
Preparation of “Constrained Geometry” Titanium Complexes of [1,2]Azasilinane Framework for Ethylene/1-Octene Copolymerization



Abstract
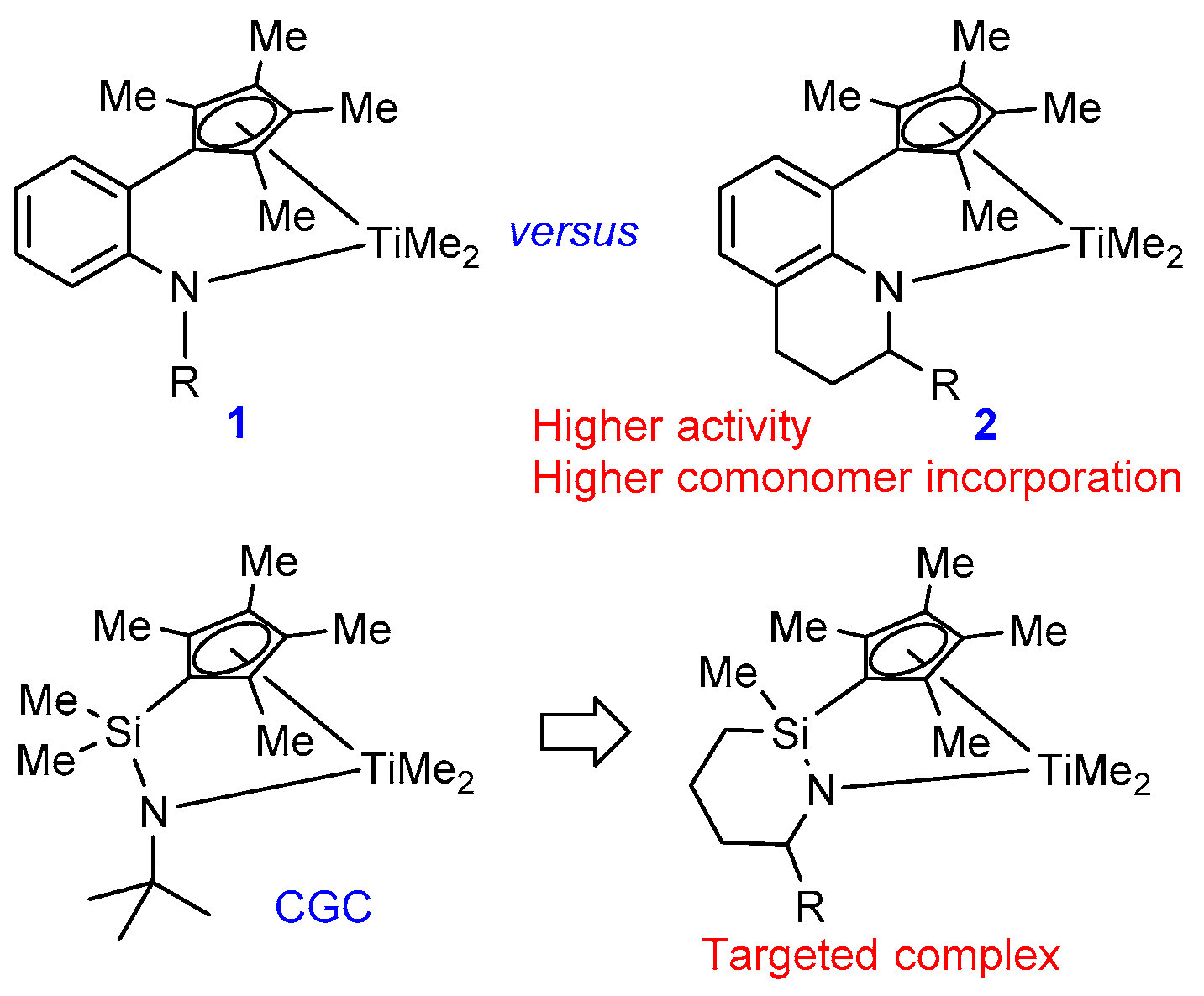
:1. Introduction
2. Results
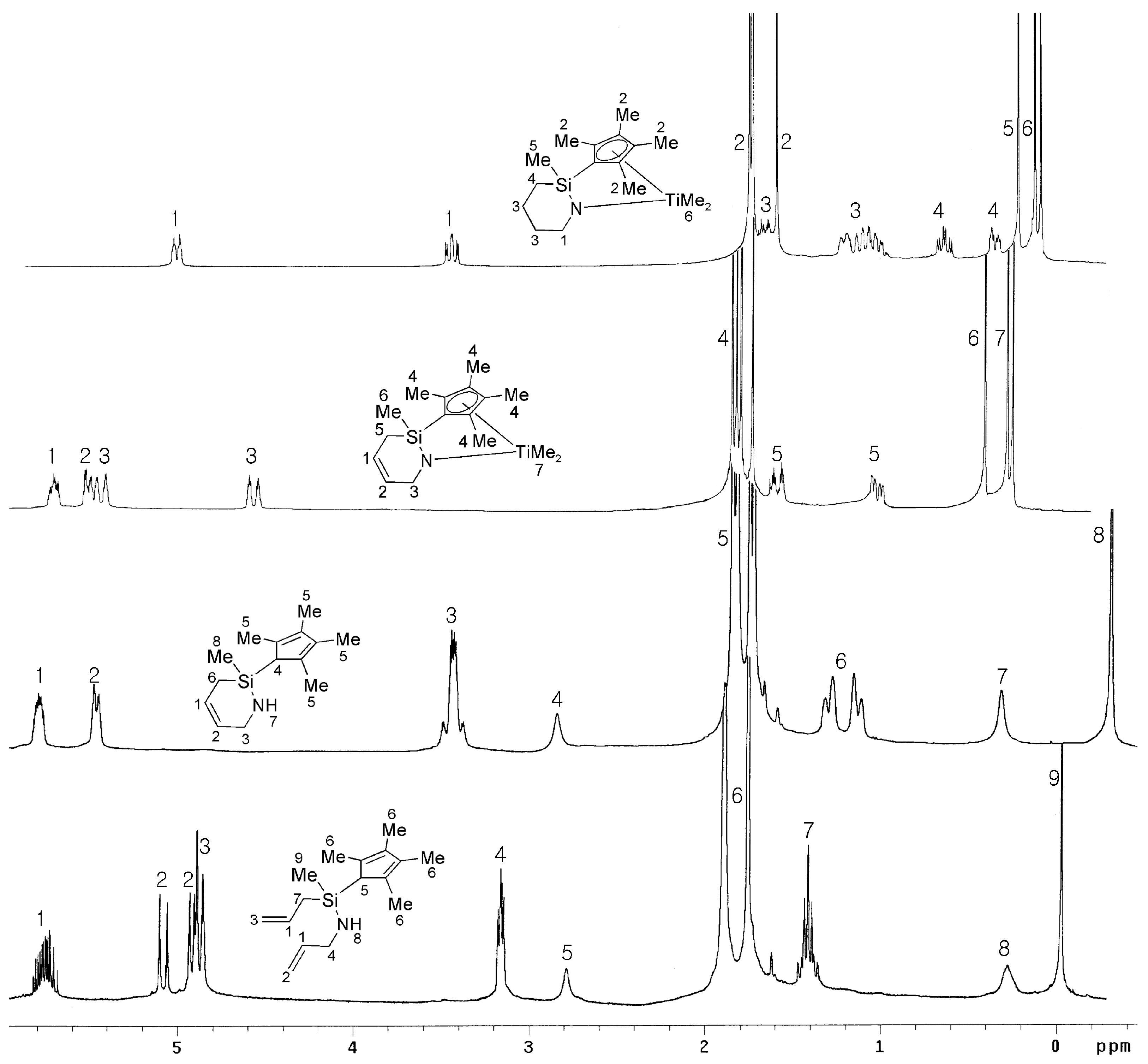
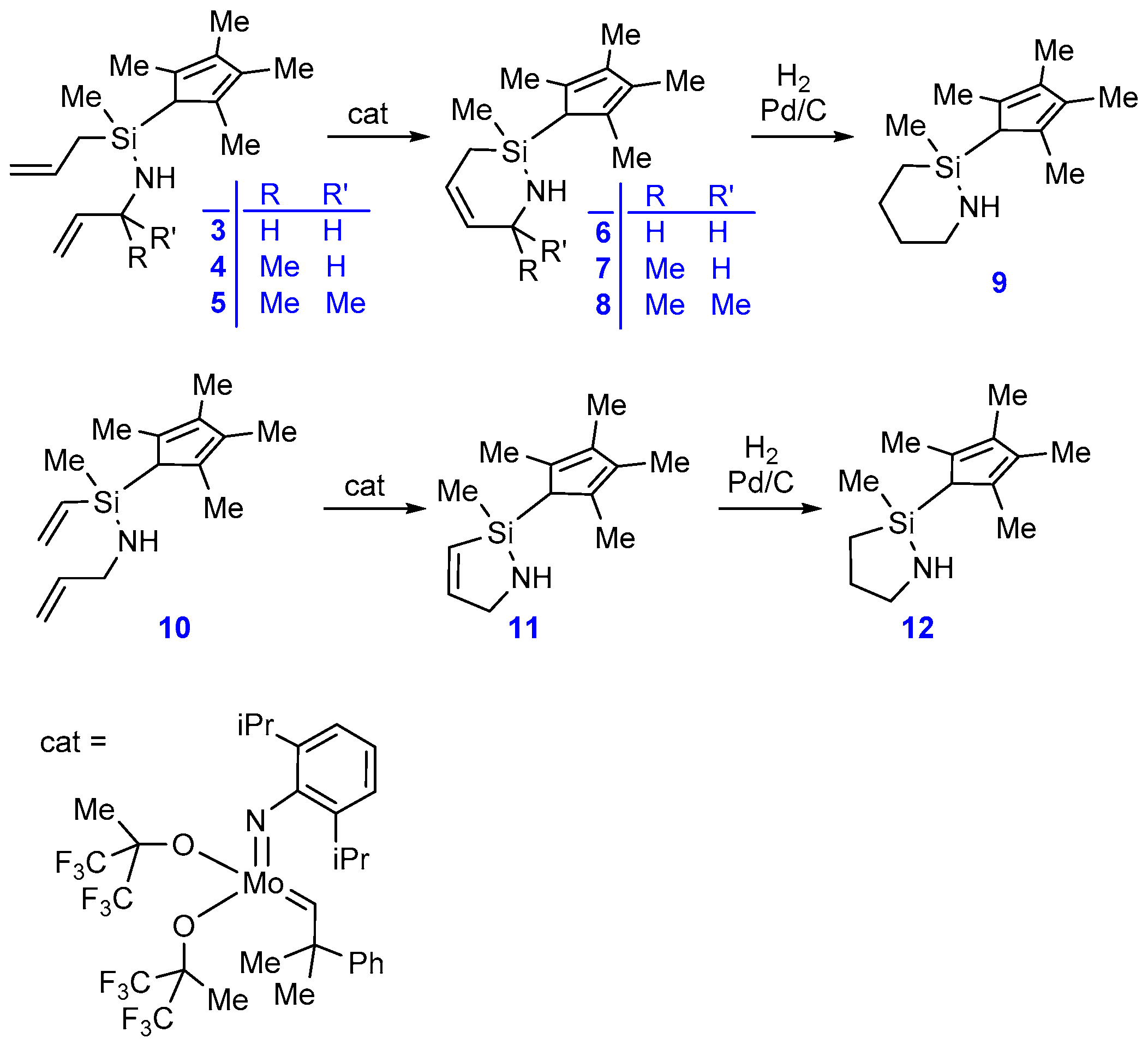
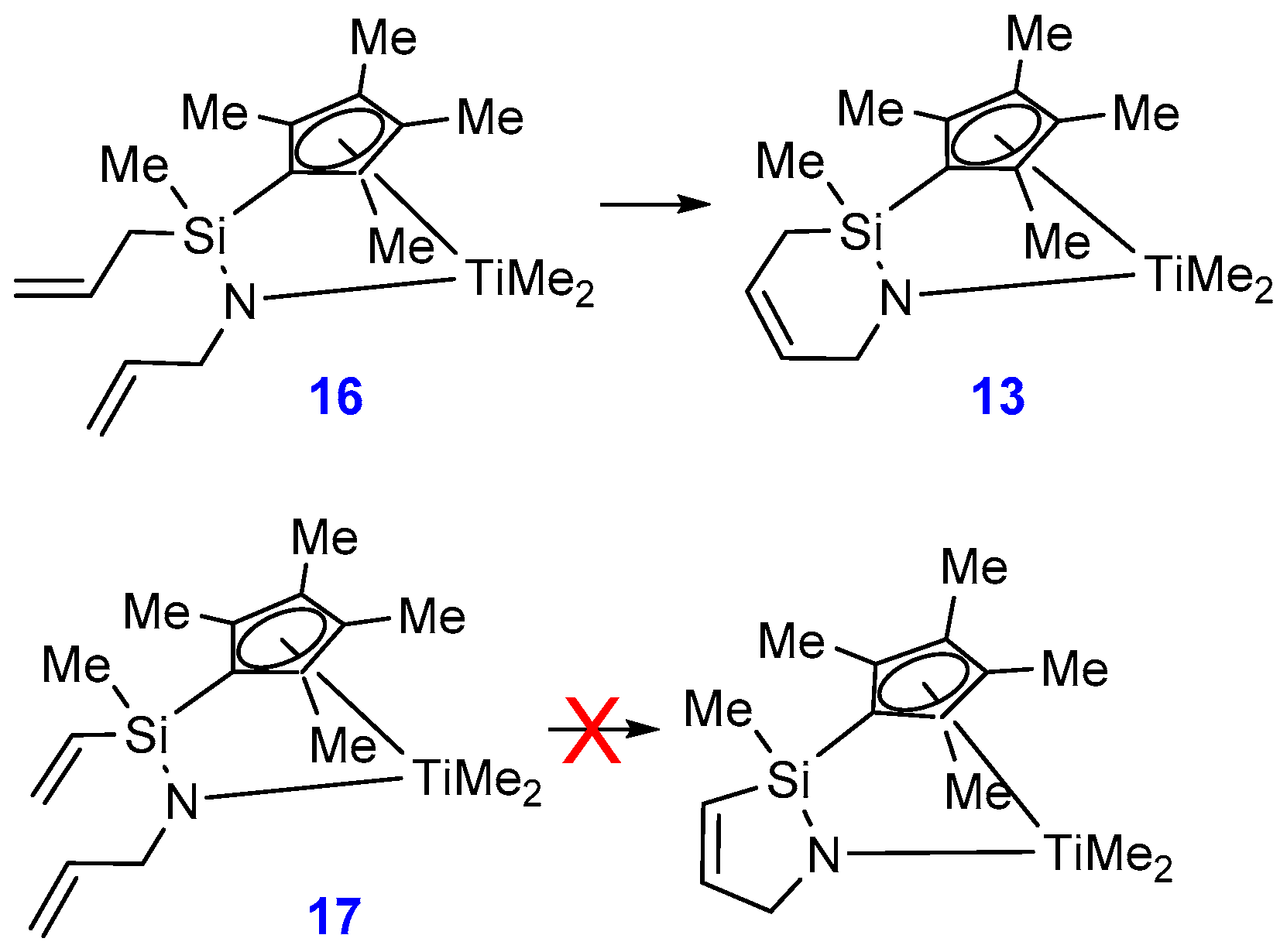
2.1. Synthesis and Characterization
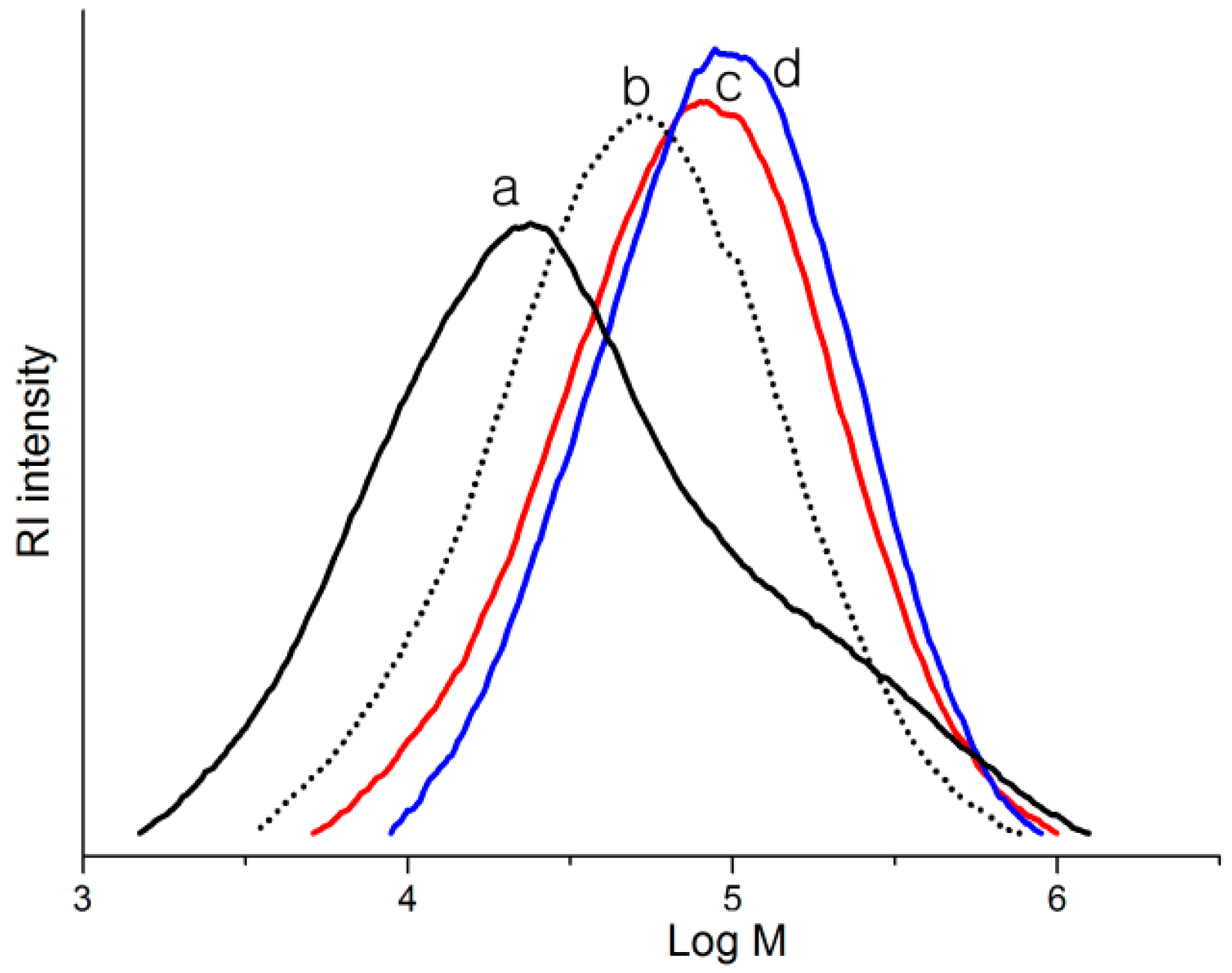
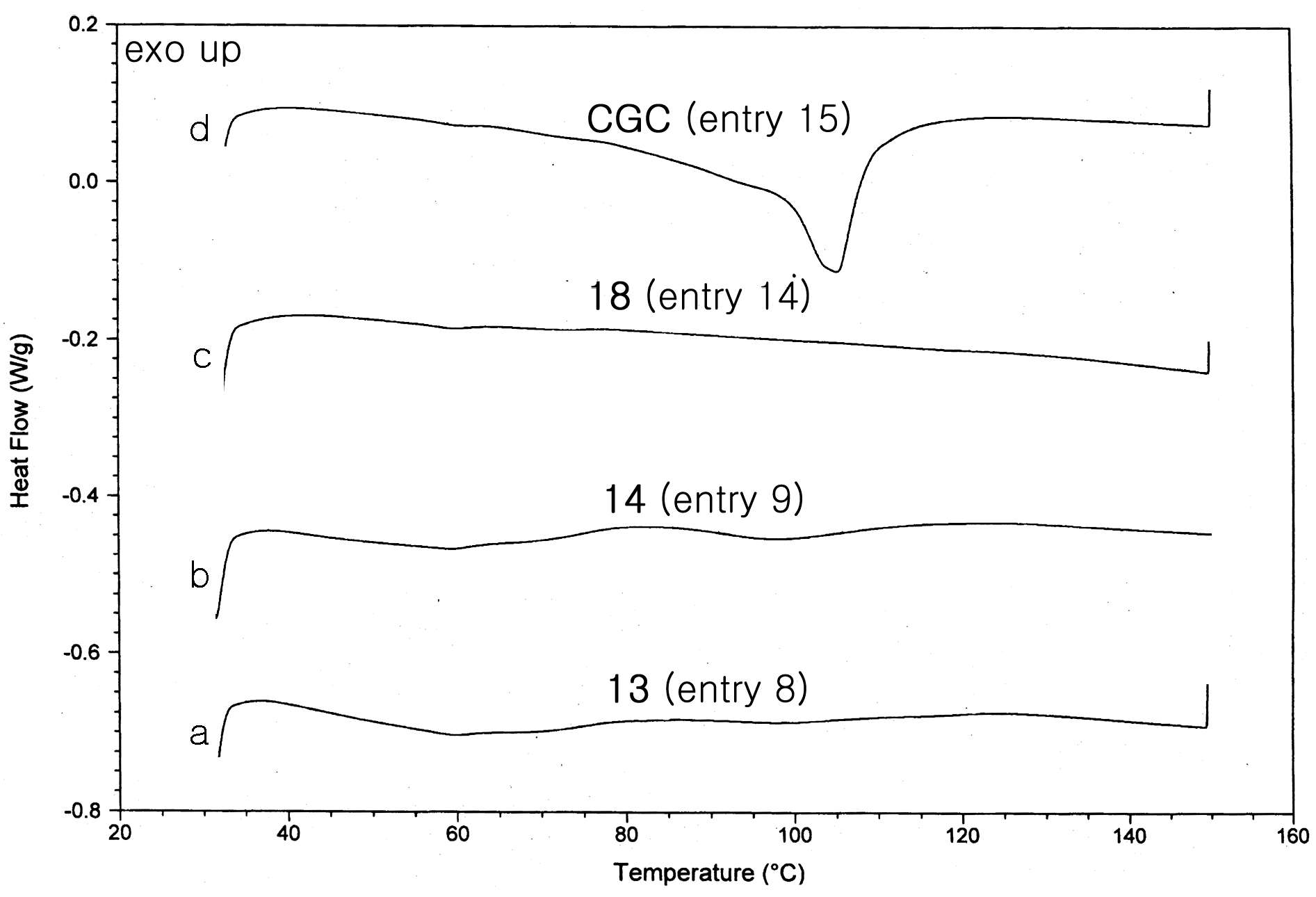
2.2. Polymerization Studies
3. Experimental Section
3.1 General Remarks
3.2. Compound 3
3.3. Compound 4
3.4. Compound 5
3.5. Compound 6
3.6. Compound 7
3.7. Compound 8
3.8. Compound 9
3.9. Compound 10
3.10. Compound 11
3.11. Compound 12
3.12. Compound 13
3.13. Compound 14
3.14. Compound 15
3.15. Compound 16
3.16. Compound 17
3.17. Compound 18
3.18. Ethylene/1-Octene Copolymerization
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Kaminsky, W. Discovery of methylaluminoxane as cocatalyst for olefin polymerization. Macromolecules 2012, 45, 3289–3297. [Google Scholar] [CrossRef]
- Stürzel, M.; Mihan, S.; Mülhaupt, R. From multisite polymerization catalysis to sustainable materials and all-polyolefin composites. Chem. Rev. 2016, 116, 1398–1433. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Liu, W.; Wang, W.-J.; Li, B.-G.; Zhu, S. A Comprehensive review on controlled synthesis of long-chain branched polyolefins: Part 1, Single catalyst systems. Macromol. React. Eng. 2016, 10, 156–179. [Google Scholar] [CrossRef]
- Tsurugi, H.; Yamamoto, K.; Rochat, R.; Mashima, K. Non-bridged half-metallocene complexes of group 4–6 metals with chelating ligands as well-defined catalysts for α-olefin polymerization. Polym. J. 2015, 47, 2–17. [Google Scholar] [CrossRef]
- Shiono, T. Living polymerization of olefins with ansa-dimethylsilylene(fluorenyl)(amido)dimethyltitanium-based catalysts. Polym. J. 2011, 43, 331–351. [Google Scholar] [CrossRef]
- Kim, T.-J.; Kim, S.-K.; Kim, B.-J.; Hahn, J.S.; Ok, M.-A.; Song, J.H.; Shin, D.-H.; Ko, J.; Cheong, M.; Kim, J.; et al. Half-metallocene titanium(IV) phenyl phenoxide for high temperature olefin polymerization: Ortho-substituent effect at ancillary o-phenoxy ligand for enhanced catalytic performance. Macromolecules 2009, 42, 6932–6943. [Google Scholar] [CrossRef]
