-3-methyl-1-methoxy anthraquinone, 2-hydroxy-3-methoxy-7-methoxy anthraquinone, 2-methyl-3-hydroxy anthraquinone, 2-methyl-3-hydroxy-4-methoxy anthraquinone, deacetyl asperulosidic acid, scandoside, E-6-O-p-coumaroyl scandoside methyl ester [9]. Various hepatoprotective, immunoloregulation, anti-tumor, anti-inflammatory, antibacterial, analgesia, sedative, and anti-oxidant activities can be found in pharmacological studies of H. diffusa Willd [3,6,10,11,12,13], but more studies found that the extracts from H. diffusa Willd. possessed evident anticancer activities [1,2,14,15,16,17,18,19,20]. H. diffusa Willd. has also been used to treat cancers adjuvantly for a long time in China. With increasing incidence and mortality in China, cancer has become the leading cause of death and caused serious public health problems. According to the latest report, in 2015, about 4,292,000 new cancer cases and 2,814,000 cancer deaths occurred in China, with lung cancer being the most common incident cancer and the leading cause of cancer death. Stomach, esophageal, and liver cancers were also commonly diagnosed and identified as leading causes of cancer death [21]. Therefore, screened active components from H. diffusa Willd. might be helpful. In this study, we carried out chemical study on H. diffusa Willd., and four new (1–4) along with six known (5–10) compounds were obtained. The structures of known compounds were determined by detailed NMR and ESI-MS spectra analyses, as well as comparing the data with the literature. In this paper, we describe the isolation of compounds 1–4 and elucidate their structures. The cytotoxic activity of all compounds against tumor cell lines of HL-60 (human leukemia cells), HeLa (human cervical cancer cells), HCT15 (human colon cancer cells), A459 (human lung cancer cells), HepG2 (human hepatoma cells), PC-3 (human prostate cancer cells), CNE-2 (human nasopharyngeal cancer cells), and BCG-823 (human gastric gland carcinoma cells) were also investigated in vitro. As a result, some active compounds will be screened, and the therapeutic basis of H. diffusa Willd. on tumors will also be revealed.
2. Results and Discussion
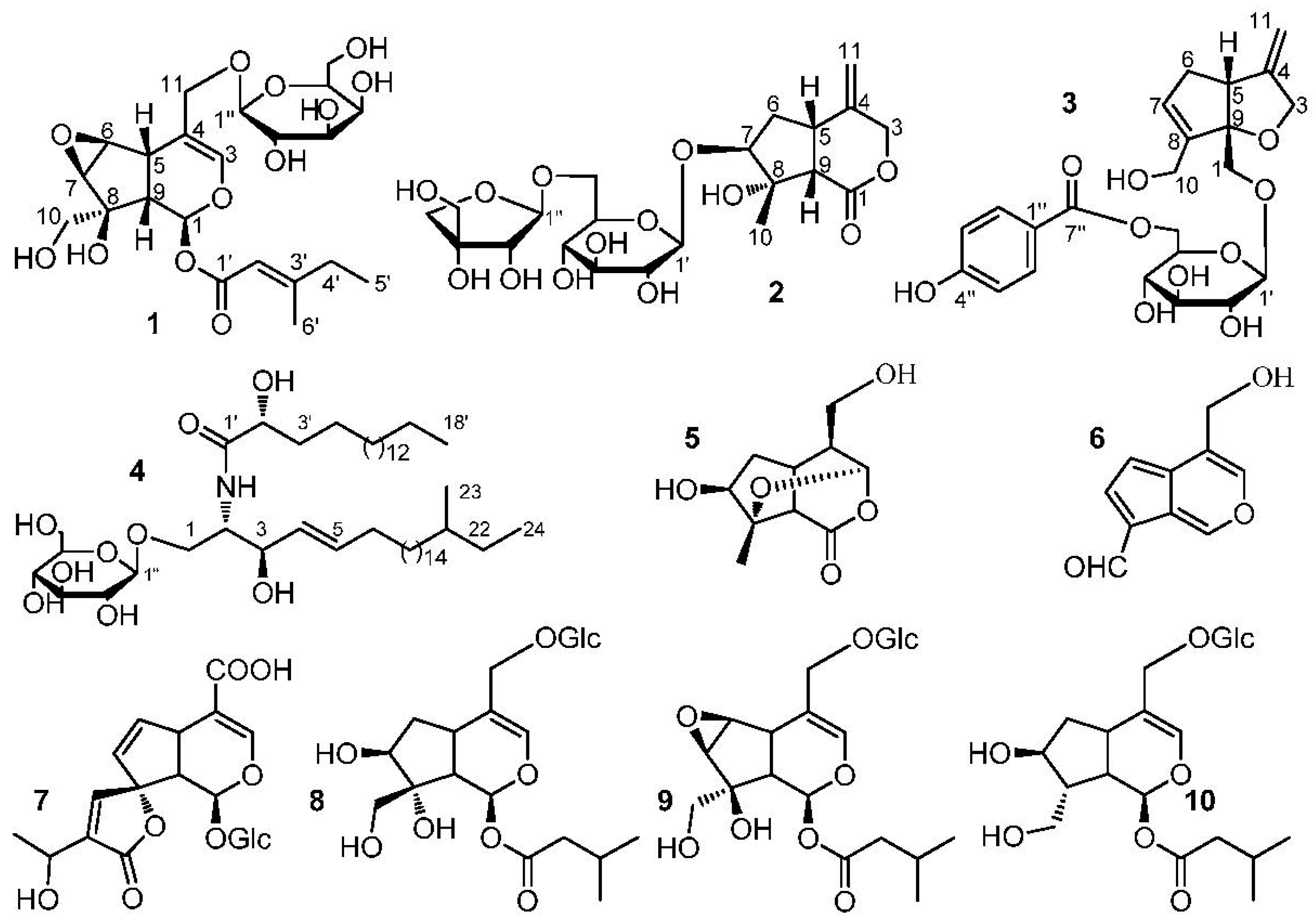
Compound 1 was obtained as a white amorphous powder. HRESIMS ([M + Na]+ m/z 511.1788, calc. for 511.1791) established the molecular formula of 1 as C22H32O12. Hydrolysis experiment of 1 liberated d-galactose which determined by GC-MS analysis. In the 1H-NMR spectrum of 1 (Table 1), signals of two methyl groups at δH 0.89 (3H, t, J = 7.4 Hz, H-5′) and 2.15 (3H, s, H-6′) could be observed. The β-configuration of galactopyranosyl moiety was confirmed by the coupling constant of H-1” (J = 8.1 Hz). The 13C-NMR and DEPT spectra of 1 (Table 2) showed 22 carbon signals, including six carbon signals for a β-d-galactopyranosyl moiety at δC 100.4, 72.6, 73.2, 69.2, 75.6, and 63.4, and a 4-methylsenecioyloxy group at δC 165.8, 114.6, 162.1, 33.8, 11.7, and 19.0. The left carbon signals were further identified by the 2D-NMR spectra of 1. The HSQC and 1H−1H COSY spectra of 1 showed the coupling sequences of C(1)−C(9)−C(5)−C(6)−C(7) (Figure 1). The iridoids structure for 1 was established by the HMBC spectrum (Figure 1). The HMBC correlations from H-1” to C-11 and H-1 to C-1′ suggested that the galactopyranosyl moiety was located at C-11 and the 4-methylsenecioyloxy group was located at C-1.
