
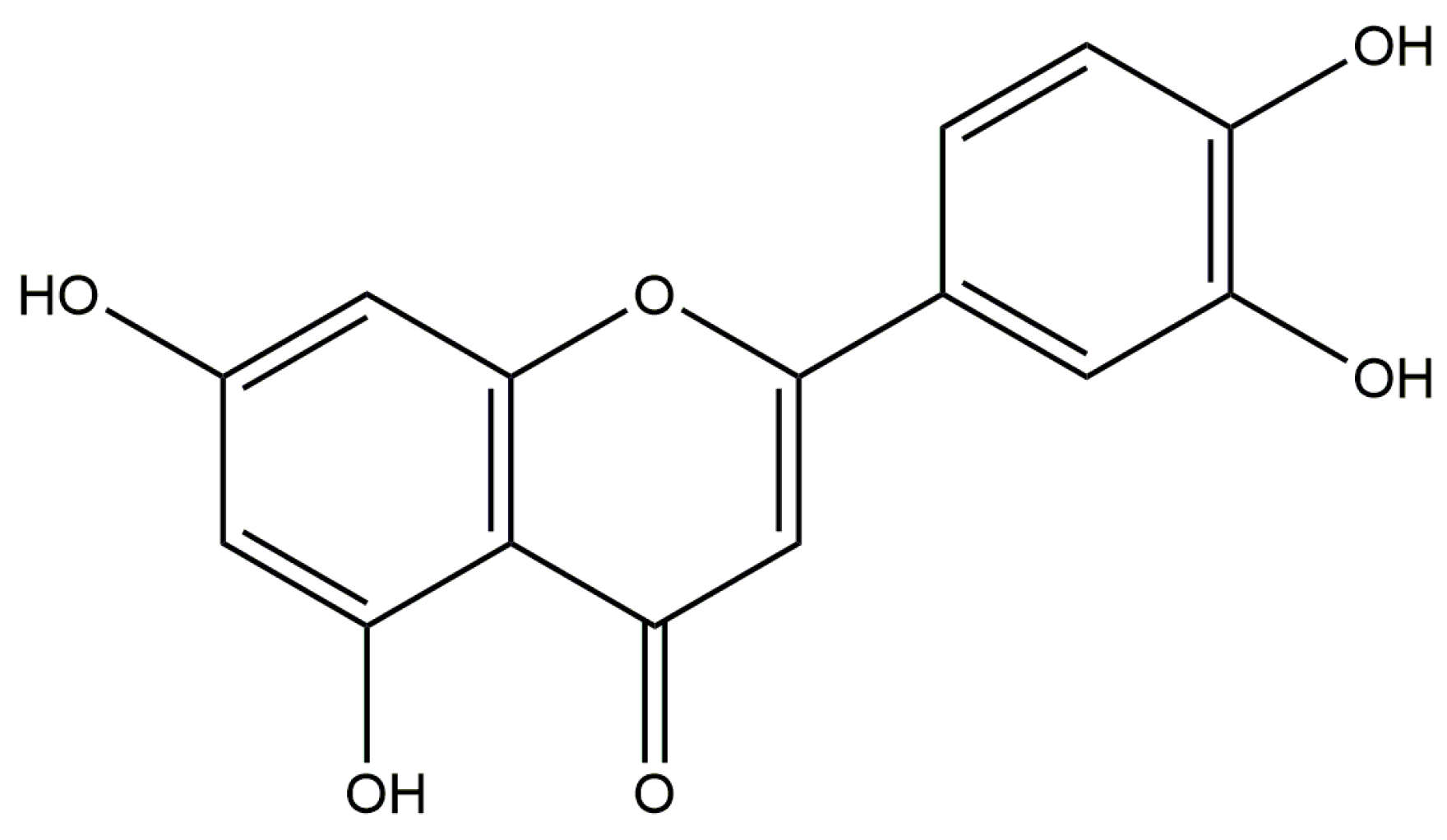
Luteolin Inhibits Fibrillary β-Amyloid1–40-Induced Inflammation in a Human Blood-Brain Barrier Model by Suppressing the p38 MAPK-Mediated NF-κB Signaling Pathways

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
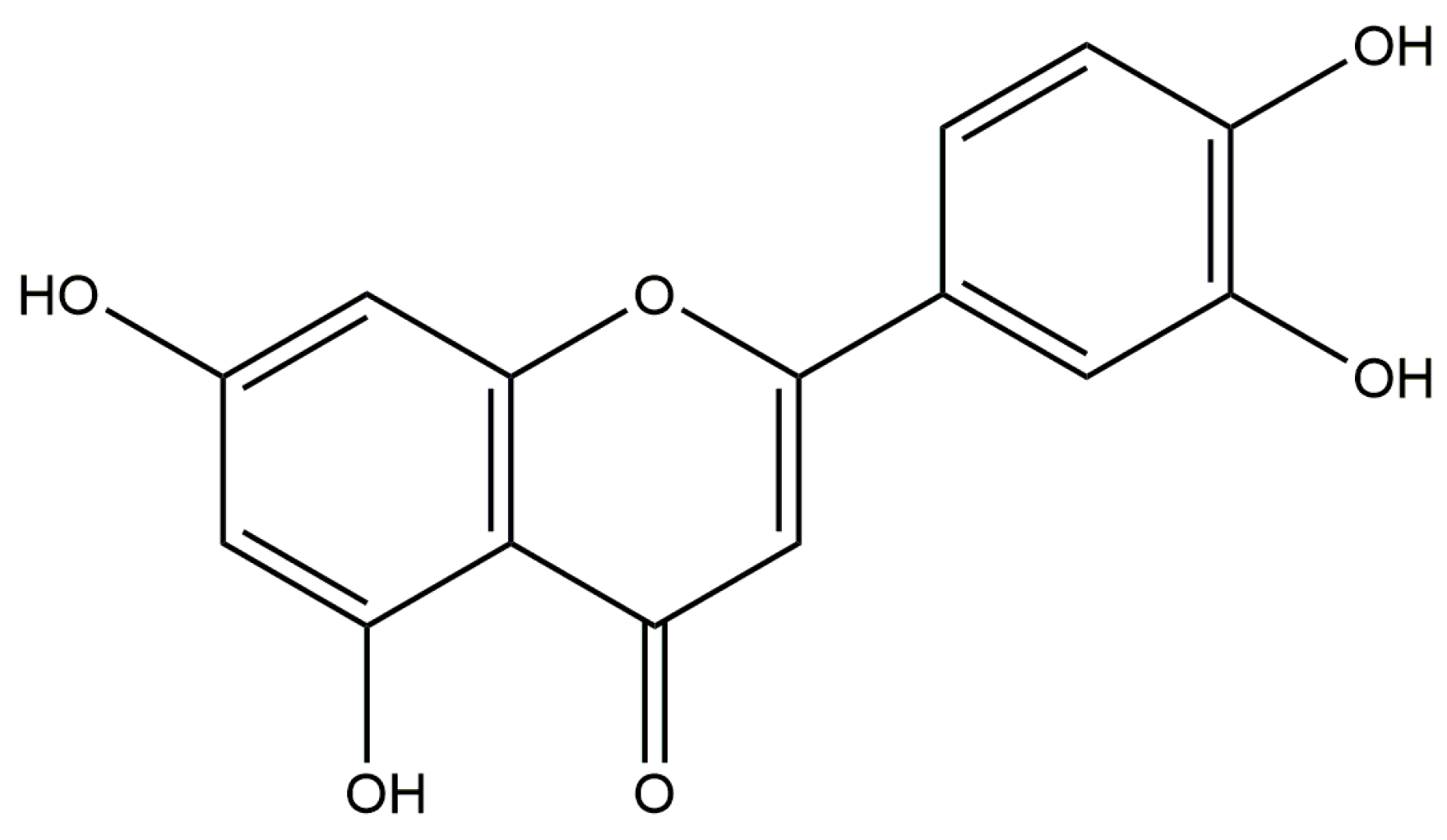
:1. Introduction
2. Results
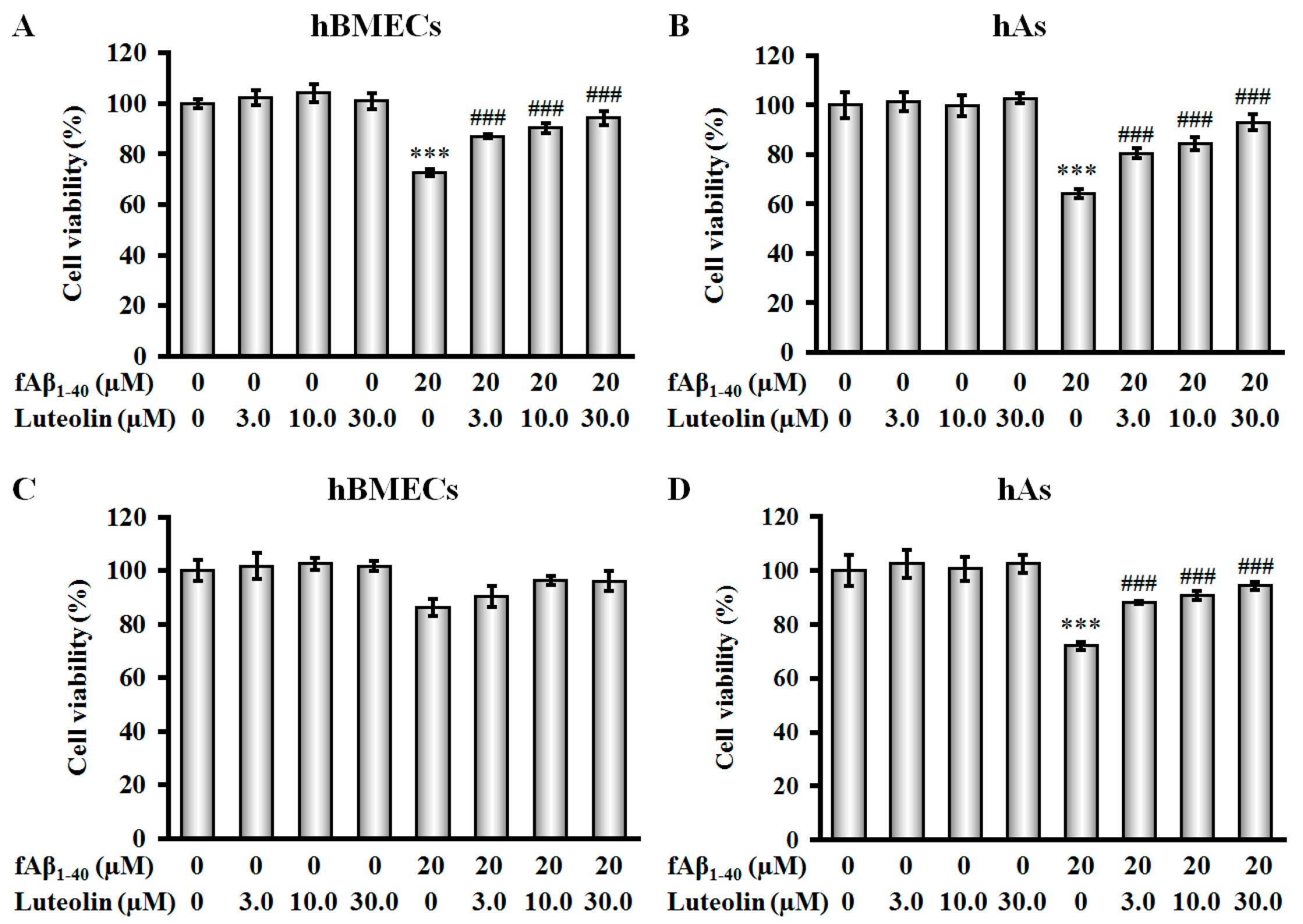
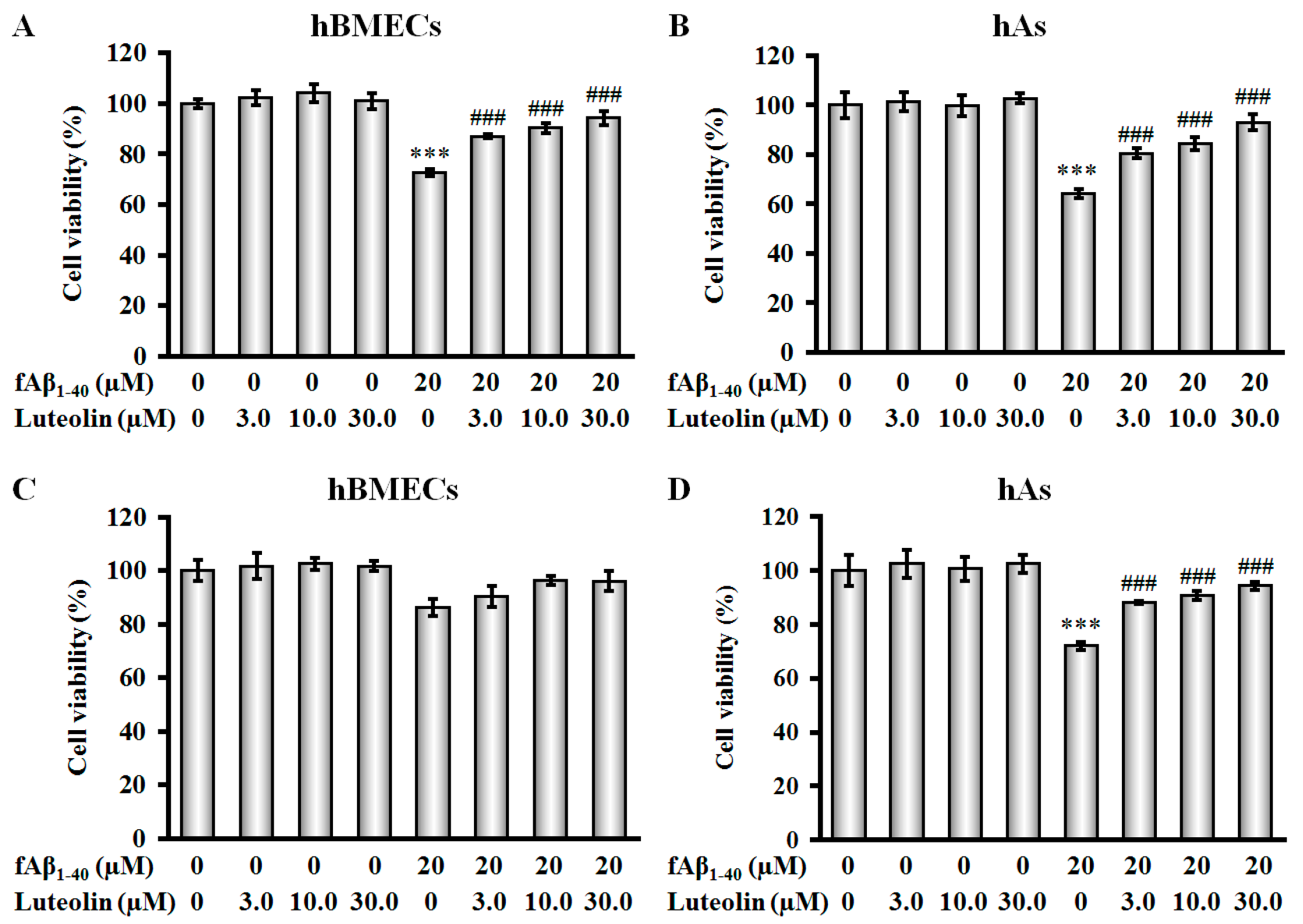
2.1. Luteolin Protects hBMECs, hAs, and Co-Culture against fAβ1–40-Induced Toxicity
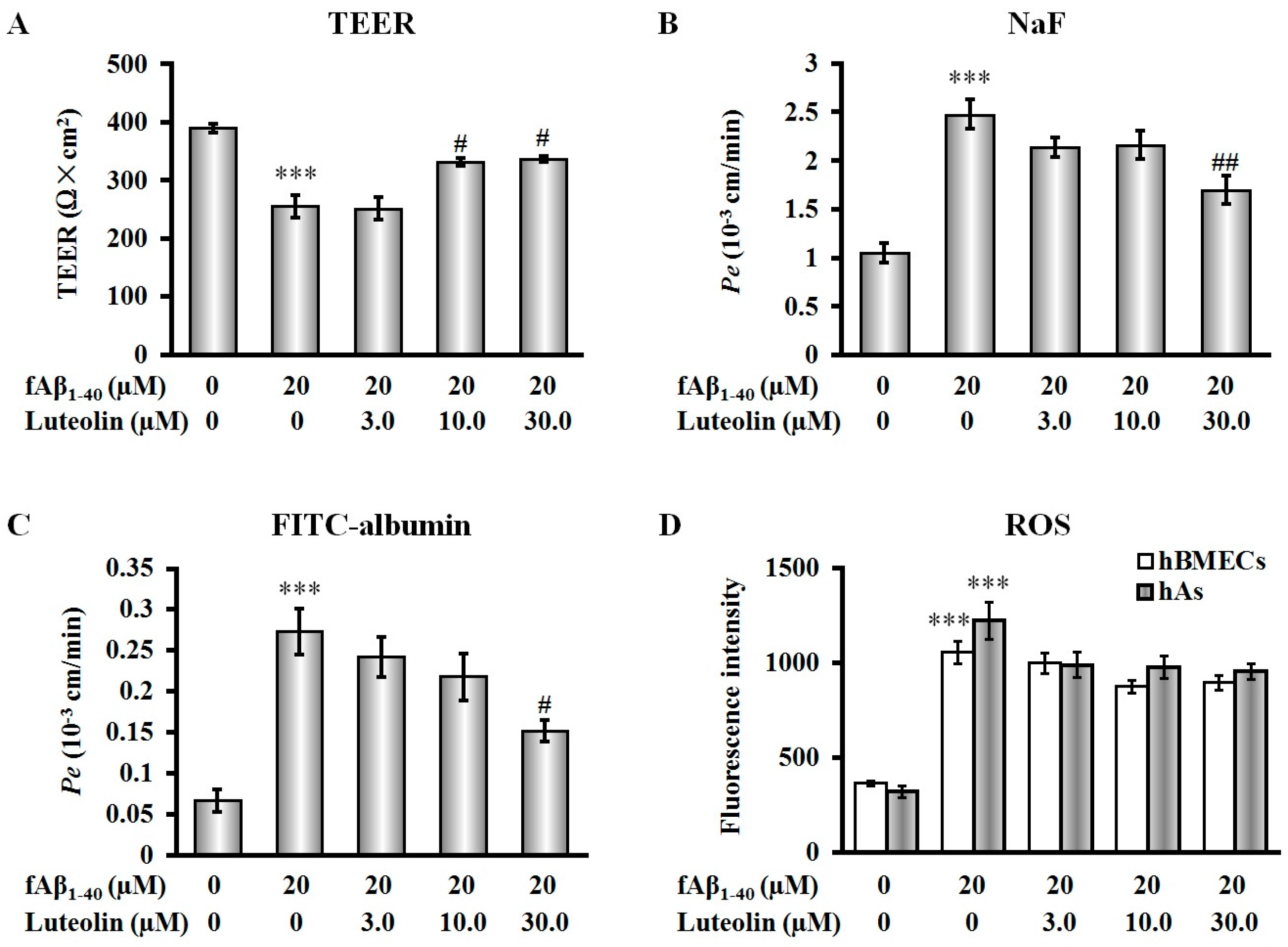
2.2. Luteolin Improves BBB Function But Does Not Scavenge Intracellular Reactive Oxygen Species against fAβ1–40-Induced Toxicity
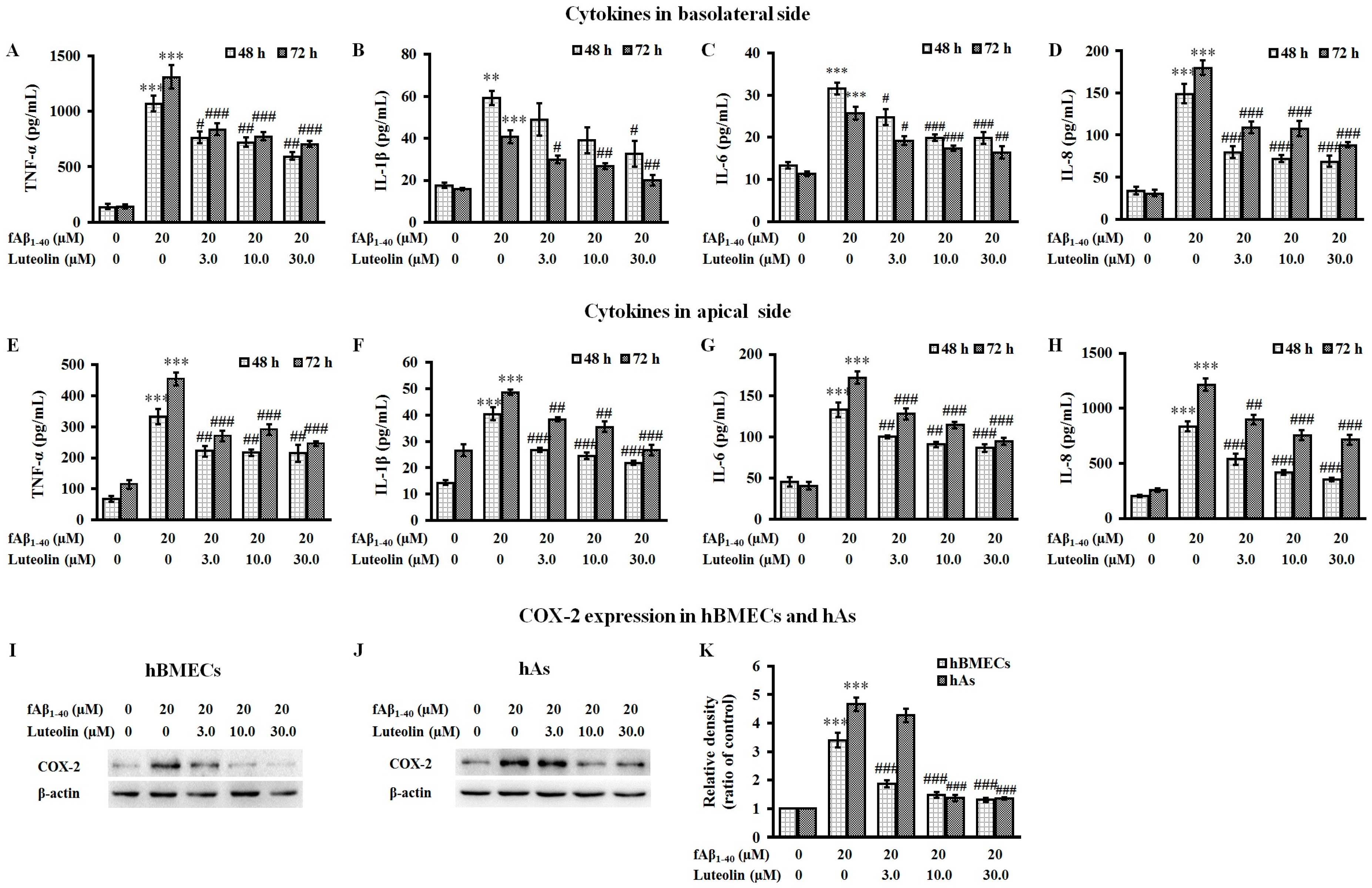
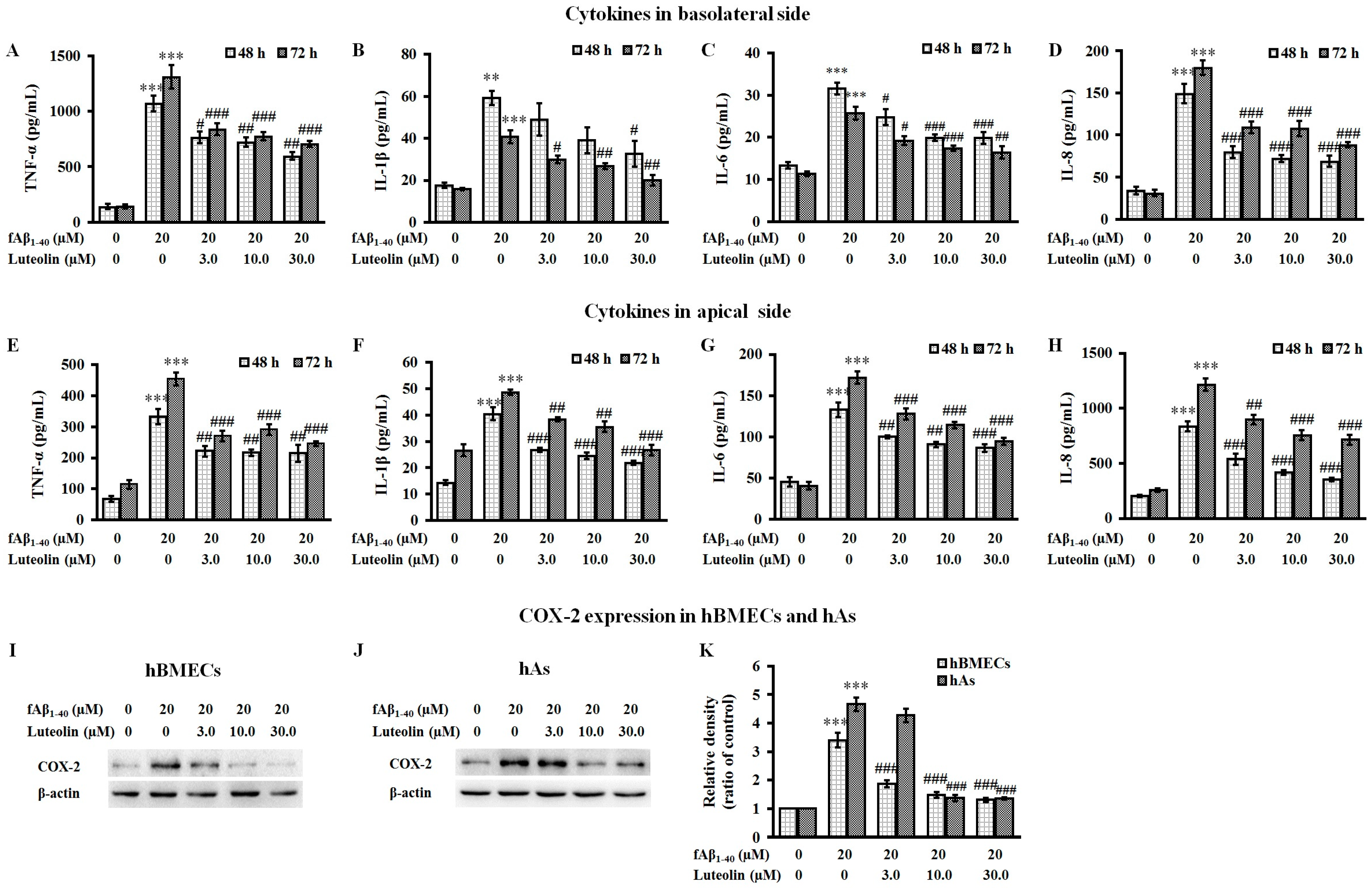
2.3. Luteolin Inhibits the Release of Inflammatory Cytokines and the Expression of COX-2 Against fAβ1–40-Induced Toxicity
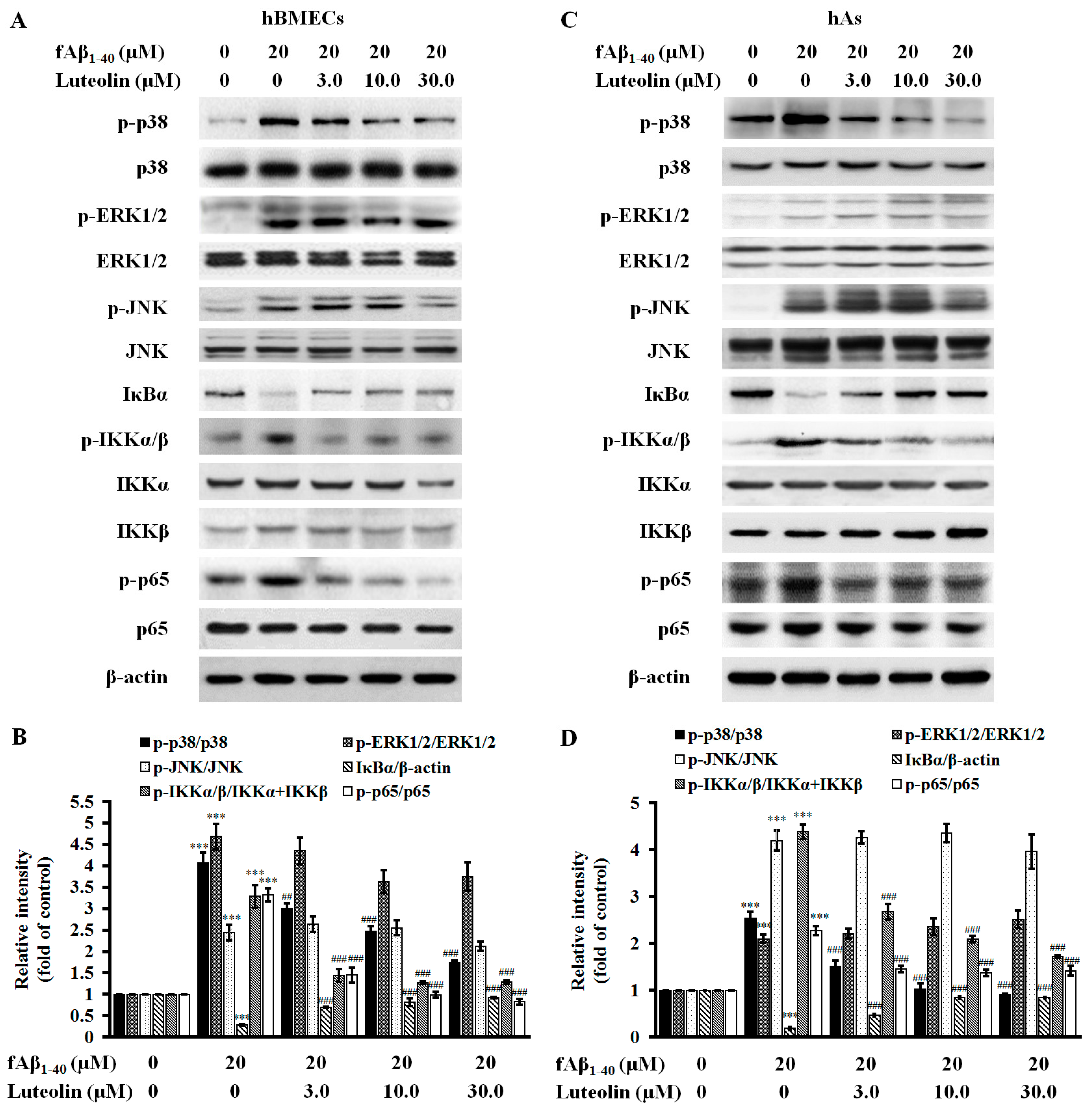
2.4. Luteolin Inhibits NF-κB and MAPK Signal Pathways against fAβ1–40-Induced Toxicity
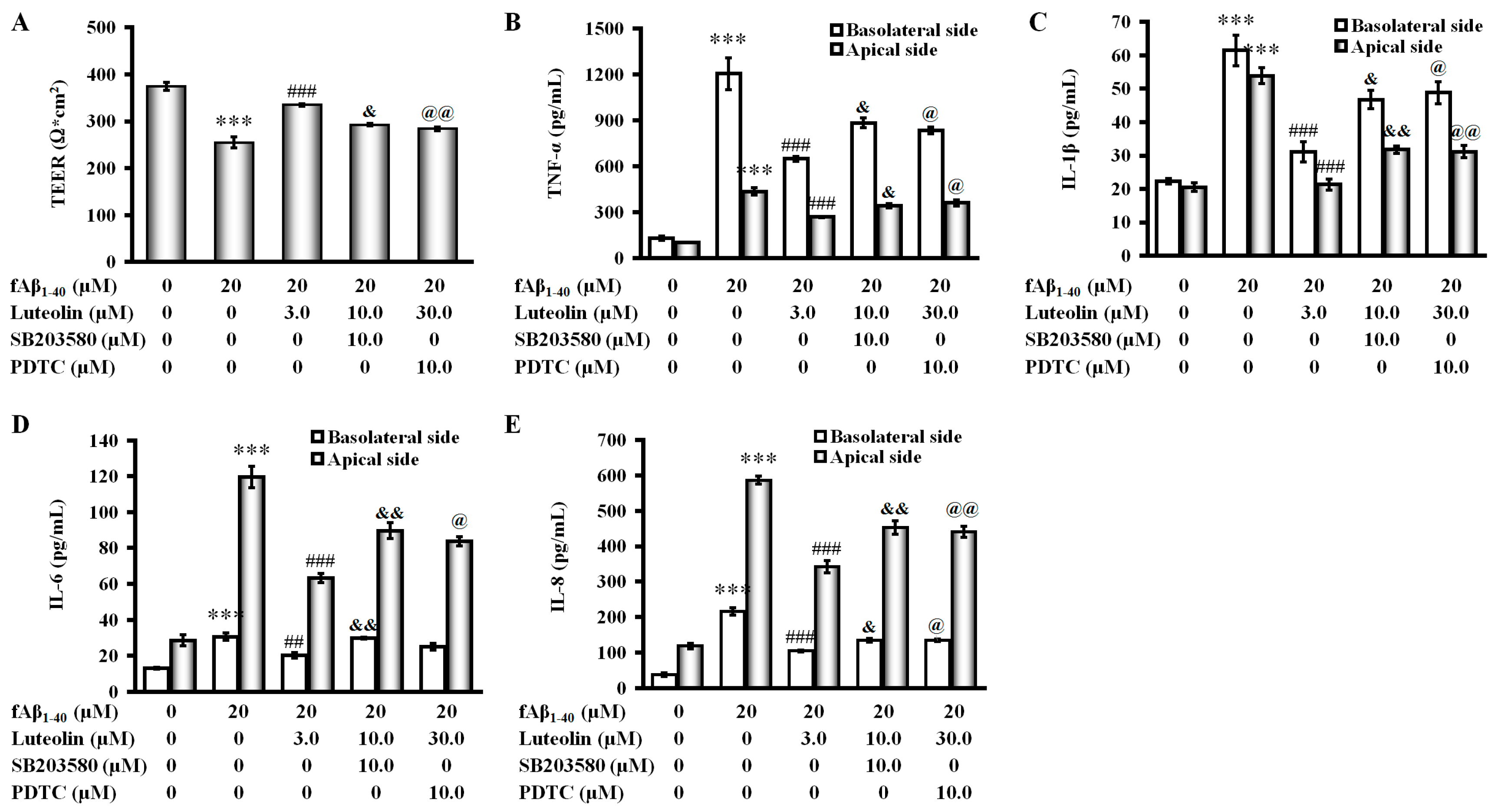
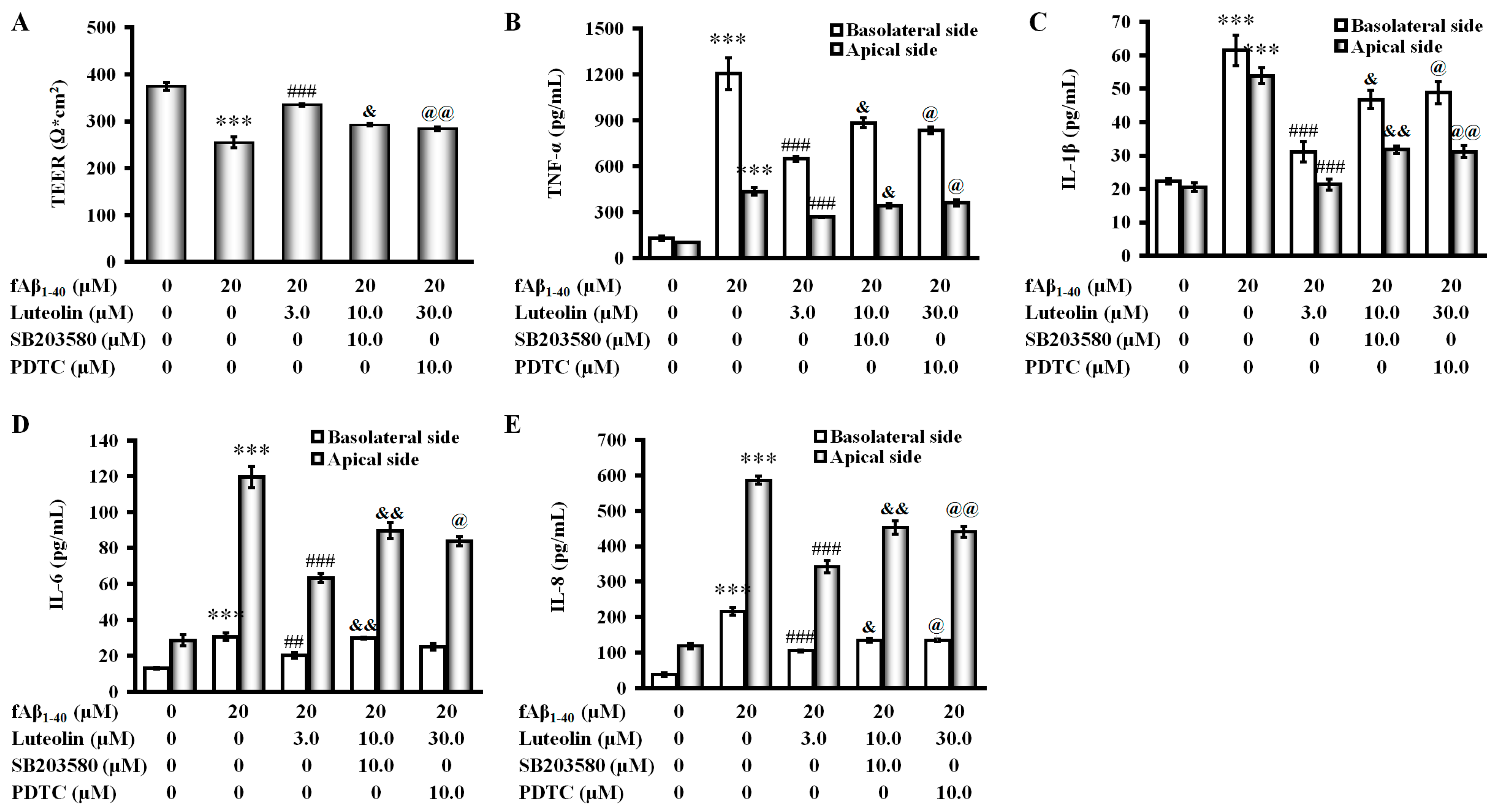
2.5. Involvement of the p38 MAPK/NF-κB Pathway in the Protective Effect of Luteolin on fAβ1–40-Induced Barrier Function and Inflammation
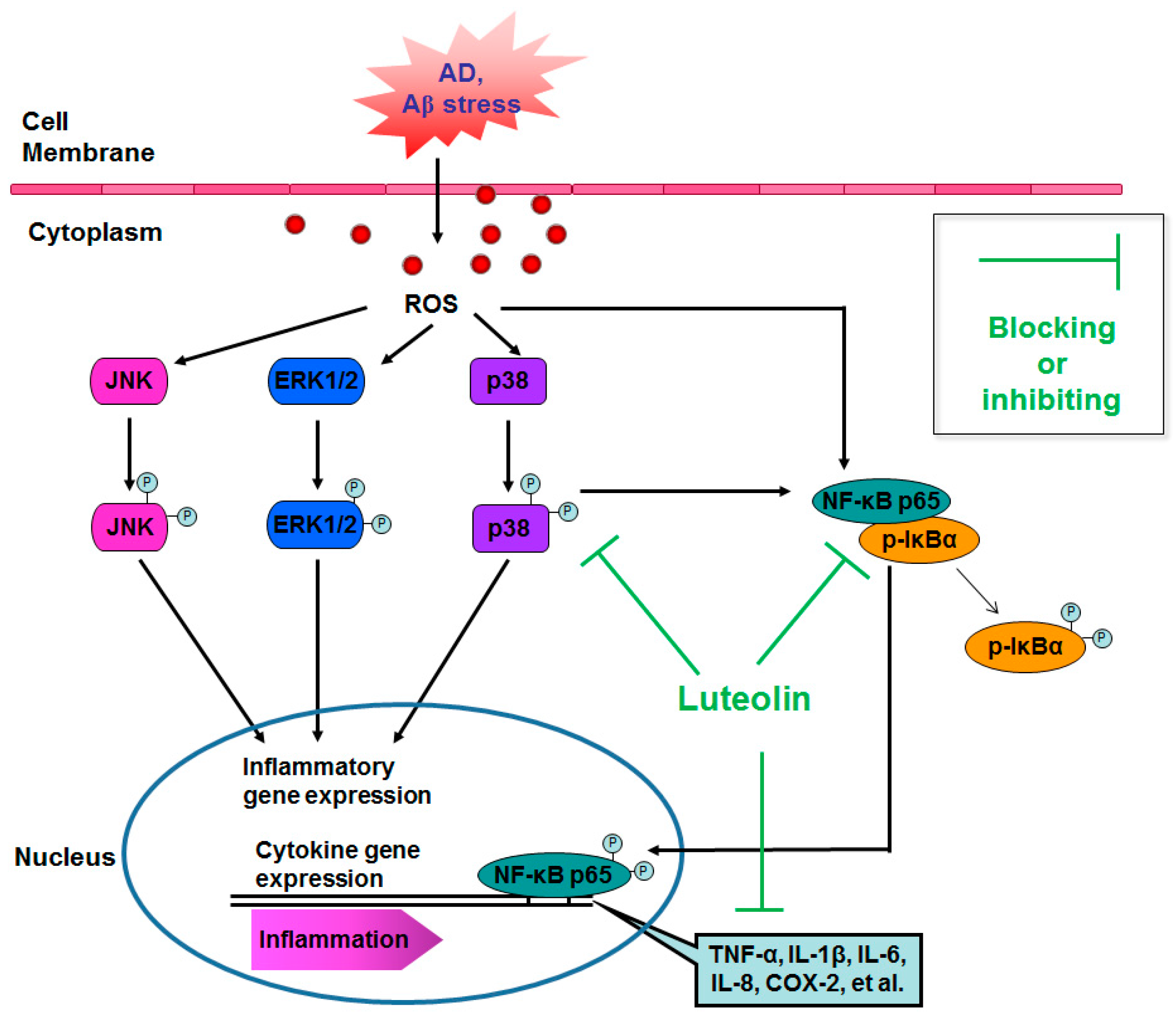
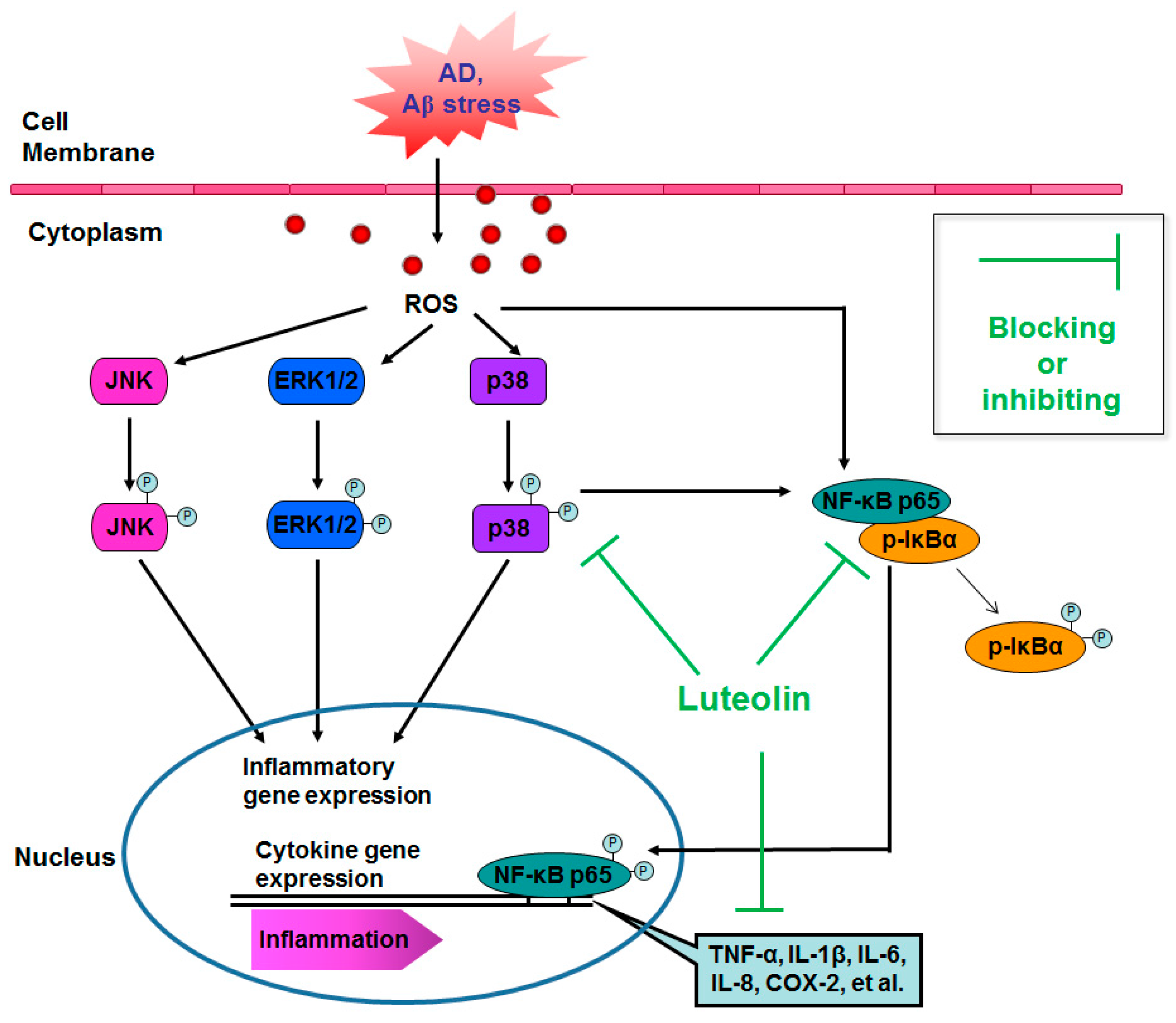
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Cell Culture and Treatment
4.3. Co-Culture of hBMECs and hAs
4.4. MTS Cell Viability Assay
4.5. Alamar Blue Cell Viability Assay
4.6. TEER Measurement
4.7. Transendotheial Permeability Measurements
4.8. Enzyme-Linked Immunosorbent Assay for Proinflammatory Cytokines
4.9. Western Blot Analysis
4.10. Intracellular ROS Detection
4.11. Statistical Analysis
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Selkoe, D.J. Translating cell biology into therapeutic advances in Alzheimer’s disease. Nature 1999, 399, A23–A31. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.; Selkoe, D.J. The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef] [PubMed]
- Verdile, G.; Fuller, S.; Atwood, C.S.; Laws, S.M.; Gandy, S.E.; Martins, R.N. The role of beta amyloid in Alzheimer’s disease: Still a cause of everything or the only one who got caught? Pharmacol. Res. 2004, 50, 397–409. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Li, J.Z.; Song, J.K.; Sun, J.L.; Li, Y.J.; Zhou, S.B.; Zhang, T.T.; Du, G.H. Pinocembrin protects human brain microvascular endothelial cells against fibrillar amyloid-β1–40 injury by suppressing the MAPK/NF-κB inflammatory pathways. Biomed. Res. Int. 2014, 14, 370–393. [Google Scholar]
- Giulian, D.; Haverkamp, L.J.; Li, J.; Karshin, W.L.; Yu, J.; Tom, D.; Li, X.; Kirkpatrick, J.B. Senile plaques stimulate microglia to release a neurotoxin found in Alzheimer brain. Neurochem. Int. 1995, 27, 119–137. [Google Scholar] [CrossRef]