- Stephan, D.W. The road to early-transition-metal phosphinimide olefin polymerization catalysts. Organometallics 2005, 24, 2548–2560. [Google Scholar] [CrossRef]
- Senda, T.; Hanaoka, H.; Hino, T.; Oda, Y.; Tsurugi, H.; Mashima, K. Substituent effects on silicon of bridged tetramethylcyclopentadienyl-phenoxy titanium complexes for controlling the regiochemistry and molecular weight in 1-olefin polymerization. Macromolecules 2009, 42, 8006–8009. [Google Scholar] [CrossRef]
- Itagaki, K.; Hasumi, S.; Fujiki, M.; Nomura, K. Ethylene polymerization and ethylene/1-octene copolymerization using group 4 half-metallocenes containing aryloxo ligands, Cp*MCl2(OAr) [M = Ti, Zr, Hf; Ar = O-2,6-R2C6H3, R = tBu, Ph]—MAO catalyst systems. J. Mol. Catal. A Chem. 2009, 303, 102–109. [Google Scholar] [CrossRef]
- Nomura, K. Half-titanocenes containing anionic ancillary donor ligands as promising new catalysts for precise olefin polymerisation. Dalton Trans. 2009, 8811–8823. [Google Scholar] [CrossRef] [PubMed]
- Lamberti, M.; Mazzeo, M.; Pappalardo, D.; Pellecchia, C. Mechanism of stereospecific polymerization of α-olefins by late-transition metal and octahedral group 4 metal catalysts. Coord. Chem. Rev. 2009, 253, 2082–2097. [Google Scholar] [CrossRef]
- Makio, H.; Terao, H.; Iwashita, A.; Fujita, T. FI catalysts for olefin polymerization—A comprehensive treatment. Chem. Rev. 2011, 111, 2363–2449. [Google Scholar] [CrossRef] [PubMed]
- Boussie, T.R.; Diamond, G.M.; Goh, C.; Hall, K.A.; LaPointe, A.M.; Leclerc, M.; Lund, C.; Murphy, V.; Shoemaker, J.A.W.; Tracht, U.; et al. A fully integrated high-throughput screening methodology for the discovery of new polyolefin catalysts: Discovery of a new class of high temperature single-site group (IV) copolymerization catalysts. J. Am. Chem. Soc. 2003, 125, 4306–4317. [Google Scholar] [CrossRef] [PubMed]
- Jun, S.H.; Park, J.H.; Lee, C.S.; Park, S.Y.; Go, M.J.; Lee, J.; Lee, B.Y. Preparation of phosphine-amido hafnium and zirconium complexes for olefin polymerization. Organometallics 2013, 32, 7357–7365. [Google Scholar] [CrossRef]