The stereo-configuration of 1 was determined by NOESY spectrum. The NOE correlations of H-5/H-9, H-7/H-10 and H-6/H-10, but the absence of correlations of H-5/H-1, H-5/H-7, H-5/H-10 and H-7/H-9 suggested that 8-OH, H-5 and H-9 were β-oriented, while H-1, H-6, H-7 and 8-CH2OH were α-oriented. Thus, the structure of 1 was established as (1S,5S,9S,6S,7R,8S)-8-hydroxy-8-hydroxymethyl-6,7-epoxylcyclopenta[c]pyran-1-O-4-methylsenecioyloxyl-11-hydroxymethyl-3-en 11-O-β-d-galactopyranoside and named as Shecaoiridoidside A (Figure 2).
Compound 2 was isolated as a white amorphous powder. HRESIMS ([M + Na]+ m/z 515.1737, calcd. 515.1741) determined the molecular formula of 2 as C21H32O13. Hydrolysis experiment of 2 liberated d-glucose and d-apiose which determined by GC-MS analysis. In the 1H-NMR spectrum of 2 (Table 1), signals of three methenyl groups at δ 3.34 (1H, m, H-5), 3.87 (1H, m, H-7), 3.08 (1H, d, J = 10.7 Hz, H-9), three methylene groups at δ 5.10 (1H, d, J = 11.0 Hz, H-3a), 4.44 (1H, d, J = 11.6 Hz, H-3b), 2.34 (1H, dd, J = 8.4, 13.4 Hz, H-6a), 2.19 (1H, m, H-6b), 5.08 (s, H-11a), 5.011 (s, H-11b), and a methyl group at δ 1.59 (3H, s, H-10) could be observed. The β-configuration of glucopyranosyl moiety was confirmed by the coupling constant of H-1′ (J = 7.8 Hz). The 13C-NMR and DEPT spectra of 2 (Table 2) showed 21 carbon signals, except for the 6 carbon signals at δC 99.9, 75.5, 78.7, 72.3, 78.3, 68.3 belong to a C-6′ substituted β-d-glucopyranosyl moiety and 5 carbon signals at δC 111.5, 76.2, 80.8, 75.4, 65.8 belong to a terminal β-d-apiofuranosyl moiety [22]. The left 10 carbon signals were similar to those of jatamanin A [23]. The main difference lies in the chemical shift value of C-7 in 2 was shifted downfield by 8.3 compared to that of jatamanin A, which confirmed that the β-d-glucopyranosyl moiety was located at C-7. The HSQC and 1H−1H COSY spectra of 2 showed the coupling sequence of C(9)−C(5)−C(6)−C(7) (Figure 1). The cyclopenta[c]pyran-type iridoid structure for 2 was established by the HMBC correlations from H-7 to C-8, H-7 to C-9, H-5 to C-1, and H-3 to C-5. The HMBC correlations from H-1” to C-6′ and H-1′ to C-8 suggested that the apiofuranosyl moiety was located at C-6′ and glucopyranosyl moieties was located at C-7 (Figure 1).
The stereo-configuration of 2 was determined by NOESY spectrum. The NOE correlations (Figure 2) of H-9/CH3-10 and H-5/CH3-10, but absence of the correlations of H-9/H-7 and H-5/H-7, suggested that H-5, H-9 and CH3-10 were β-oriented, while 8-OH and H-7 was α-oriented. Therefore, the structure of 2 was founded to be (5S,7S,8S,9S)-8-hydroxy-8-methyl-4-methylenehexahydrocyclo-penta[c]pyran-1(3H)-one 7-O-(6-O-β-d-apiofuranosyl)-β-d-glucopyranoside and named Shecaoiridoidside B (Figure 2).
Compound 3 was obtained as a white amorphous powder. HRESIMS ([M + Na]+ m/z 487.1576, calcd. 487.1580) established the molecular formula of 3 as C23H28O10. Hydrolysis experiment of 3 liberated d-glucose which determined by GC-MS analysis. In the 1H-NMR of 3 (Table 1), signals of two oxygenated methylenes at δ 3.96 (1H, d, J = 10.4 Hz, H-1a), 3.76(1H, d, J = 10.4 Hz, H-1b), 4.37 (1H, d, J = 12.6 Hz, H-3a) and 4.18 (1H, d, J = 12.6 Hz, H-3b), a nonoxygenated methylene at δ 2.74 (1H, m, H-6a) and 2.26 (1H, brd, J = 16.4 Hz, H-6b), a nonoxygenatedmethine at δ 3.25 (1H, m, H-5), and three olefinic protons at δ 5.78 (1H, brs, H-7), 4.91 (1H, d, J = 2.0 Hz, H-11a) and 4.92 (1H, d, J = 2.0 Hz, H-11b), and a p-substituted benzene protons at 7.88 (2H, d, J = 8.8 Hz) and 6.81 (2H, d, J = 8.8 Hz) were observed. The β-configuration of glucopyranosyl moiety was confirmed by coupling constant of H-1′ (J = 7.8 Hz). The 13C-NMR and DEPT spectra of 3 (Table 2) showed 23 carbon signals, except for the 6 carbon signals at δC 103.9, 74.8, 77.7, 72.3, 76.0, 65.2 belong to a C-6′ substituted β-d-glucopyranosyl moiety and 7 carbon signals at δC 122.4, 132.9 × 2, 116.6 × 2, 164.2, 167.8 belong to a p-hydroxybenzoyl moiety, the left 10 carbon signals were similar with those of patriridoside G [24]. The main difference lies in the signal at δC 12.1 (CH3-10) in patriridoside G was substituted by the signal at δC 59.3 (CH2-10) in 3, which indicated that CH3-10 of patriridoside G was substituted by a hydroxyl group.