- Combs, C.K.; Johnson, D.E.; Karlo, J.C.; Cannady, S.B.; Landreth, G.E. Inflammatory mechanisms in Alzheimer’s disease: Inhibition of beta-amyloid-stimulated proinflammatory responses and neurotoxicity by PPARγ agonists. J. Neurosci. 2000, 20, 558–567. [Google Scholar] [PubMed]
- Li, Y.; Zhou, S.; Li, J.; Sun, Y.; Hasimu, H.; Liu, R.; Zhang, T. Quercetin protects human brain microvascular endothelial cells from fibrillar β-amyloid1–40-induced toxicity. Acta Pharm. Sin. B 2015, 5, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Zhang, T.T.; Wu, C.X.; Lan, X.; Du, G.H. Targeting the neurovascular unit: Development of a new model and consideration for novel strategy for Alzheimer’s disease. Brain Res. Bull. 2011, 86, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.F.; Li, N.; Wang, Q.; Cheng, X.J.; Li, X.M.; Liu, T.T. Resveratrol decreases the insoluble Aβ1–42 level in hippocampus and protects the integrity of the blood-brain barrier in AD rats. Neuroscience 2015, 310, 641–649. [Google Scholar] [CrossRef] [PubMed]
- Abbott, N.J.; Patabendige, A.A.; Dolman, D.E.; Yusof, S.R.; Begley, D.J. Structure and function of the blood-brain barrier. Neurobiol. Dis. 2010, 37, 13–25. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Hou, L.; Sun, H.; Sun, X.; Li, J.; Bian, Y.; Chu, Y.; Liu, Q. Apigenin Isolated from the Medicinal Plant Elsholtzia rugulosa Prevents β-Amyloid 25–35-Induces Toxicity in Rat Cerebral Microvascular Endothelial Cells. Molecules 2011, 16, 4005–4019. [Google Scholar] [CrossRef]
- Keaney, J.; Walsh, D.M.; O’Malley, T.; Hudson, N.; Crosbie, D.E.; Loftus, T.; Sheehan, F.; McDaid, J.; Humphries, M.M.; Callanan, J.J.; et al. Autoregulated paracellular clearance of amyloid-beta across the blood-brain barrier. Sci. Adv. 2015, 1, e1500472. [Google Scholar] [CrossRef] [PubMed]
- Qosa, H.; LeVine, H.; Keller, J.N.; Kaddoumi, A. Mixed oligomers and monomeric amyloid-β disrupts endothelial cells integrity and reduces monomeric amyloid-β transport across hCMEC/D3 cell line as an in vitro blood-brain barrier model. Biochim. Biophys. Acta 2014, 1842, 1806–1815. [Google Scholar] [CrossRef] [PubMed]
- Tai, L.M.; Holloway, K.A.; Male, D.K.; Loughlin, A.J.; Romero, I.A. Amyloid-β-induced occludin down-regulation and increased permeability in human brain endothelial cells is mediated by MAPK activation. J. Cell. Mol. Med. 2010, 14, 1101–1112. [Google Scholar] [CrossRef] [PubMed]
- White, J.A.; Manelli, A.M.; Holmberg, K.H.; Van Eldik, L.J.; Ladu, M.J. Differential effects of oligomeric and fibrillary amyloid-β1–42 on astrocyte-mediated inflammation. Neurobiol. Dis. 2005, 18, 459–465. [Google Scholar] [CrossRef] [PubMed]
- Griffin, J.M.; Kho, D.; Graham, E.S.; Nicholson, L.F.; O’Carroll, S.J. Statins Inhibit Fibrillary β-Amyloid Induced Inflammation in a Model of the Human Blood Brain Barrier. PLoS ONE 2016, 11, e0157483. [Google Scholar] [CrossRef] [PubMed]