- Hwang, E.Y.; Park, G.H.; Lee, C.S.; Kang, Y.Y.; Lee, J.; Lee, B.Y. Preparation of octahydro- and tetrahydro-[1,10]phenanthroline zirconium and hafnium complexes for olefin polymerization. Dalton Trans. 2015, 44, 3845–3855. [Google Scholar] [CrossRef] [PubMed]
- Pérez, M.A.; Quijada, R.; Ortega-Jiménez, F.; Alvarez-Toledano, C. Cyclopalladated complexes derivates of phenylhydrazones and their use as catalysts in ethylene polymerization. J. Mol. Catal. A Chem. 2005, 226, 291–295. [Google Scholar] [CrossRef]
- Delferro, M.; Marks, T.J. Multinuclear olefin polymerization catalysts. Chem. Rev. 2011, 111, 2450–2485. [Google Scholar] [CrossRef] [PubMed]
- Cano, J.; Kunz, K. How to synthesize a constrained geometry catalyst (CGC)—A survey. J. Organomet. Chem. 2007, 692, 4411–4423. [Google Scholar] [CrossRef]
- Klosin, J.; Fontaine, P.P.; Figureueroa, R. Development of group IV molecular catalysts for high temperature ethylene-α-olefin copolymerization reactions. Acc. Chem. Res. 2015, 48, 2004–2016. [Google Scholar] [CrossRef] [PubMed]
- Chai, J.; Abboud, K.A.; Miller, S.A. Sterically expanded CGC catalysts: Substituent effects on ethylene and α-olefin polymerization. Dalton Trans. 2013, 42, 9139–9147. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.J.; Nayab, S.; Woo, H.Y.; Hahn, J.S.; Lee, H.S.; Kang, S.O.; Lee, H. Synthesis and X-ray crystal structure of [Me2Si(C5Me2H2)(tBuN)]MCl2 (M = Ti, Zr) bearing a 3,4-dimethylcyclopentadienyl ring: Investigation of the substitution effect on the cyclopentadienyl (Cp) ring for catalytic performance in ethylene/1-octene (co)polymerization. Polyhedron 2014, 67, 199–204. [Google Scholar]
- Hong, M.; Cui, L.; Liu, S.; Li, Y. Synthesis of novel cyclic olefin copolymer (COC) with high performance via effective copolymerization of ethylene with bulky cyclic olefin. Macromolecules 2012, 45, 5397–5402. [Google Scholar] [CrossRef]
- Cho, D.J.; Wu, C.J.; Sujith, S.; Han, W.S.; Kang, S.O.; Lee, B.Y. o-Phenylene-bridged Cp/amido titanium complexes for ethylene/1-hexene copolymerizations. Organometallics 2006, 25, 2133–2134. [Google Scholar] [CrossRef]