The HSQC and 1H−1H COSY spectra of 3 showed the coupling sequences of C-3/C-4/C-11, C-11/C-4/C-5/C-6/C-7/C-8/C-10, C-2”/C-3”, and C-5”/C-6” (Figure 2). HMBC correlations of from H-10 to C-7, C-8, and C-9, H-1 to C-5 and C-9, H-11 to C-3, C-4, and C-5, H-3 to C-9, H-1′ to C-1, H-6′ to C-7” and H-2”/H-6” to C-7” established the structure of 3 (Figure 1). The NOE correlation of H-5β/H-1 in NOESY spectra suggested a β-orientation for C-1 (Figure 1). As a result, the structure of 3 was identified as (5R,9S)-6-O-(6-O-4-hydroxybenzoyl-β-d-glucopyranosyl)-8-hydroxymethyl-4-methylene-4,5,6,9-tetrahydro-3H-cyclopenta[b]furan-9-yl-methanol and named Shecaoiridoidside C (Figure 2).
Compound 4 was obtained as white amorphous powder. HRESIMS m/z 828.6924 [M + H]+ (calc. for 828.6929) determined the molecular formula of 4 as C48H93NO9. Methanolysis experiment of 4 liberated d-glucose which determined by GC-MS analysis. In 1H- and 13C-NMR spectra of 4, signals of anomeric proton δH (4.90, 1H, d, J = 7.6 Hz) and δC (105.6, 75.2, 78.6, 71.5, 78.7, and 62.6) indicated the presence of a β-d-glucopyranosyl moiety. The characteristics of a cerebroside with a 2-hydroxy fatty acid fraction in 4 could be confirmed by analyzing its 1H- and 13C-NMR data (Table 1 and Table 2). A fatty acid methyl ester (FAME) and a long-chain base (LCB) were obtained respectively by methanolysis of 4. GC-MS analysis determined the structure of FAM as 2-hydroxyoctadecanoic acid methyl ester. The absolute configuration of C-2′R was determined by the specific rotation = −4.8° (c 0.03, CHCl3) of the FAM [25]. The NMR data of C-2 and C-3 were compared with those of in literatures [26,27] and determined their stereo-configurations as 2S and 3R, respectively. The correlations of δH 4.77 (1H, m, H-3) with 131.6 (C-4) and 132.7 (C-5) in HMBC spectrum of 4 confirmed the olefinic bond was located in the LCB (Figure 2). The signals at δC 11.8 and 19.6 in 13C-NMR spectrum of 4 indicated the presence of a branched methyl group in 3. To determine the position of the branched methyl group, the 1D-TOCSY spectrum was used and correlations of δH 4.22 (1H, m, H-1) with 5.86 (1H, m, H-4), 0.88 (3H, d, J = 6.4 Hz, CH3-23), and 0.86 (3H, t, J = 6.4 Hz, CH3-24) could be observed. Therefore, the branched methyl group was located in the LCB. The 1H- and 13C-NMR data (Table 1 and Table 2) were further assigned by the spectra of DEPT, HSQC, 1H-1H COSY, and HMBC. Thus, 4 was established as 1-O-β-d-glucopyranosyl-(2S,3R,4E)-2-[(2′R)-2-hydroxyloctadecanamideamino]-21-methyl-4-tetracosene-1,3-diol which was named as shecaocerenoside A (Figure 1).
The known compounds were identified as jatamanin E (5) [28], 11-methoxyviburtinal (6) [29], 15-Demethylisoplumieride (7) [28], suspensolide F (8) [30], kanokoside A (9) [31], and patrinoside (10) [32] by comparing their physico-chemical constants and NMR spectroscopic data with those of in literatures (Figure 2).
The cytotoxicity of compounds 1–10 against tumor human cell lines of HL-60, HeLa, HCT15, A459, HepG2, PC-3, CNE-2, and BCG-823 were investigated in vitro. The MTT method was used to determine the IC50 values. New compound 3 exhibited evident cytotoxicity to all tumor cell lines except the Hela, and the IC50 values are from 9.6 μM to 62.2 μM, while new compound 4 showed moderate cytotoxicity to all the cell lines and the IC50 values are from 33.6 μM to 89.3 μM. By contrast, new compound 1 and known compound 9 showed moderate cytotoxicity to HCT15, A459, and HepG2 selectively. Known compound 7 also exhibited moderate cytotoxicity to HCT15 and A459 selectively (Table 3). Compounds 1 and 9 with the structural stem-nucleus 8-hydroxy-8-hydroxymethyl-6,7-epoxylcyclopenta[c]pyran-1-O-4-methylsenecioyloxyl-11-hydroxymethyl-3-en 11-O-β-d-glycoside were tend to show cytotoxicity to HepG2, which was consist with the reference reported [33]. While HCT15 was tend to sensitive to compound 3. The cytotoxicity of sfingolipids has been reported in many references, and depend on its LCB, FAM, double bonds and glycosyl group to show moderate or weak activity to most of tumor cell lines [34,35,36,37], as well as compound 4.
3. Materials and Methods
3.1. General
Column chromatographies such as Macroporous resin (AB-8 Crosslinked Polystyrene, Shanxi Lanshen Resin, Xi’an, China), silica gel (200–300 mesh, Hejie Technology Co. Ltd., Shanghai, China), and ODS-A (120A, 50 mm; DAISO, Kyoto, Japan) were used for isolations. Compounds were prepared on a preparative HPLC (Waters, Milford, MA, USA). Bruker AVANCE 400 MHz NMR instrument (Bruker SpectroSpin, Karlsruhe, Germany) was used to measure all the NMR spectra, including 1D-NMR and 2D-NMR spectra. Measured and analyzed the HRESIMS data was conducted on a Xero Q Tof MS spectrometer (Waters, Milford, MA, USA). IR Spectra data was recorded on FTIR-8400S (Shimadzu, Kyoto, Japan). The GC-MS (Angilent, Palo Alto, CA, USA) instrument was used to analysis the volatile derivatives from compounds. The growth of the tumor cell lines was monitored with a microplate reader (BMG FLUOStar OPTIMA, Ortenberg, Germany).
3.2. Plant Materials
The aerial part of H. diffusa Willd. was collected from Guangdong province of China and identified by Shuyuan Li of Guangdong Pharmaceutical University. The voucher specimen (No. 20160987) is deposited at the Herbarium of Guangdong Pharmaceutical University, Guangzhou, China.