- Zenaro, E.; Piacentino, G.; Constantin, G. The blood-brain barrier in Alzheimer’s disease. Neurobiol. Dis. 2016, 95, 232–242. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.R.; Xu, X.Z.; Wang, Y.H.; Chen, J.W.; Xu, S.W.; Gu, L.Q.; Liu, P.Q. Icariin Derivative Inhibits Inflammation through Suppression of p38 Mitogen-Activated Protein Kinase and Nuclear Factor-κB Pathways. Biol. Pharm. Bull. 2011, 33, 1307–1313. [Google Scholar] [CrossRef]
- Uto, T.; Suangkaew, N.; Morinaga, O.; Kariyazono, H.; Oiso, S.; Shoyama, Y. Eriobotryae Folium Extract Suppresses LPS-Induced iNOS and COX-2 Expression by Inhibition of NF-κB and MAPK Activation in Murine Macrophages. Am. J. Chin. Med. 2011, 38, 985–994. [Google Scholar] [CrossRef] [PubMed]
- Park, E.; Kum, S.; Wang, C.; Park, S.Y.; Kim, B.S.; Schuller-Levis, G. Anti-inflammatory Activity of Herbal Medicines: Inhibition of Nitric Oxide Production and Tumor Necrosis Factor-α Secretion in an Activated Macrophage-like Cell Line. Am. J. Chin. Med. 2005, 33, 415–424. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.S.; Jobin, C. The flavonoid luteolin prevents lipopolysaccharide-induced NF-κB signaling and gene expression by blocking IκB kinase activity in intestinal epithelial cells and bone-marrow derived dendritic cells. Immunology 2005, 115, 375–387. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Shin, K.J.; Kim, D.; Kim, Y.H.; Han, M.S.; Lee, T.G. Luteolin inhibits the nuclear factor-κB transcriptional activity in Rat-1 fibroblasts. Biochem. Pharmacol. 2003, 66, 955–963. [Google Scholar] [CrossRef]
- Gutiérrez-Venegas, G.; Kawasaki-Cárdenas, P.; Arroyo-Cruz, S.R.; Maldonado-Frías, S. Luteolin inhibits lipopolysaccharide actions on human gingival fibroblasts. Eur. J. Pharmacol. 2006, 541, 95–105. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Gao, M.; Qiang, G.F.; Zhang, T.T.; Lan, X.; Ying, J.; Du, G.H. The anti-amnesic effects of luteolin against amyloid β25–35 peptide-induced toxicity in mice involve the protection of neurovascular unit. Neuroscience 2009, 162, 1232–1243. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.X.; Zhang, P.; Guan, Y.; Wang, M.; Zhen, M.Q. Protective effects of luteolin against cognitive impairment induced by infusion of Aβ peptide in rats. Int. J. Clin. Exp. Pathol. 2015, 8, 6740–6747. [Google Scholar] [PubMed]
- Wang, H.; Wang, H.; Cheng, H.; Che, Z. Ameliorating effect of luteolin on memory impairment in an Alzheimer’s disease model. Mol. Med. Rep. 2016, 13, 4215–4220. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Lan, X.; Ying, J.; Du, G. Protective Effects of Luteolin against Amyloid β25–35-induced Toxicity on Rat Cerebral Microvascular Endothelial Cells. Chin. J. Nat. Med. 2010, 8, 223–227. [Google Scholar]