- Lee, S.H.; Wu, C.J.; Joung, U.G.; Lee, B.Y.; Park, J. Bimetallic phenylene-bridged Cp/amide titanium complexes and their olefin polymerization. Dalton Trans. 2007, 4608–4614. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.J.; Lee, S.H.; Yun, H.; Lee, B.Y. Ortho lithiation of tetrahydroquinoline derivatives and its use for the facile construction of polymerization catalysts. Organometallics 2007, 26, 6685–6687. [Google Scholar] [CrossRef]
- Wu, C.J.; Lee, S.H.; Yu, S.T.; Na, S.J.; Yun, H.; Lee, B.Y. CO2-Mediated ortho-lithiation of N-alkylanilines and its use for the construction of polymerization catalysts. Organometallics 2008, 27, 3907–3917. [Google Scholar] [CrossRef]
- Park, J.H.; Do, S.H.; Cyriac, A.; Yun, H.; Lee, B.Y. Preparation of half-metallocenes of thiophene-fused and tetrahydroquinoline-linked cyclopentadienyl ligands for ethylene/α-olefin copolymerization. Dalton Trans. 2010, 39, 9994–10002. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.T.; Na, S.J.; Lim, T.S.; Lee, B.Y. Preparation of a bulky cycloolefin/ethylene copolymer and its tensile properties. Macromolecules 2010, 43, 725–730. [Google Scholar] [CrossRef]
- Lee, C.H.; Lee, E.J.; Jung, S.W.; Lee, J.A.; Lee, B.R.; Lee, B.Y. Method for Preparing Transition Metal Complexes, Transition Metal Complexes Prepared Using the Method, Catalyst Composition Containing the Complexes. U.S. Patent 7932207B2, 26 April 2011. [Google Scholar]
- Tremblay, J.-F. Dow chemical loses elastomers patent lawsuit against LG Chem. Chem. Eng. News 2012, 90, 7. [Google Scholar]
- Lin, F.; Wang, X.; Pan, Y.; Wang, M.; Liu, B.; Luo, Y.; Cui, D. Nature of the entire range of rare earth metal-based cationic catalysts for highly active and syndioselective styrene polymerization. ACS Catal. 2016, 6, 176–185. [Google Scholar] [CrossRef]
- Ryabov, A.N.; Voskoboynikov, A.Z. Constrained geometry complexes of titanium (IV) and zirconium (IV) involving cyclopentadienyl fused to thiophene ring. J. Organomet. Chem. 2005, 690, 4213–4221. [Google Scholar] [CrossRef]
- Cai, Z.; Ohmagari, M.; Nakayama, Y.; Shiono, T. Highly active syndiospecific living polymerization of higher 1-alkene with ansa-fluorenylamidodimethyltitanium complex. Macromol. Rapid Commun. 2009, 30, 1812–1816. [Google Scholar] [CrossRef] [PubMed]