3.3. Extraction and Isolation
The dried H. diffusa Willd. (10.0 Kg) were extracted two times (each for 2 h) with 75% EtOH (100 L) under reflux. The extract (1611 g) was suspended in water (15 L), and then extracted with petroleum ether (60–90 °C), EtOAc and n-butanol, respectively. Solvents were removed under vacuum to give extracts of petroleum ether (74.3 g), EtOAc (135.3 g), n-butanol (196.5 g), and remained water (1152.4 g). The EtOAc fraction (150.0 g) was subject to silica gel column column and eluted with a gradient of CH2Cl2/MeOH (30:1 to 0:1) to yield fractions of F1–F6. F2 (28.4 g) was further chromatographed on silica gel column and eluted with petroleum ether/EtOAc (15:1 to 1:1) to yield subfractions of A1–A4. The sub-fraction A2 (6.2 g) was repeated chromatographed on silica gel column and eluted with petroleum ether/EtOAc (5:1) to yield compound 4 (58 mg). F3 (30.6 g) was chromatographed on silica gel column and eluted with a gradient of CH2Cl2/MeOH (20:1 to 5:1) to yield sub-fractions B1–B5. B2 (10.4 g) was repeated chromatographed on silica gel column and eluted with CH2Cl2/MeOH (15:1) to yield compound 6 (46 mg). B4 (5.6 g) was repeated chromatographed on silica gel column and eluted with CH2Cl2/MeOH (8:1) to yield compound 5 (41 mg). F4 (62.4 g) was chromatographed on silica gel column and eluted with a gradient of CH2Cl2/MeOH (15:1–1:1) to yield sub-fractions C1–C6. C3 (11.2 g) was chromatographed on silica gel column and eluted with a gradient of CH2Cl2/MeOH (10:1 to 3:1), and then purified on a preparative HPLC with Hypersil-ODS II column (10 μm, 20 × 300 mm) eluted with MeOH/H2O (18%, flow rate 8 mL/min) to yield compounds 8 (48 mg, tR = 15 min), 10 (57 mg, tR = 27 min), 1 (62 mg, tR = 31 min), and 9 (53 mg, tR = 35 min). C5 (14.4 g) was chromatographed on silica gel column and eluted with CH2Cl2/MeOH (5:1), and then purified on a preparative HPLC with Hypersil-ODS II column (10 μm, 20 × 300 mm) eluted with MeOH/H2O (8%, flow rate 8 mL/min) to yield compounds 2 (48 mg, tR = 11 min), 7 (43 mg, tR = 18 min), and 3 (55 mg, tR = 23 min).
Shecaoiridoidside A (1). white amorphous powder; = −25.4 (c = 0.20, CH3OH); IR (KBr) νmax 3433, 3384, 2921, 2871, 1723, 1648, 1455, 1353, 1252, 1082, 880 cm−1; ESIMS m/z 511 (100) [M + Na]+; HRESIMS [M + Na]+ m/z 511.1788 calc. 511.1791 for C22H32O12Na; 1H- and 13C-NMR data, see Table 1 and Table 2.
Shecaoiridoidside B (2). white amorphous powder, + 109.4° (c 0.10, MeOH); IR (KBr) νmax 3462, 3430, 2974, 2858, 1712, 1648, 1428, 1373, 1235, 1104 cm−1; ESIMS m/z 515 (100) [M + Na]+; HRESIMS [M + Na]+ m/z 515.1737, calcd. 515.1741 for C21H32O13Na; 1H- and 13C-NMR data, see Table 1 and Table 2.
3.4. Acid Hydrolysis of 1–3
Acid hydrolysis experiment was carried out as the method in reference [24]. Briefly, the sugar residues were obtained by hydrolyzing of compounds 1–3 (2.0 mg) with 2 mol/L H2SO4 (2.0 mL), and then treated with trimethylchlorosilane, respectively. The sugar derivatives were further analyzed by GC-MS. As a result, the sugar derivatives from compounds 1 and 3 were determined to be d-galactose (tR = 19.46 min) and d-glucose (tR = 11.33 min), respectively. The sugar derivatives from compound 2 was determined to be d-glucose (tR = 11.33 min) and d-apiose (tR = 14.53 min).
3.5. Methanolysis of 4
Methanolysis of 4 was carried out according to the previous study [38]. In short, compound 4 (5.0 mg) was dissolved in in 82 % aqueous MeOH (20 mL) with 5% HCl and refluxed for 18 h. The FAME of 4 was obtained by extracting the reaction mixture with n-hexane. The FAME of 4 was a white amorphous powder, = −4.8° (c 0.02, CHCl3). Analyzed the FAME by GC-MS and the characteristic fragment ions (m/z 314 [M]+, 256 [M − COOMe]+) were obtained. As a result, the FAME of 4 was identified as 2-hydroxyoctadecanoic acid methyl ester. The remained solution was analyzed by GC-MS and the monosaccharide of 4 was identified as d-glucose (tR = 11.33 min). After that the remained solution was evaporated MeOH and the aqueous ammonia was added to adjust pH 9.0, and then extracted the solution with Et2O to obtain the LCB. The fragment ions of m/z 384 [M + H]+ and 366 [M − H2O + H]+ from ESIMS analysis led the LCB of 4 was identified as 2-aminotetracos-7-ene-1,3-diol (Figure 2).
3.6. Cytotoxicity Assay of Compounds 1–10
The cytotoxicity of all compounds against human tumor cell lines of HL-60, HeLa, HCT15, A459, HepG2, PC-3, CNE-2 and BCG-823 was assayed by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) method in vitro. The assay protocol was conducted by previous published paper [24,39]. The tested compounds 1–10 were dissolved in DMSO and adjusted to the final concentrations from 1.0 μM to 300 μM by diluting with the growth medium. 5-Fluorouracil was used as the positive drug.
4. Conclusions
We investigated the chemical constituents of H. diffusa Willd. based on its clinical application of treating malignant tumors and 10 compounds were obtained, including three new iridoid glycosides and a new cerebroside. The structures of new compounds were identified as (1S,5S,9S,6S,7R,8S)-8-hydroxy-8-hydroxymethyl-6,7-epoxylcyclopenta[c]pyran-1-O-4-methylsenecioyl-oxyl-11-hydroxymethyl-3-en 11-O-β-d-glucopyranoside (1), (5S,7S,8S,9S)-8-hydroxy-8-methyl-4-methylenehexahydrocyclopenta[c]pyran-1(3H)-one 7-O-(6-O-β-d-apiofuranosyl)-β-d-glucopyrano-side (2), (5R,9S)-6-O-(6-O-4-hydroxybenzoyl-β-d-glucopyranosyl)-8-hydroxymethyl-4-methylene-4,5,6,9-tetrahydro-3H-cyclopenta[b]furan-9-yl-methanol (3), and 1-O-β-d-glucopyranosyl-(2S,3R,4E) -2-[(2′R)-2-hydroxyloctadecanamideamino]-21-methyl-4-tetracosene-1,3-diol (4), respectively. Antitumor assays in vitro discovered cytotoxic compounds 1, 3, 4, 7, and 9, especially found that new compound 3 exhibited evident cytotoxicity to all tumor cell lines except the Hela.
Acknowledgments
This research was supported by the Program of Natural Science Foundation of Heilongjiang (No. H2015040) and the Postgraduate Tutor Foundation of Harbin Medical University Daqing (DSJJ2017001).