- Zhu, L.H.; Bi, W.; Qi, R.B.; Wang, H.D.; Lu, D.X. Luteolin inhibits microglial inflammation and improves neuron survival against inflammation. Int. J. Neurosci. 2011, 121, 329–336. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Bi, W.; Lu, D.; Zhang, C.; Shu, X.; Lu, D. Luteolin inhibits SH-SY5Y cell apoptosis through suppression of the nuclear transcription factor-κB, mitogen-activated protein kinase and protein kinase B pathways in lipopolysaccharide-stimulated cocultured BV2 cells. Exp. Ther. Med. 2014, 7, 1065–1070. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Deng, Y.; Gao, J.; Liu, Y.; Li, W.; Shi, J.; Gong, Q. Sodium Hydrosulfide Attenuates Beta-Amyloid-Induced Cognitive Deficits and Neuroinflammation via Modulation of MAPK/NF-κB Pathway in Rats. Curr. Alzheimer Res. 2015, 12, 673–683. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; Wang, H.; Ying, H.; Yu, Y.; Chen, D.; Ge, W.; Shi, L. Daphnetin attenuates microglial activation and proinflammatory factor production via multiple signaling pathways. Int. Immunopharmacol. 2014, 21, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, B.T.; Davis, T.P. The blood-brain barrier/neurovascular unit in health and disease. Pharmacol. Rev. 2005, 57, 173–185. [Google Scholar] [CrossRef] [PubMed]
- Cecchelli, R.; Berezowski, V.; Lundquist, S.; Culot, M.; Renftel, M.; Dehouck, M.P.; Fenart, L. Modelling of the blood–brain barrier in drug discovery and development. Nat. Rev. Drug Discov. 2007, 6, 650–661. [Google Scholar] [CrossRef] [PubMed]
- Bell, R.D.; Winkler, E.A.; Sagare, A.P.; Singh, I.; LaRue, B.; Deane, R.; Zlokovic, B.V. Pericytes control key neurovascular functions and neuronal phenotype in the adult brain and during brain aging. Neuron 2010, 68, 409–427. [Google Scholar] [CrossRef] [PubMed]
- Merlini, M.; Meyer, E.P.; Ulmann-Schuler, A.; Nitsch, R.M. Vascular β-amyloid and early astrocyte alterations impair cerebrovascular function and cerebral metabolism in transgenic arcAβ mice. Acta Neuropathol. 2011, 122, 293–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, S.D.; Chen, X.; Fu, J.; Chen, M.; Zhu, H.; Roher, A.; Slattery, T.; Zhao, L.; Nagashima, M.; Morser, J.; et al. RAGE and amyloid-β peptide neurotoxicity in Alzheimer’s disease. Nature 1996, 382, 685–691. [Google Scholar] [CrossRef] [PubMed]
- Deane, R.; Du, Y.S.; Submamaryan, R.K.; LaRue, B.; Jovanovic, S.; Hogg, E.; Welch, D.; Manness, L.; Lin, C.; Yu, J.; et al. RAGE mediates amyloid-β peptide transport across the blood-brain barrier and accumulation in brain. Nat. Med. 2003, 9, 907–913. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Li, J.Z.; Song, J.K.; Zhou, D.; Huang, C.; Bai, X.Y.; Xie, T.; Zhang, X.; Li, Y.J.; Wu, C.X.; et al. Pinocembrin improves cognition and protects the neurovascular unit in Alzheimer related deficits. Neurobiol. Aging 2014, 35, 1275–1285. [Google Scholar] [CrossRef] [PubMed]
- Shibata, M.; Yamada, S.; Kumar, S.R.; Calero, M.; Bading, J.; Frangione, B.; Holtzman, D.M.; Miller, C.A.; Strickland, D.K.; Ghiso, J.; et al. Clearance of Alzheimer’s amyloid-β1–40 peptide from brain by LDL receptor-related protein-1 at the blood-brain barrier. J. Clin. Investig. 2000, 106, 1489–1499. [Google Scholar] [CrossRef] [PubMed]