- Schwerdtfeger, E.D.; Irwin, L.J.; Miller, S.A. Highly branched polyethylene from ethylene alone via a single zirconium-based catalyst. Macromolecules 2008, 41, 1080–1085. [Google Scholar] [CrossRef]
- Ramos, C.; Royo, P.; Lanfranchi, M.; Pellinghelli, M.A.; Tiripicchio, A. [(Allylsilyl)(η-amidosilyl)-η5-cyclopentadienyl] Group 4 Metal Complexes: Synthesis, Reactivity, and Olefin Polymerization. Organometallics 2007, 26, 445–454. [Google Scholar] [CrossRef]
- Klosin, J.; Kruper, W.J.; Nickias, P.N.; Roof, G.R.; De Waele, P.; Abboud, K.A. Heteroatom-substituted constrained-geometry complexes. Dramatic substituent effect on catalyst efficiency and polymer molecular weight. Organometallics 2001, 20, 2663–2665. [Google Scholar] [CrossRef]
- Van Leusen, D.; Beetstra, D.J.; Hessen, B.; Teuben, J.H. Ethylene-bridged tetramethylcyclopentadienylamide titanium complexes: Ligand synthesis and olefin polymerization properties. Organometallics 2000, 19, 4084–4089. [Google Scholar] [CrossRef]
- Altenhoff, G.; Bredeau, S.; Erker, G.; Kehr, G.; Kataeva, O.; Fröhlich, R. Structural features of Me2Si-bridged Cp/phosphido group 4 metal complexes, “CpSiP” constrained-geometry Ziegler−Natta catalyst precursors. Organometallics 2002, 21, 4084–4089. [Google Scholar] [CrossRef]
- Dureen, M.A.; Brown, C.C.; Morton, J.G.M.; Stephan, D.W. Titanium “constrained geometry” complexes with pendant arene groups. Dalton Trans. 2011, 40, 2861–2867. [Google Scholar] [CrossRef] [PubMed]
- Piola, L.; Nahra, F.; Nolan, S.P. Olefin metathesis in air. Beil. J. Org. Chem. 2015, 11, 2038–2056. [Google Scholar] [CrossRef] [PubMed]
- Schrock, R.R.; Murdzek, J.S.; Bazan, G.C.; Robbins, J.; DiMare, M.; O’Regan, M. Synthesis of molybdenum imido alkylidene complexes and some reactions involving acyclic olefins. J. Am. Chem. Soc. 1990, 112, 3875–3886. [Google Scholar] [CrossRef]
- Wang, C.; Yu, M.; Kyle, A.F.; Jakubec, P.; Dixon, D.J.; Schrock, R.R.; Hoveyda, A.H. Efficient and selective formation of macrocyclic disubstituted Z alkenes by ring-closing metathesis (RCM) reactions catalyzed by Mo- or W-based monoaryloxide pyrrolide (MAP) complexes: Applications to total syntheses of epilachnene, yuzu lactone, ambrettolide, epothilone C, and nakadomarin A. Chem. Eur. J. 2013, 19, 2726–2740. [Google Scholar] [PubMed]