Author Contributions
Changfu Wang designed and performed the experiments; Xuegang Zhou contributed literature search, figures preparation and data collection; Youzhi Wang and Donghua Wei contributed data analysis and data interpretation; Chengjie Deng and Xiaoyun Xu helped Changfu Wang to perform the experiments; Shiqin Sun contributed reagents/materials/analysis tools and Ping Xin wrote the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Cai, Q.Y.; Lin, J.M.; Wei, L.H.; Zhang, L.; Wang, L.L.; Zhan, Y.Z.; Zeng, J.W.; Xu, W.; Shen, A.L.; Hong, Z.F.; et al. Hedyotis diffusa Willd Inhibits Colorectal Cancer Growth in Vivo via Inhibition of STAT3 Signaling Pathway. Int. J. Mol. Sci. 2012, 13, 6117–6128. [Google Scholar] [CrossRef] [PubMed]
- Niu, Y.; Meng, Q.X. Chemical and preclinical studies on Hedyotis diffusa with anticancer potential. J. Asian Nat. Prod. Res. 2013, 15, 550–565. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W. Preparation of Graphene-Multi-Walled Carbon Nanotube Composite for Quantitive Determination of 2-hydroxy-3- Methylanthraquinone in Hedyotis diffusa. Int. J. Electrochem. Sci. 2017, 12, 629–638. [Google Scholar]
- Chao, T.H.; Fu, P.K.; Chang, C.H.; Chang, S.N.; Chiahung, M.F.; Lin, C.H. Prescription patterns of Chinese herbal products for post-surgery colon cancer patients in Taiwan. J. Ethnopharmacol. 2014, 155, 702–708. [Google Scholar] [CrossRef] [PubMed]
- Yeh, Y.C.; Chen, H.Y.; Yang, S.H.; Lin, Y.H.; Chiu, J.H.; Lin, Y.H.; Chen, J.L. Hedyotis diffusa Combined with Scutellaria barbata Are the Core Treatment of Chinese Herbal Medicine Used for Breast Cancer Patients: A Population-Based Study. Evid. Based Complement. Altern. Med. 2014, 2014, 202378. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; He, J.; Tong, X.; Tang, L.; Liu, M. The Hedyotis diffusa willd. (rubiaceae): A review on phytochemistry, pharmacology, quality control and pharmacokinetics. Molecules 2016, 21, 710. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.H.; Liu, M.H.; Zhang, X.L.; He, J.Y. Chemical profiles and protective effect of Hedyotis diffusa willd in lipopolysaccharide-induced renal inflammation mice. Int. J. Mol. Sci. 2015, 16, 27252–27269. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Schmitz, O.J. Comprehensive two-dimensional liquid chromatography tandem diode array detector (DAD) and accurate mass QTOF-MS for the analysis of flavonoids and iridoid glycosides in Hedyotis diffusa. Anal. Bioanal. Chem. 2015, 407, 231–240. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Q.Q.; Cheng, C.S.; Liu, Z.Z.; Ouyang, Y.; Lin, J.R.; Zhang, Z.F.; Liu, Z.Q.; Zhou, H. Comparison of identification and medicinal progress of Hedyotis diffusa and H. corymbosa. Chin. Tradit. Herb. Drugs 2017, 48, 4328–4338. [Google Scholar]
- Chen, Y.; Lin, Y.; Yachan, L.I.; Candong, L.I. Total flavonoids of Hedyotis diffusa willd inhibit inflammatory responses in lps-activated macrophages via suppression of the nf-κb and mapk signaling pathways. Exp. Ther. Med. 2016, 11, 1116–1122. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Bai, Y.; Huo, Z. The protective effect of Hedyotis diffusa on collagen induced arthritis rats. Int. J. Clin. Exp. Med. 2016, 9, 12880–12887. [Google Scholar]
- Gao, X.; Li, C.; Tang, Y.L.; Zhang, H. Chan, S.W. Effect of Hedyotis diffusa water extract on protecting human hepatocyte cells (LO) from HO-induced cytotoxicity. Pharm. Biol. 2015, 1, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Kuo, Y.J.; Lin, J.P.; Hsiao, Y.T.; Chou, G.L.; Tsai, Y.H.; Chiang, S.Y.; Lin, J.G.; Chung, J.G. Ethanol Extract of Hedyotis diffusa Willd Affects Immune Responses in Normal Balb/c Mice In Vivo. In Vivo 2015, 29, 453–460. [Google Scholar] [PubMed]
- Hu, E.; Wang, D.; Chen, J.; Tao, X. Novel cyclotides from Hedyotis diffusa induce apoptosis and inhibit proliferation and migration of prostate cancer cells. Int. J. Clin. Exp. Med. 2015, 8, 4059–4065. [Google Scholar] [PubMed]
- Zhang, Y.; Xie, R.F.; Xiao, Q.G.; Li, R.; Shen, X.L.; Zhu, X.G. Hedyotis diffusa Willd extract inhibits the growth of human glioblastoma cells by inducing mitochondrial apoptosis via AKT/ERK pathways. J. Ethnopharmacol. 2014, 158, 404–411. [Google Scholar] [CrossRef] [PubMed]
- Kuo, Y.J.; Liu, Y.J.; Way, T.D. Synergistic inhibition of leukemia wehi-3 cell growth by arsenic trioxide and Hedyotis diffusa willd extract in vitro and in vivo. Exp. Ther. Med. 2017, 13, 3388–3396. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Wang, B.; Li, J.; Ye, B.; Lin, S.; Qian, W.; Shan, L.; Efferthd, T. Total coumarins of Hedyotis diffusa, induces apoptosis of myelodysplastic syndrome skm-1 cells by activation of caspases and inhibition of pi3k/akt pathway proteins. J. Ethnopharmacol. 2017, 196, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhang, J.; Qi, B.; Jiang, G.; Liu, J.; Zhang, P.; Ma, Y.; Li, W. The anti-tumor effect and bioactive phytochemicals of Hedyotis diffusa willd on ovarian cancer cells. J. Ethnopharmacol. 2016, 192, 132–139. [Google Scholar] [CrossRef] [PubMed]