- Matsubara, E.; Ghiso, J.; Frangione, A.M.; Tomidokoro, Y.; Ikeda, Y.; Harigaya, Y.; Okamoto, K.; Shoji, M. Lipoprotein-free amyloidogenic peptides in plasma are elevated in patients with sporadic Alzheimer’s disease and Down’s syndrome. Ann. Neurol. 1999, 45, 537–541. [Google Scholar] [CrossRef]
- Kawarabayashi, T.; Younkin, L.H.; Saido, T.C.; Shoji, M.; Ashe, K.H.; Younkin, S.G. Age-Dependent Changes in Brain, CSF, and Plasma Amyloid β Protein in the Tg2576 Transgenic Mouse Model of Alzheimer’s disease. J. Neurosci. 2001, 21, 372–381. [Google Scholar] [PubMed]
- Selkoe, D.J. Alzheimer’s disease: Genes, proteins, and therapy. Physiol. Rev. 2001, 81, 741–766. [Google Scholar] [CrossRef] [PubMed]
- Thal, D.R. The role of astrocytes in amyloid beta-protein toxicity and clearance. Exp. Neurol. 2012, 236, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Grosso, C.; Valentão, P.; Ferreres, F.; Andrade, P.B. The use of flavonoids in central nervous system disorders. Curr. Med. Chem. 2013, 20, 4694–4719. [Google Scholar] [CrossRef] [PubMed]
- Alonzo, N.C.; Hyman, B.T.; Rebeck, G.W.; Greenberg, S.M. Progression of cerebral amyloid angiopathy: Accumulation of amyloid-beta40 in affected vessels. J. Neuropathol. Exp. Neurol. 1998, 57, 353–359. [Google Scholar] [CrossRef] [PubMed]
- Salvarani, C.; Hunder, G.G.; Morris, J.M.; Brown, R.D.; Jr Christianson, T.; Giannini, C. Aβ-related angiitis: Comparison with CAA without inflammation and primary CNS vasculitis. Neurology 2013, 81, 1596–1603. [Google Scholar] [CrossRef] [PubMed]
- Carrano, A.; Hoozemans, J.J.; Vies, S.M.; van Horssen, J.; Vries, H.E.; Rozemuller, A.J. Neuroinflammation and blood-brain barrier changes in capillary amyloid angiopathy. Neurodegener. Dis. 2012, 10, 329–331. [Google Scholar] [CrossRef] [PubMed]
- Zlokovic, B.V. The blood-brain barrier in health and chronic neurodegenerative disorders. Neuron 2008, 57, 178–201. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Ramirez, M.A.; Reijerkerk, A.; de Vries, H.E.; Romero, I.A. Regulation of brain endothelial barrier function by microRNAs in health and neuroinflammation. FASEB J. 2016, 30, 2662–2672. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Shi, X.Y.; Bi, D.C.; Fang, W.S.; Wei, G.B.; Xu, X. Alginate-Derived Oligosaccharide Inhibits Neuroinflammation and Promotes Microglial Phagocytosis of β-Amyloid. Mar. Drugs 2015, 13, 5828–5846. [Google Scholar] [CrossRef] [PubMed]
- Hoozemans, J.J.; Rozemuller, A.J.; Janssen, I.; De Groot, C.J.; Veerhuis, R.; Eikelenboom, P. Cyclooxygenase expression in microglia and neurons in Alzheimer’s disease and control brain. Acta Neuropathol. 2001, 101, 2–8. [Google Scholar] [PubMed]