- Ogasawara, M.; Arae, S.; Watanabe, S.; Nakajima, K.; Takahashi, T. Kinetic resolution of planar-chiral ferrocenylphosphine derivatives by molybdenum-catalyzed asymmetric ring-closing metathesis and their application in asymmetric catalysis. ACS Catal. 2016, 6, 1308–1315. [Google Scholar] [CrossRef]
- Ogasawara, M.; Arae, S.; Watanabe, S.; Nakajima, K.; Takahashi, T. Kinetic resolution of planar-chiral 1,2-disubstituted ferrocenes by molybdenum-catalyzed asymmetric intraannular ring-closing metathesis. Chem. Eur. J. 2013, 19, 4151–4154. [Google Scholar] [CrossRef] [PubMed]
- Grandini, C.; Camurati, I.; Guidotti, S.; Mascellani, N.; Resconi, L.; Nifant’ev, I.E.; Kashulin, I.A.; Ivchenko, P.V.; Mercandelli, P.; Sironi, A. Heterocycle-fused indenyl silyl amido dimethyl titanium complexes as catalysts for high molecular weight syndiotactic amorphous polypropylene. Organometallics 2004, 23, 344–360. [Google Scholar] [CrossRef]
- Kim, S.; Park, J.; Song, B.; Yoon, S.-W.; Go, M.; Lee, J.; Lee, B. Preparation of thiophene-fused and tetrahydroquinoline-linked cyclopentadienyl titanium complexes for ethylene/α-olefin copolymerization. Catalysts 2013, 3, 104–124. [Google Scholar] [CrossRef]
- Wang, W.-J.; Kolodka, E.; Zhu, S.; Hamielec, A.E.; Kostanski, L.K. Temperature rising elution fractionation and characterization of ethylene/octene-1 copolymers synthesized with constrained geometry catalyst. Macromol. Chem. Phys. 1999, 200, 2146–2151. [Google Scholar] [CrossRef]
- Theurkauff, G.; Bondon, A.; Dorcet, V.; Carpentier, J.-F.; Kirillov, E. Heterobi- and -trimetallic Ion Pairs of Zirconocene-Based Isoselective Olefin Polymerization Catalysts with AlMe3. Angew. Chem. Int. Ed. 2015, 54, 6343–6346. [Google Scholar] [CrossRef] [PubMed]
- Song, F.; Cannon, R.D.; Bochmann, M. Zirconocene-catalyzed propene polymerization: A quenched-flow kinetic study. J. Am. Chem. Soc. 2003, 125, 7641–7653. [Google Scholar] [CrossRef] [PubMed]