- Xin, G.; Chang, L.; Tang, Y.L.; Zhang, H.; Chan, S.W. Effect of Hedyotis diffusa water extract on protecting human hepatocyte cells (lo2) from H2O2-induced cytotoxicity. Pharm. Biol. 2016, 54, 1148–1155. [Google Scholar]
- Sun, G.; Wei, L.; Feng, J.; Lin, J.; Peng, J. Inhibitory effects of Hedyotis diffusa willd on colorectal cancer stem cells. Oncol. Lett. 2016, 11, 3875–3881. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Zheng, R.; Baade, P.D.; Zhang, S.; Zeng, H.; Bray, F.; Ahmedin, J.; Xue, Q.Y.; Jie, H. Cancer statistics in China, 2015. CA Cancer J. Clin. 2016, 66, 115–132. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.Y.; Li, S.Y.; Li, W.J.; Guo, J.M.; Tian, K.; Hu, Q.F.; Huang, X.Z. Phenolic glycosides from Ficus tikoua and their cytotoxic activities. Carbohydr. Res. 2013, 382C, 19–24. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Chen, T.; Liu, X.H.; Shen, Y.H.; Li, H.L.; Shan, L.; Liu, R.H.; Xu, X.K.; Zhang, W.D.; Wang, H. Iridoids and lignans from Valeriana jatamansi. J. Nat. Prod. 2010, 73, 632–638. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Di, L.; Gao, W.C.; Wang, K.J.; Zu, L.B. Cytotoxic iridoids from the roots of Patrinia scabra. J. Nat. Prod. 2012, 75, 1723–1728. [Google Scholar] [CrossRef] [PubMed]
- Shibuya, H.; Kawashima, K.; Sakagami, M.; Shimomura, M.; Ohashi, K.; Kitagawa, I. Sphingolipids and glycerolipids. Ι. Chemical structures and ionophoretic activities of soyacerebrosides Ι and ΙΙ form soybean. Chem. Pharm. Bull. 1990, 38, 2933–2938. [Google Scholar] [CrossRef] [PubMed]
- Satoshi, K.; Kazufumi, N.; Masanori, I.; Ryuichi, H. Isolation and Structure Determination of Six Glucocerebrosides from the Starfish Luidia maculate. Chem. Pharm. Bull. 2002, 50, 1091–1096. [Google Scholar]
- Koji, Y.; Noriko, W.; Hiroyuki, O.; Rei, M.; Ryuichi, I.; Masanori, I.; Ryuichi, H. Constituents of Holothuroidea, Isolation of Ante-iso Type Regio isomer on Long Chain Base Moiety of Glucocerebroside from the Sea Cucumber Holothuria leucospilota. Chem. Pharm. Bull. 2005, 53, 788–791. [Google Scholar]
- Dinda, B.; Debnath, S.; Banik, R. Naturally occurring iridoids and secoiridoids. An updated review, part 4. Chem. Pharm. Bull. 2011, 59, 803–833. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.G.; Yu, L.L.; Huang, R.; Lv, Y.P.; Gui, S.H. 11-methoxyviburtinal, a new iridoid from Valeriana jatamansi. Arch. Pharmacal. Res. 2005, 28, 1161–1163. [Google Scholar] [CrossRef]
- Iwagawa, T.; Yaguchi, S.; Hase, T. Iridoid glucosides from viburnum suspensum. Phytochemistry 1990, 29, 310–312. [Google Scholar] [CrossRef]
- Zhou, Y.; Fang, Y.; Gong, Z.F.; Duan, X.Y.; Liu, Y.W. Two new terpenoids from Valeriana officinalis. Chin. J. Nat. Med. 2009, 7, 270–273. [Google Scholar] [CrossRef]
- Nishiya, K.; Kimura, T.; Takeya, K.; Itokawa, H. Sesquiterpenoids and iridoid glycosides from Valeriana fauriei. Phytochemistry 1992, 31, 3511–3514. [Google Scholar] [CrossRef]
- Dinda, B.; Debnath, S.; Harigaya, Y. Naturally occurring secoiridoids and bioactivity of naturally occurring iridoids and secoiridoids. A review, part 2. Chem. Pharm. Bull. 2007, 38, 689–728. [Google Scholar] [CrossRef]
- Teinkela, J.E.; Noundou, X.S.; Nguemfo, E.L.; Meyer, F.; Djoukoue, A.; Van Antwerpen, P.; Ngouela, S.; Tsamo, E.; Mpondo, E.A.; Vardamides, J.C.; et al. Identification of compounds with anti-proliferative activity from the wood of Ficus elastica Roxb. ex Hornem. aerial roots. Fitoterapia 2016, 112, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Murshid, S.S.A.; Badr, J.M.; Youssef, D.T.A. Penicillosides A and B: New cerebrosides from the marine-derived fungus Penicillium, species. Rev. Bras. Farmacogn. 2016, 26, 29–33. [Google Scholar] [CrossRef]
- Tian, X.R.; Tang, H.F.; Feng, J.T.; Li, Y.S.; Lin, H.W.; Fan, X.P.; Zhang, X. Neritinaceramides A–E, New Ceramides from the Marine Bryozoan Bugula neritina inhabiting South China Sea and their cytotoxicity. Mar. Drugs 2014, 12, 1987–2003. [Google Scholar] [CrossRef] [PubMed]
- Cateni, F.; Zilic, J.; Zacchigna, M.; Procida, G. Cerebrosides with antiproliferative activity from Euphorbia peplis L. Fitoterapia 2010, 81, 97–103. [Google Scholar] [CrossRef] [PubMed]
- Lei, X.; Li, J.; Li, B.; Zhang, N.; Liu, H. Separation and identification of four new compounds with antibacterial activity from Portulaca oleraceal. Molecules 2015, 20, 16375–16387. [Google Scholar] [CrossRef] [PubMed]
- Nguen, T.H.; Pham, H.V.; Pham, N.K.; Quach, N.D.; Pudhom, K.; Hansen, P.E.; Phung Nguyen, K.P. Chemical constituents from Sonneratia ovata backer and their in vitro cytotoxicity and acetylcholinesterase inhibitory activities. Bioorg. Med. Chem. Lett. 2015, 25, 2366–2371. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the Shecaoiridoidside A–C and shecaocerenoside A are available from the authors. |
Figure 1.
The 1H−1H Correlation Spectroscopy (COSY), key Heteronuclear Multiple Bond Correlation (HMBC) correlations of 1–4, and Nuclear Overhauser Effect (NOE) correlations of 1–3.
Figure 1.
The 1H−1H Correlation Spectroscopy (COSY), key Heteronuclear Multiple Bond Correlation (HMBC) correlations of 1–4, and Nuclear Overhauser Effect (NOE) correlations of 1–3.