- Lamoke, F.; Mazzone, V.; Persichini, T.; Maraschi, A.; Harris, M.B.; Venema, R.C.; Colasanti, M.; Gliozzi, M.; Muscoli, C.; Bartoli, M.; et al. Amyloid beta peptide-induced inhibition of endothelial nitric oxide production involves oxidative stress-mediated constitutive eNOS/HSP90 interaction and disruption of agonist-mediated Akt activation. J. Neuroinflamm. 2015, 12, 84–92. [Google Scholar] [CrossRef] [PubMed]
- Gimbrone, M.A., Jr. Vascular Endothelium, Hemodynamic Forces and Atherogenesis. Am. J. Pathol. 1999, 155, 1–5. [Google Scholar] [CrossRef]
- Grammas, P.; Ovase, R. Inflammatory factors are elevated in brain microvessels in Alzheimer’s disease. Neurobiol. Aging 2001, 22, 837–842. [Google Scholar] [CrossRef]
- Dorheim, M.A.; Tracey, W.R.; Pollock, J.S.; Grammas, P. Nitric oxide is elevated in Alzheimer’s brain microvessels. Biochem. Biophys. Res. Commun. 1994, 205, 659–665. [Google Scholar] [CrossRef] [PubMed]
- Sung, J.; Lee, J. Anti-Inflammatory Activity of Butein and Luteolin through Suppression of NFκB Activation and Induction of Heme Oxygenase-1. J. Med. Food 2015, 18, 557–564. [Google Scholar] [CrossRef] [PubMed]
- Hendriks, J.J.; Alblas, J.; van der Pol, S.M.; van Tol, E.A.; Dijkstra, C.D.; de Vries, H.E. Flavonoids influence monocytic GTPase activity and are protective in experimental allergic encephalitis. J. Exp. Med. 2004, 200, 1667–1672. [Google Scholar] [CrossRef] [PubMed]
- Sawmiller, D.; Li, S.; Shahaduzzaman, M.; Smith, A.J.; Obregon, D.; Giunta, B.; Borlongan, C.V.; Sanberg, P.R.; Tan, J. Luteolin reduces Alzheimer’s disease pathologies induced by traumatic brain injury. Int. J. Mol. Sci. 2014, 15, 895–904. [Google Scholar] [CrossRef] [PubMed]
- Paterniti, I.; Cordaro, M.; Campolo, M.; Siracusa, R.; Cornelius, C.; Navarra, M.; Cuzzocrea, S.; Esposito, E. Neuroprotection by Association of Palmitoylethanolamide with Luteolin in Experimental Alzheimer’s Disease Models: The Control of Neuroinflammation. CNS Neurol. Disord. Drug Targets 2014, 13, 1530–1541. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.C.; Kao, M.C.; Fu, M.T.; Lin, C.T. Modulation of NO and cytokines in microglial cells by Cu/Zn-superoxide dismutase. Free Radic. Biol. Med. 2001, 31, 1084–1089. [Google Scholar] [CrossRef]
- Rojo, A.I.; McBean, G.; Cindric, M.; Egea, J.; Lopez, M.G.; Rada, P.; Zarkovic, N.; Cuadrado, A. Redox control of microglial function: Molecular mechanisms and functional significance. Antioxid. Redox Signal. 2014, 21, 1766–1801. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, I.N.; Chang, A.S.; Teng, C.M.; Chen, C.C.; Yang, C.R. Aciculatin inhibits lipopolysaccharide-mediated inducible nitric oxide synthase and cyclooxygenase-2 expression via suppressing NF-κB and JNK/p38 MAPK activation pathways. J. Biomed. Sci. 2011, 18, 28. [Google Scholar] [CrossRef] [PubMed]