- Shreve, A.P.; Mulhaupt, R.; Fultz, W.; Calabrese, J.; Robbins, W.; Ittel, S.D. Sterically hindered aryloxide-substituted alkylaluminum compounds. Organometallics 1988, 7, 409–416. [Google Scholar] [CrossRef]
- Busico, V.; Cipullo, R.; Cutillo, F.; Friederichs, N.; Ronca, S.; Wang, B. Improving the performance of methylalumoxane: A facile and efficient method to trap “free” trimethylaluminum. J. Am. Chem. Soc. 2003, 125, 12402–12403. [Google Scholar] [CrossRef] [PubMed]
- Mehdiabadi, S.; Soares, J.B.P.; Bilbao, D.; Brinen, J. Ethylene polymerization and ethylene/1-octene copolymerization with rac-dimethylsilylbis(indenyl)hafnium dimethyl using trioctyl aluminum and borate: A polymerization kinetics investigation. Macromolecules 2013, 46, 1312–1324. [Google Scholar] [CrossRef]
- Wang, Q.; Li, L.; Fan, Z. Effects of tetraalkylaluminoxane activators on ethylene polymerization catalyzed by iron-based complexes. Eur. Polym. J. 2005, 41, 1170–1176. [Google Scholar] [CrossRef]
- Wang, Q.; Li, L.; Fan, Z. Effect of alkylaluminum on ethylene polymerization catalyzed by 2,6-bis(imino)pyridyl complexes of Fe(II). J. Polym. Sci. Part A Polym. Chem. 2005, 43, 1599–1606. [Google Scholar] [CrossRef]
- Dong, J.J.; Harvey, E.C.; Fañanás-Mastral, M.; Browne, W.R.; Feringa, B.L. Palladium-catalyzed anti-markovnikov oxidation of allylic amides to protected β-amino aldehydes. J. Am. Chem. Soc. 2014, 136, 17302–17307. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, A.J.; Bax, M.N.; Willis, M.C. Palladium-catalysed N-annulation routes to indoles: The synthesis of indoles with sterically demanding N-substituents, including demethylasterriquinone A1. Chem. Commun. 2007, 45, 4764–4766. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Sample of the compound 14 is available from the authors.


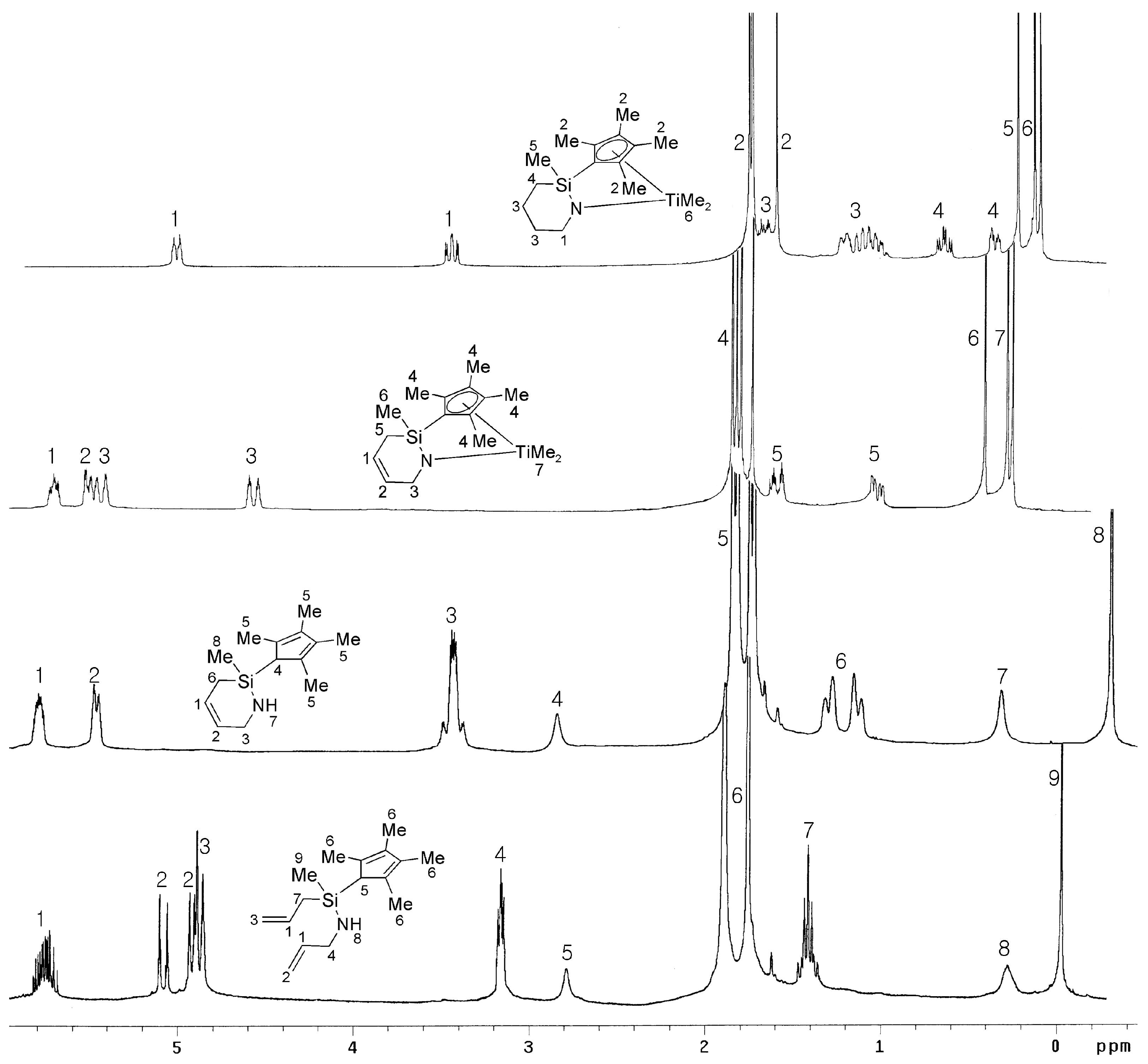
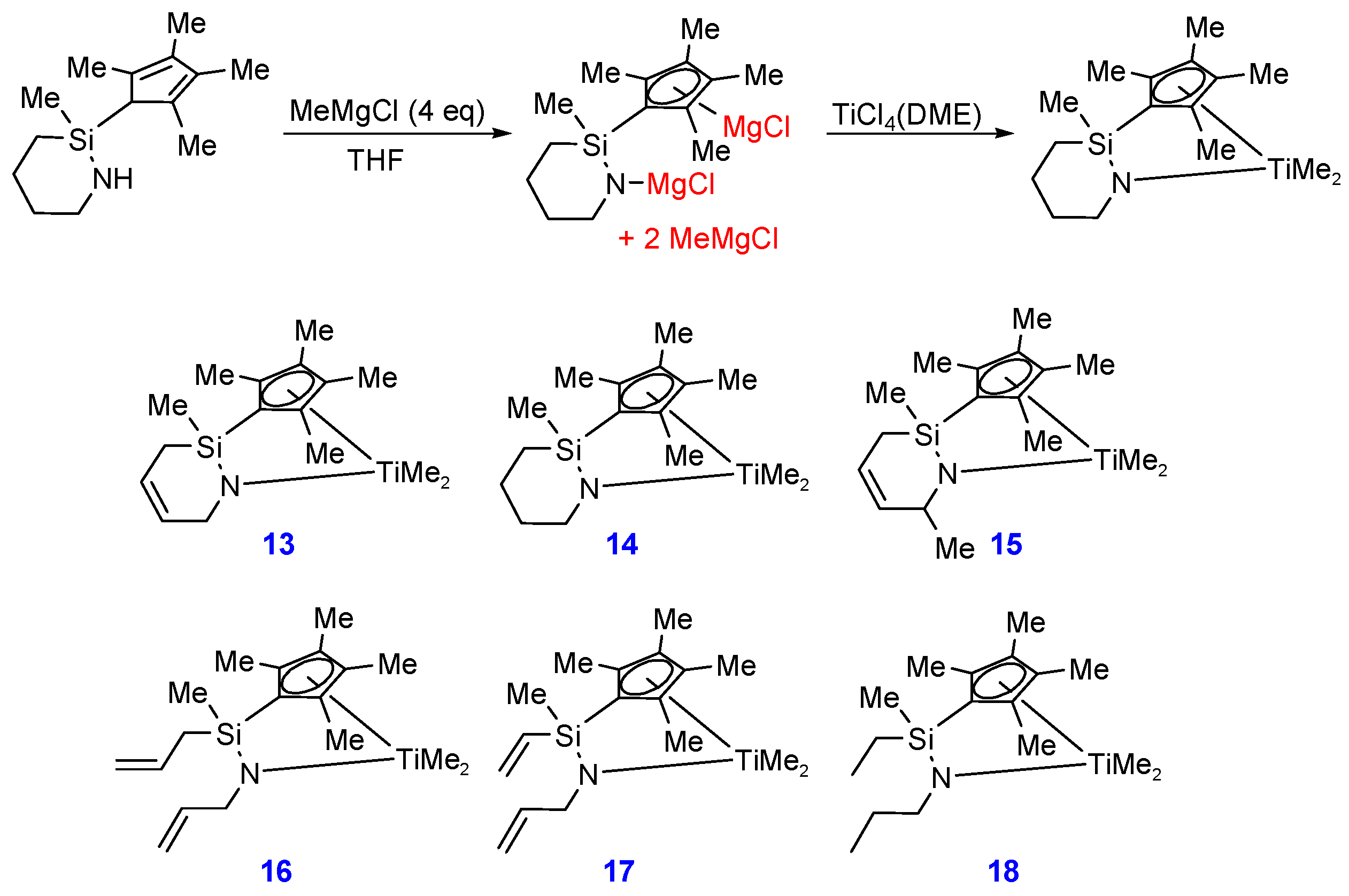

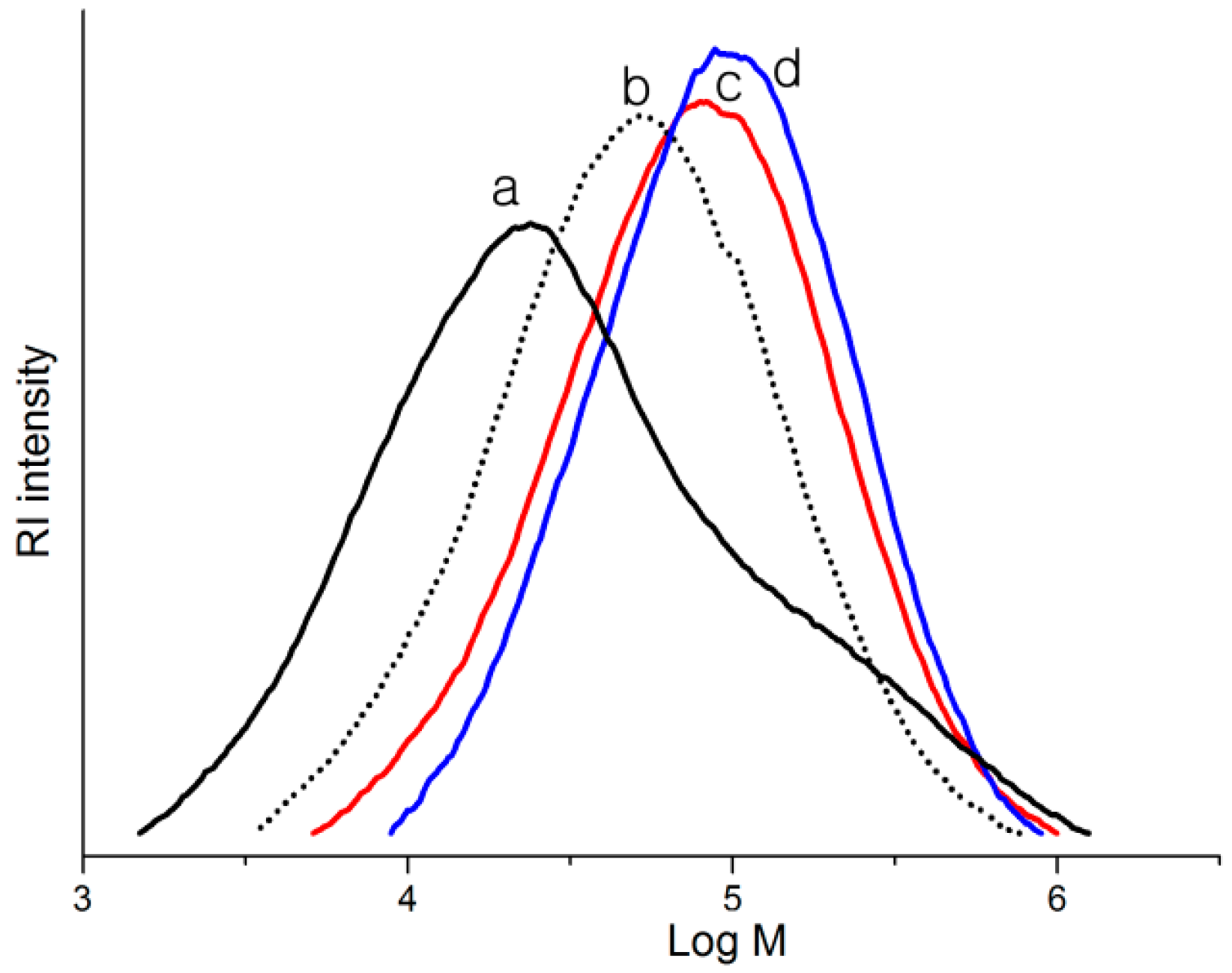
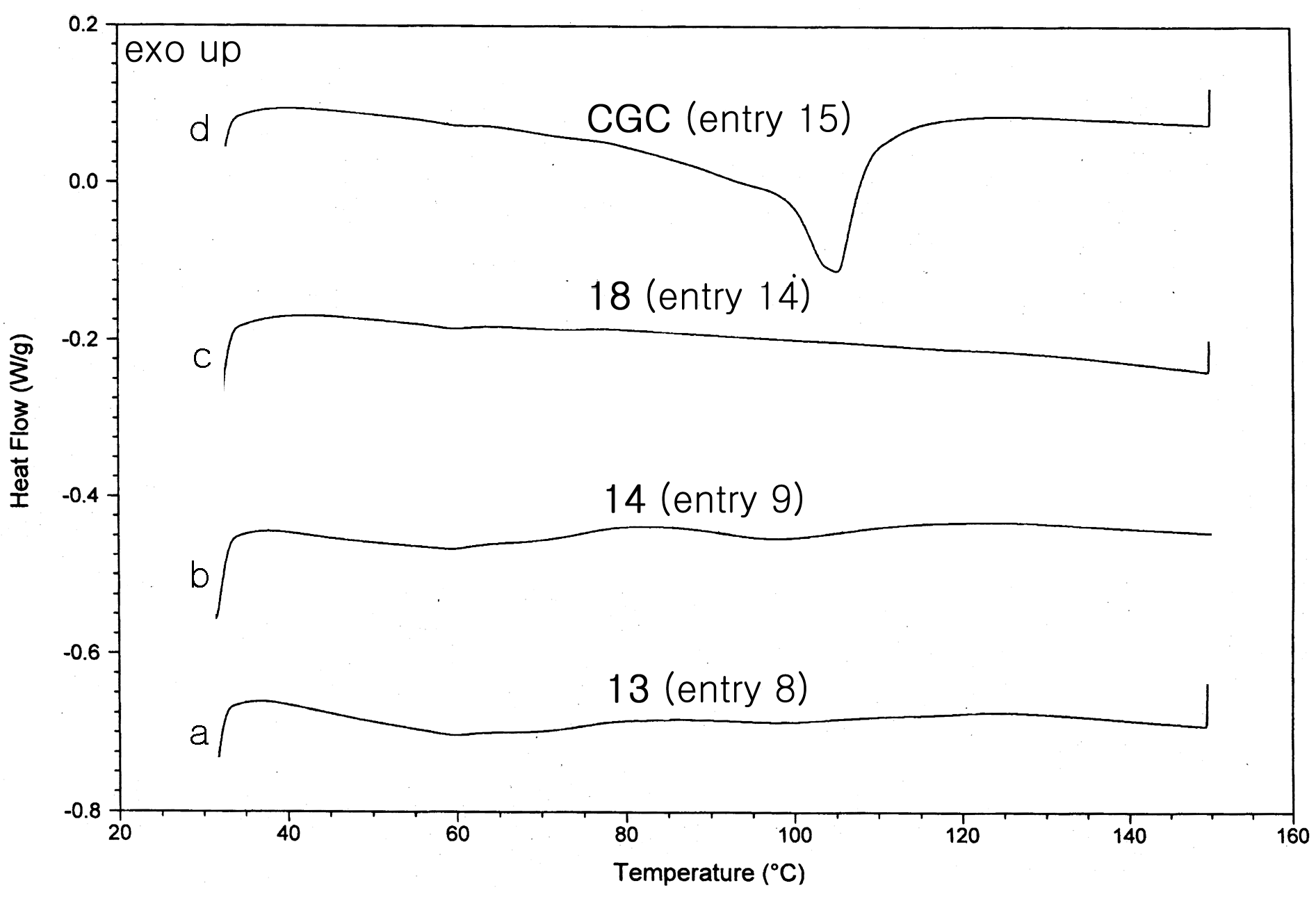

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Cat | Scavenger | Temperature (°C) | Yield (g) | Activity b | [Oct] c (mol %) | Mn (kDa) | Mw/Mn |
|---|---|---|---|---|---|---|---|---|
| 1 | 13 | iBu3Al | 129-139-134 | 0.91 | 1.82 | 12 | 69.4 | 2.02 |