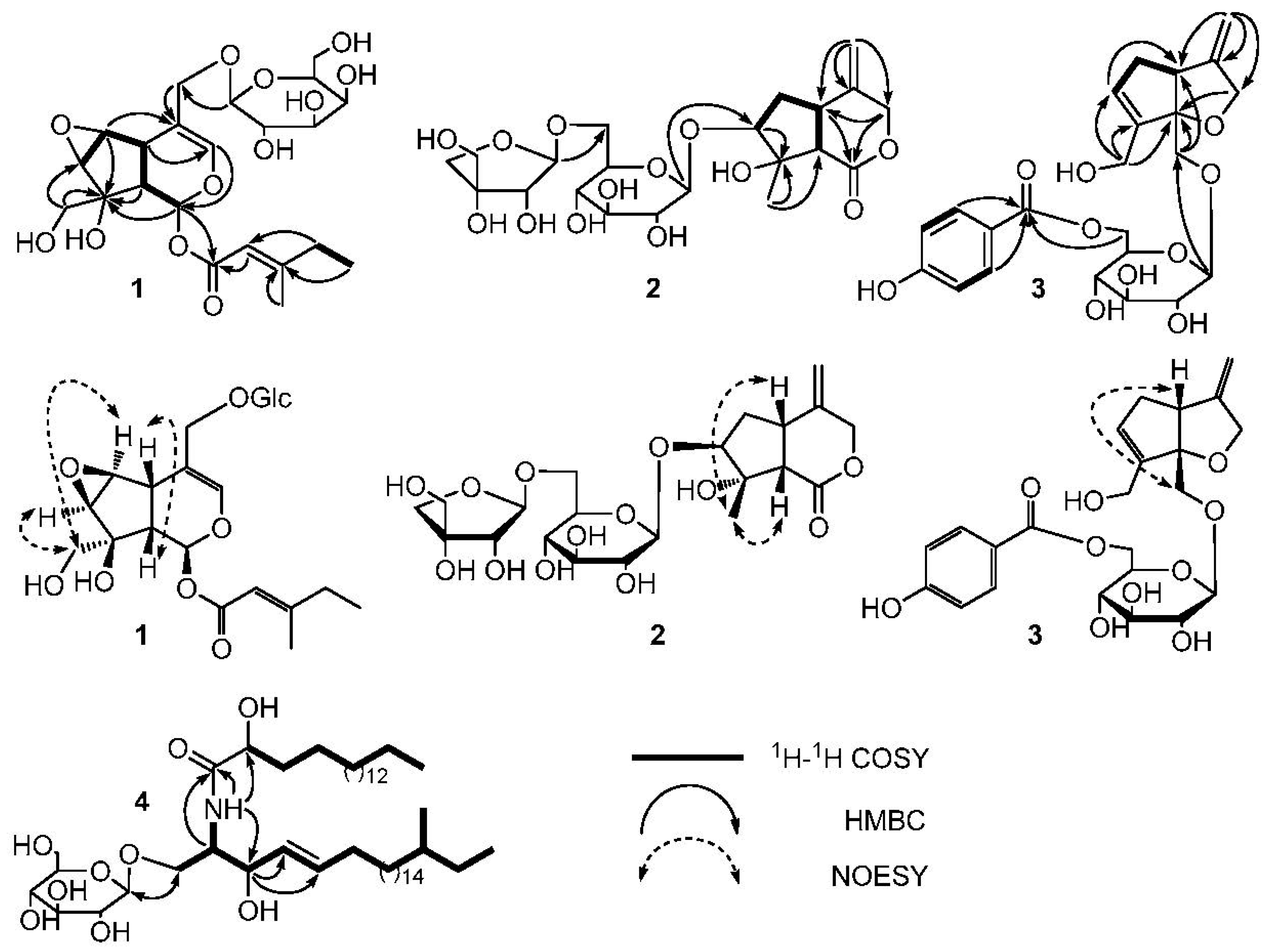
Figure 2.
The structures of compounds 1–10.


{kind=link}
{kind=link}
Table 1.
1H-NMR data of compounds 1–4 (400 MHz, δ in ppm, J in Hz).
| 1 a | 2 a | 3 a | 4 b | ||
|---|---|---|---|---|---|
| H | δH (J, Hz) | δH (J, Hz) | δH (J, Hz) | H | δH (J, Hz) |
| 1 | 6.41, d (2.0) | 3.96, d (10.4); 3.76, d (10.4) | NH | 8.35, d, (8.4) | |
| 3 | 6.40, brs | 5.10, d (11.0) 4.44, d (11.6) | 4.37, d (12.6); 4.18, d (12.6) | 1 | 4.22, m 4.72, m |
| 5 | 3.09, brd (8.5) | 3.34, m | 3.25, m | 2 | 4.78, m |
| 6 | 4.04, d (2.5) | 2.34, dd (8.4, 13.4); 2.19, m | 2.74, m; 2.26, brd (16.4) | 3 | 4.77, m |
| 7 | 3.36, d (2.5) | 3.87, m | 5.78, brs | 4 | 5.86, m |
| 9 | 2.05, m | 3.08, d (10.7) | 5 | 5.98, m | |
| 10 | 3.69, d (2.8) | 1.59, s | 4.11, brd (10.0) | 6 | 2.06, m |
| 11 | 4.21, d (11.6); 4.35, d (11.6) | 5.08, 5.11, s | 4.92, 4.91, d (2.0) | 7–22 | 1.16–1.42, brs |
| 1′ | 4.41, d (7.8) | 4.72, d (7.8) | 23 | 0.88, d (6.4) | |
| 2′ | 5.62, s | 3.16, t (8.2) | 3.27, m | 24 | 0.86, t (6.4) |
| 3′ | 3.30, m | 3.43, m | 2′ | 4.61, m | |
| 4′ | 2.16, m | 3.29, m | 3.40, m | 3′ | 1.85, m |
| 5′ | 0.89, t (7.4) | 3.34, m | 3.62, m | 4′ | 1.73, m; 1.16–1.42, brs |
| 6′ | 2.15, s | 3.59, 3.95, m | 4.60, brd, (11.6); 4.42, dd, (11.8, 4.8) | 5′–17′ | 1.16–1.42, brs |
| 1”” | 4.72, d (8.1) | 5.01, d (1.7) | 18′ | 0.88, t (6.4) | |
| 2” | 3.35, m | 3.87, m | 7.88, d (8.8) | 1” | 4.90, d, (7.6) |
| 3” | 4.05, m | 6.81, d (8.8) | 2” | 4.02, m | |
| 4” | 3.49, m | 3.95, 3.75, m | 3” | 4.22, m | |
| 5” | 3.59 (1H, m) | 3.58, s | 6.81, d (8.8) | 4” | 4.22, m |
| 6” | 3.67, m; 3.86, dd (1.5, 11.5) | 7.88, d (8.8) | 5” | 3.88, m | |
| 6” | 4.36, 4.50, m | ||||
a Measured in CD3OD at 30 °C; b Measured in C5D5N-d5 at 30 °C.
Table 2.