- Kaur, U.; Banerjee, P.; Bir, A.; Sinha, M.; Biswas, A.; Chakrabarti, S. Reactive oxygen species, redox signaling and neuroinflammation in Alzheimer’s disease: The NF-κB connection. Curr. Top. Med. Chem. 2015, 15, 446–457. [Google Scholar] [CrossRef] [PubMed]
- Caughey, G.E.; Cleland, L.G.; Penglis, P.S.; Gamble, J.R.; James, M.J. Roles of Cyclooxygenase (COX)-1 and COX-2 in Prostanoid Production by Human Endothelial Cells: Selective Up-Regulation of Prostacyclin Synthesis by COX-2. J. Immunol. 2001, 167, 2831–2838. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.D.; Chen, X.C.; Zhu, Y.G.; Chen, L.M.; Zhang, J.; Huang, T.W.; Ye, Q.Y.; Huang, H.P. Tripchlorolide protects neuronal cells from microglia-mediated βa-amyloid neurotoxicity through inhibiting NF-κB and JNK signaling. Glia 2009, 57, 1227–1238. [Google Scholar] [CrossRef] [PubMed]
- Jang, J.H.; Surh, Y.J. β-Amyloid-induced apoptosis is associated with cyclooxygenase-2 up-regulation via the mitogen-activated protein kinase-NF-κB signaling pathway. Free Radic. Biol. Med. 2005, 38, 1604–1613. [Google Scholar] [CrossRef] [PubMed]
- Gu, B.H.; Minh, N.V.; Lee, S.H.; Lim, S.W.; Lee, Y.M.; Lee, K.S.; Kim, D.K. Deoxyschisandrin inhibits H2O2-induced apoptotic cell death in intestinal epithelial cells through nuclear factor-κB. Int. J. Mol. Med. 2010, 26, 401–406. [Google Scholar] [PubMed]
- Nepali, S.; Son, J.S.; Poudel, B.; Lee, J.H.; Lee, Y.M.; Kim, D.K. Luteolin is a bioflavonoid that attenuates adipocyte-derived inflammatory responses via suppression of nuclear factor-κB/mitogen-activated protein kinases pathway. Pharmacogn. Mag. 2015, 11, 627–635. [Google Scholar] [PubMed]
- Liu, C.W.; Lin, H.W.; Yang, D.J.; Chen, S.Y.; Tseng, J.K.; Chang, T.J.; Chang, Y.Y. Luteolin inhibits viral-induced inflammatory response in RAW264.7 cells via suppression of STAT1/3 dependent NF-κB and activation of HO-1. Free Radic. Biol. Med. 2016, 95, 180–189. [Google Scholar] [PubMed]
- Fiebich, B.L.; Mueksch, B.; Boehringer, M.; Hull, M. Interleukin-1β induces cyclooxygenase-2 and prostaglandin E2 synthesis in human neuroblastoma cells: Involvement of p38 mitogen-activated protein kinase and nuclear factor-κB. J. Neurochem. 2000, 75, 2020–2028. [Google Scholar] [CrossRef] [PubMed]
- Singer, C.A.; Baker, K.J.; McCaffrey, A.; AuCoin, D.P.; Dechert, M.A.; Gerthoffer, W.T. p38 MAPK and NF-κB mediate COX-2 expression in human airway myocytes. Am. J. Physiol. Lung Cell. Mol. Physiol. 2003, 285, L1087–L1098. [Google Scholar] [CrossRef] [PubMed]
- Gloire, G.; Legrand-Poels, S.; Piette, J. NF-κB activation by reactive oxygen species: Fifteen years later. Biochem. Pharmacol. 2006, 72, 1493–1505. [Google Scholar] [CrossRef] [PubMed]
- Cainelli, G.; Angeloni, C.; Cervellati, R.; Galletti, P.; Giacomini, D.; Hrelia, S.; Sinisi, R. New Polyphenolic β-lactams with antioxidant activity. Chem. Biodivers. 2008, 5, 811–829. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Stanley, R.A.; Adaim, A.; Melton, L.D.; Skinner, M.A. Free radical scavenging and cytoprotective activities of phenolic antioxidants. Mol. Nutr. Food Res. 2006, 50, 996–1005. [Google Scholar] [CrossRef] [PubMed]