| 2 | 14 | iBu3Al | 130-142-135 | 0.89 | 1.78 | 14 | 50.4 | 2.14 |
| 3 | 15 | iBu3Al | 129-147 (50 sec)-137 | 1.54 | 3.08 | 11 | 43.0 | 2.17 |
| 4 | 16 | iBu3Al | 129-132-129 | 0.42 | 0.84 | 14 | 108 | 2.18 |
| 5 | 17 | iBu3Al | 129-136-135 | 0.61 | 1.22 | 15 | 105 | 2.30 |
| 6 | 18 | iBu3Al | 129-144 (40 sec)-137 | 1.19 | 2.38 | 19 | 72.4 | 1.97 |
| 7 | CGC | iBu3Al | 120-143 (25 sec)-125 | 2.48 | 4.96 | 21 | 17.0 | 4.41 |
| 8 | 13 | [iBu2Al]2O | 129-148-142 | 1.53 | 3.06 | 14 | 44.8 | 2.17 |
| 9 | 14 | [iBu2Al]2O | 128-153 (40 sec)-146 | 2.36 | 4.72 | 15 | 35.6 | 2.27 |
| 10 | 14 | [iBu2Al]2O | 80-112 (60 sec)-113 | 3.28 | 6.55 | 15 | 203 | 2.16 |
| 11 | 15 | [iBu2Al]2O | 129-154-150 | 2.75 | 5.50 | 13 | 26.1 | 2.22 |
| 12 | 16 | [iBu2Al]2O | 128-136-133 | 0.90 | 1.80 | 14 | 107 | 2.31 |
| 13 | 17 | [iBu2Al]2O | 129-137-133 | 1.01 | 2.02 | 16 | 105 | 2.17 |
| 14 | 18 | [iBu2Al]2O | 128-147 (50 sec)-138 | 1.73 | 3.46 | 20 | 68.8 | 1.90 |
| 15 | CGC | [iBu2Al]2O | 120-154 (50 sec)-134 | 3.58 | 7.15 | 21 | 9.3 | 6.87 |
| 16 | CGC | [iBu2Al]2O | 80-121 (35 sec)-108 | 4.20 | 8.40 | 36 | 33.7 | 1.91 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, S.; Park, S.S.; Kim, J.G.; Kim, C.S.; Lee, B.Y. Preparation of “Constrained Geometry” Titanium Complexes of [1,2]Azasilinane Framework for Ethylene/1-Octene Copolymerization. Molecules 2017, 22, 258. https://doi.org/10.3390/molecules22020258
Lee S, Park SS, Kim JG, Kim CS, Lee BY. Preparation of “Constrained Geometry” Titanium Complexes of [1,2]Azasilinane Framework for Ethylene/1-Octene Copolymerization. Molecules. 2017; 22(2):258. https://doi.org/10.3390/molecules22020258
Chicago/Turabian StyleLee, Seul, Seung Soo Park, Jin Gu Kim, Chung Sol Kim, and Bun Yeoul Lee. 2017. "Preparation of “Constrained Geometry” Titanium Complexes of [1,2]Azasilinane Framework for Ethylene/1-Octene Copolymerization" Molecules 22, no. 2: 258. https://doi.org/10.3390/molecules22020258
APA StyleLee, S., Park, S. S., Kim, J. G., Kim, C. S., & Lee, B. Y. (2017). Preparation of “Constrained Geometry” Titanium Complexes of [1,2]Azasilinane Framework for Ethylene/1-Octene Copolymerization. Molecules, 22(2), 258. https://doi.org/10.3390/molecules22020258