13C-NMR data of compounds 1–4 (100 MHz, δ in ppm).
| 1 a | 2 a | 3 a | 4 b | ||||
|---|---|---|---|---|---|---|---|
| C | δC | C | δC | C | δC | C | δC |
| 1 | 90.8, CH | 1 | 175.2, C | 1 | 72.8, CH2 | 1 | 70.2, CH2 |
| 3 | 142.4, CH | 3 | 71.5, CH2 | 3 | 72.8, CH2 | 2 | 54.5, CH |
| 4 | 109.8, C | 4 | 144.5, C | 4 | 156.2, C | 3 | 72.3, CH |
| 5 | 35.4, CH | 5 | 41.2, CH | 5 | 49.8, CH | 4 | 131.6, CH |
| 6 | 59.9, CH | 6 | 40.0, CH2 | 6 | 39.2, CH2 | 5 | 132.7, CH |
| 7 | 60.3, CH | 7 | 90.1, CH | 7 | 131.4, CH | 6 | 34.2, CH2 |
| 8 | 80.2, C | 8 | 86.1, C | 8 | 144.4, C | 7-20 | 29.5–30.5, CH2 |
| 9 | 43.6, CH | 9 | 54.2, CH | 9 | 99.8, C | 21 | 35.7, CH |
| 10 | 67.2, CH2 | 10 | 22.5, CH3 | 10 | 59.3, CH2 | 22 | 30.5, CH2 |
| 11 | 69.8, CH2 | 11 | 113.8, CH2 | 11 | 105.4, CH2 | 23 | 19.6, CH3 |
| 1′ | 165.8, C | 1′ | 99.9, CH | 1′ | 103.9, CH | 24 | 11.8, CH3 |
| 2′ | 114.6, CH | 2′ | 75.5, CH | 2′ | 74.8, CH | 1′ | 175.6, C |
| 3′ | 162.1, C | 3′ | 78.7, CH | 3′ | 77.7, CH | 2′ | 72.5, CH |
| 4′ | 33.8, CH2 | 4′ | 72.3, CH | 4′ | 72.3, CH | 3′ | 35.8, CH2 |
| 5′ | 11.7, CH3 | 5′ | 78.3, CH | 5′ | 76.0, CH | 4′ | 26.2, CH2 |
| 6′ | 19.0, CH3 | 6′ | 68.3, CH2 | 6′ | 65.2, CH2 | 5′-15′ | 29.5–30.5, CH2 |
| 1” | 100.4, CH | 1” | 111.5, CH | 1” | 122.4, C | 16′ | 32.2, CH2 |
| 2” | 72.6, CH | 2” | 76.2, CH | 2” | 132.9, CH | 17′ | 22.8, CH2 |
| 3” | 73.2, CH | 3” | 80.8, C | 3” | 116.6, CH | 18′ | 14.2, CH3 |
| 4” | 69.2, CH | 4” | 75.4, CH2 | 4” | 164.2, C | 1” | 105.6, CH |
| 5” | 75.6, CH | 5” | 65.8, CH2 | 5” | 116.6, CH | 2” | 75.2, CH |
| 6” | 63.4, CH2 | 6” | 132.9, CH | 3” | 78.6, CH | ||
| 7” | 167.8, C | 4” | 71.5, CH | ||||
| 5” | 78.7, CH | ||||||
| 6” | 62.6, CH2 | ||||||
a Measured in CD3OD at 30 °C; b Measured in C5D5N-d5 at 30 °C.
Table 3.
In vitro antitumor activity of compounds 1–10 in a panel of 8 tumor cell lines.
| Compounds | HL-60 | Hela | HCT15 | A459 | HepG2 | PC-3 | CNE-2 | BGC-823 |
|---|---|---|---|---|---|---|---|---|
| 1 | >100.0 | >100.0 | 87.6 ± 1.2 | 77.7 ± 1.6 | 37.6 ± 1.4 | >100.0 | >100.0 | >100.0 |
| 2 | >100.0 | >100.0 | >100.0 | >100.0 | >100.0 | >100.0 | >100.0 | >100.0 |
| 3 | 17.1 ± 0.7 | 62.2 ± 0.5 | 9.6 ± 0.8 | 14.8 ± 0.9 | 11.4 ± 1.6 | 26.2 ± 1.3 | 21.5 ± 0.6 | 13.4 ± 1.1 |
| 4 | 74.8 ± 1.3 | 89.3 ± 1.8 | 37.3 ± 1.5 | 33.6 ± 1.1 | 49.5 ± 1.4 | 64.0 ± 0.9 | 55.2 ± 1.1 | 44.1 ± 1.7 |
| 5 | >100.0 | >100.0 | >100.0 | >100.0 | >100.0 | >100.0 | >100.0 | >100.0 |
| 6 | >100.0 | >100.0 | >100.0 | >100.0 | >100.0 | >100.0 | >100.0 | >100.0 |
| 7 | >100.0 | >100.0 | 71.3 ± 1.2 | 50.4 ± 1.1 | >100.0 | 34.2 ± 1.3 | >100.0 | >100.0 |
| 8 | >100.0 | >100.0 | 89.8 ± 1.2 | 91.3 ± 0.7 | >100.0 | >100.0 | >100.0 | >100.0 |
| 9 | >100.0 | >100.0 | 96.1 ± 1.6 | 78.3 ± 0.8 | 97.9 ± 1.4 | >100.0 | >100.0 | >100.0 |
| 10 | >100.0 | >100.0 | >100.0 | >100.0 | >100.0 | >100.0 | >100.0 | >100.0 |
| 5-Fluorouracil | 7.5 ± 0.6 | 10.4 ± 0.4 | 4.7 ± 0.4 | 14.7 ± 1.1 | 22.8 ± 1.4 | 13.2 ± 0.7 | 11.6 ± 0.8 | 17.8 ± 0.7 |
Key: All results are expressed as IC50 values in μM. Compounds with IC50 > 100 μM were inactive for the tumor cell lines.
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Wang, C.; Zhou, X.; Wang, Y.; Wei, D.; Deng, C.; Xu, X.; Xin, P.; Sun, S. The Antitumor Constituents from Hedyotis Diffusa Willd. Molecules 2017, 22, 2101. https://doi.org/10.3390/molecules22122101
AMA Style
Wang C, Zhou X, Wang Y, Wei D, Deng C, Xu X, Xin P, Sun S. The Antitumor Constituents from Hedyotis Diffusa Willd. Molecules. 2017; 22(12):2101. https://doi.org/10.3390/molecules22122101
Chicago/Turabian StyleWang, Changfu, Xuegang Zhou, Youzhi Wang, Donghua Wei, Chengjie Deng, Xiaoyun Xu, Ping Xin, and Shiqin Sun. 2017. "The Antitumor Constituents from Hedyotis Diffusa Willd" Molecules 22, no. 12: 2101. https://doi.org/10.3390/molecules22122101