- Dajas, F.; Rivera, F.; Blasina, F.; Arredondo, F.; Echeverry, C.; Lafon, L.; Morquio, A.; Heinzen, H. Cell culture protection and in vivo neuroprotective capacity of flavonoids. Neurotox. Res. 2003, 5, 425–432. [Google Scholar] [CrossRef] [PubMed]
- Brunet, A.; Datta, S.R.; Greenberg, M.E. Transcription-dependent and -independent control of neuronal survival by the PI3K-Akt signaling pathway. Curr. Opin. Neurobiol. 2001, 11, 297–305. [Google Scholar] [CrossRef]
- Hodnick, W.F.; Duval, D.L.; Pardini, R.S. Inhibition of mitochondrial respiration and cyanide-stimulated generation of reactive oxygen species by selected flavonoids. Biochem. Pharmacol. 1994, 47, 573–580. [Google Scholar] [CrossRef]
- Vauzour, D.; Vafeiadou, K.; Rodriguez-Mateos, A.; Rendeiro, C.; Spencer, J.P. The neuroprotective potential of flavonoids: A multiplicity of effects. Genes Nutr. 2008, 3, 115–126. [Google Scholar] [CrossRef] [PubMed]
- Jekabsone, A.; Mander, P.K.; Tickler, A.; Sharpe, M.; Brown, G.C. Fibrillar beta-amyloid peptide Aβ1–40 activates microglial proliferation via stimulating TNF-α release and H2O2 derived from NADPH oxidase: A cell culture study. J. Neuroinflamm. 2006, 3, 24. [Google Scholar] [CrossRef] [PubMed]
- Veszelka, S.; Pásztói, M.; Farkas, A.E.; Krizbai, I.; Ngo, T.K.; Niwa, M.; Abrahám, C.S.; Deli, M.A. Pentosan polysulfate protects brain endothelial cells against bacterial lipopolysaccharide-induced damages. Neurochem. Int. 2007, 50, 219–228. [Google Scholar] [CrossRef] [PubMed]
- Monaghan-Benson, E.; Wittchen, E.S. In vitro analyses of endothelial cell permeability. Methods Mol. Biol. 2011, 763, 281–290. [Google Scholar] [PubMed]
- Sample Availability: Samples of the compounds are available from the authors.


© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, J.-X.; Xing, J.-G.; Wang, L.-L.; Jiang, H.-L.; Guo, S.-L.; Liu, R. Luteolin Inhibits Fibrillary β-Amyloid1–40-Induced Inflammation in a Human Blood-Brain Barrier Model by Suppressing the p38 MAPK-Mediated NF-κB Signaling Pathways. Molecules 2017, 22, 334. https://doi.org/10.3390/molecules22030334
Zhang J-X, Xing J-G, Wang L-L, Jiang H-L, Guo S-L, Liu R. Luteolin Inhibits Fibrillary β-Amyloid1–40-Induced Inflammation in a Human Blood-Brain Barrier Model by Suppressing the p38 MAPK-Mediated NF-κB Signaling Pathways. Molecules. 2017; 22(3):334. https://doi.org/10.3390/molecules22030334
Chicago/Turabian StyleZhang, Jun-Xia, Jian-Guo Xing, Lin-Lin Wang, Hai-Lun Jiang, Shui-Long Guo, and Rui Liu. 2017. "Luteolin Inhibits Fibrillary β-Amyloid1–40-Induced Inflammation in a Human Blood-Brain Barrier Model by Suppressing the p38 MAPK-Mediated NF-κB Signaling Pathways" Molecules 22, no. 3: 334. https://doi.org/10.3390/molecules22030334