
Experimental and Theoretical Reduction Potentials of Some Biologically Active ortho-Carbonyl para-Quinones

 ,
,
Abstract
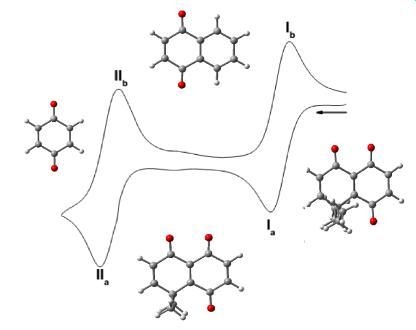
:
1. Introduction
2. Materials and Methods
2.1. Cyclic Voltammetry
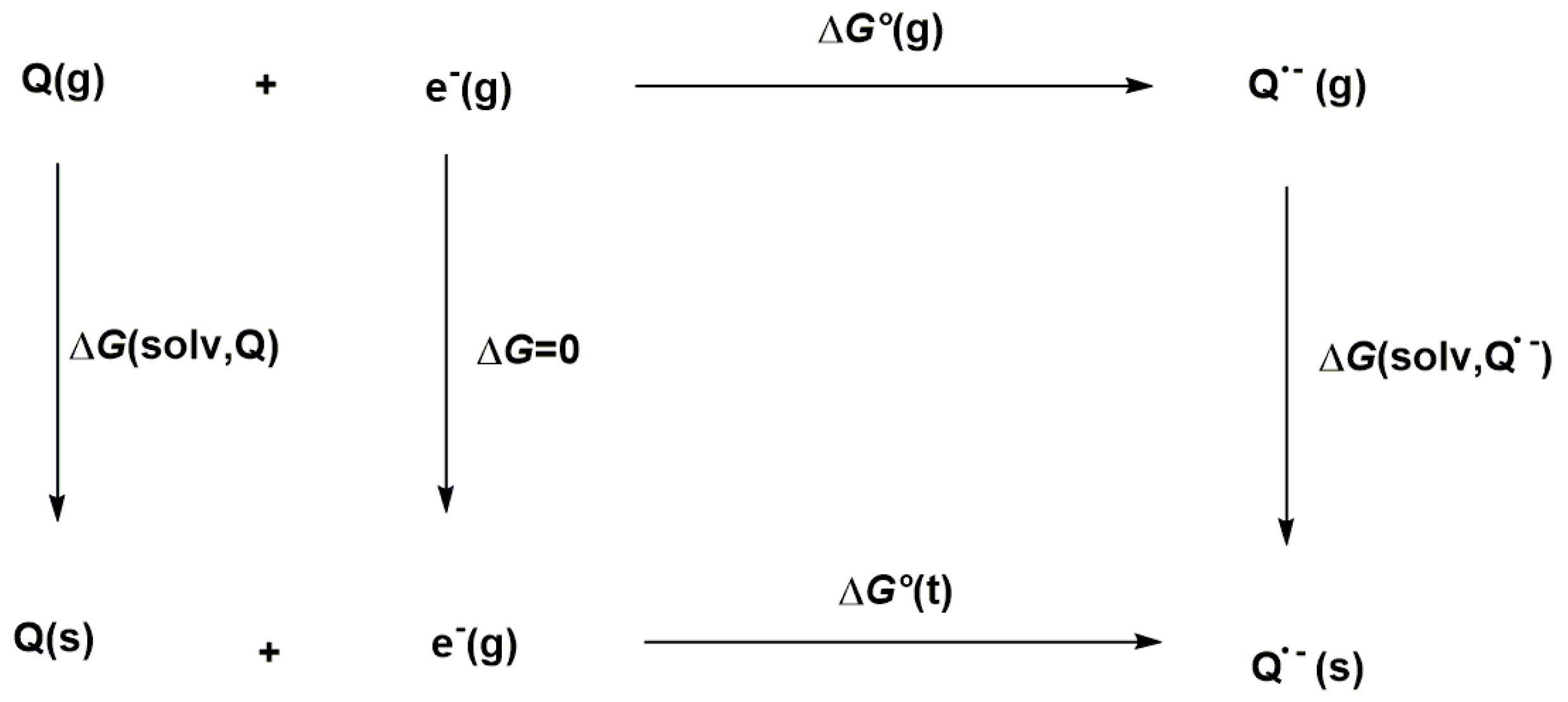
2.2. Computational Details
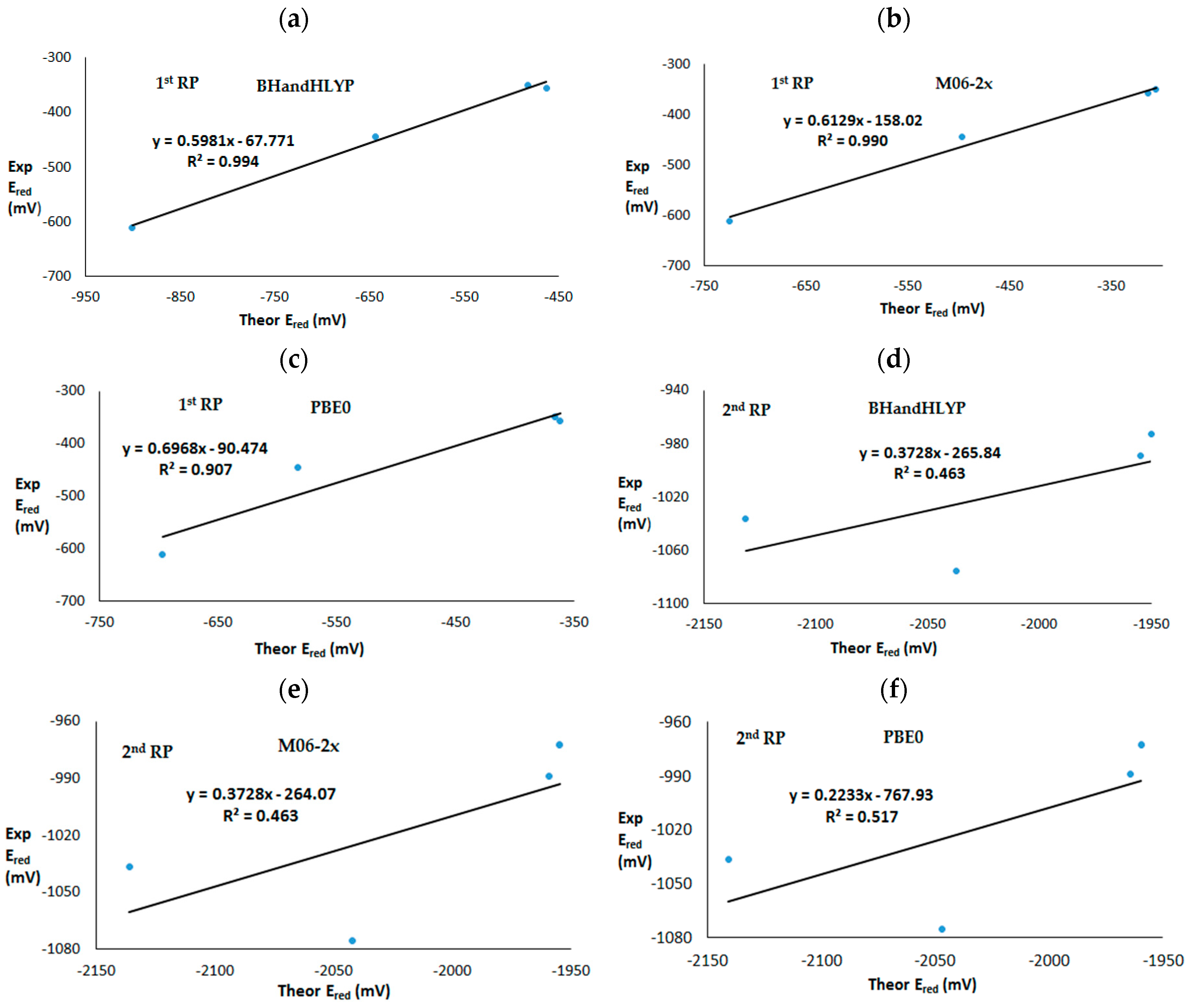
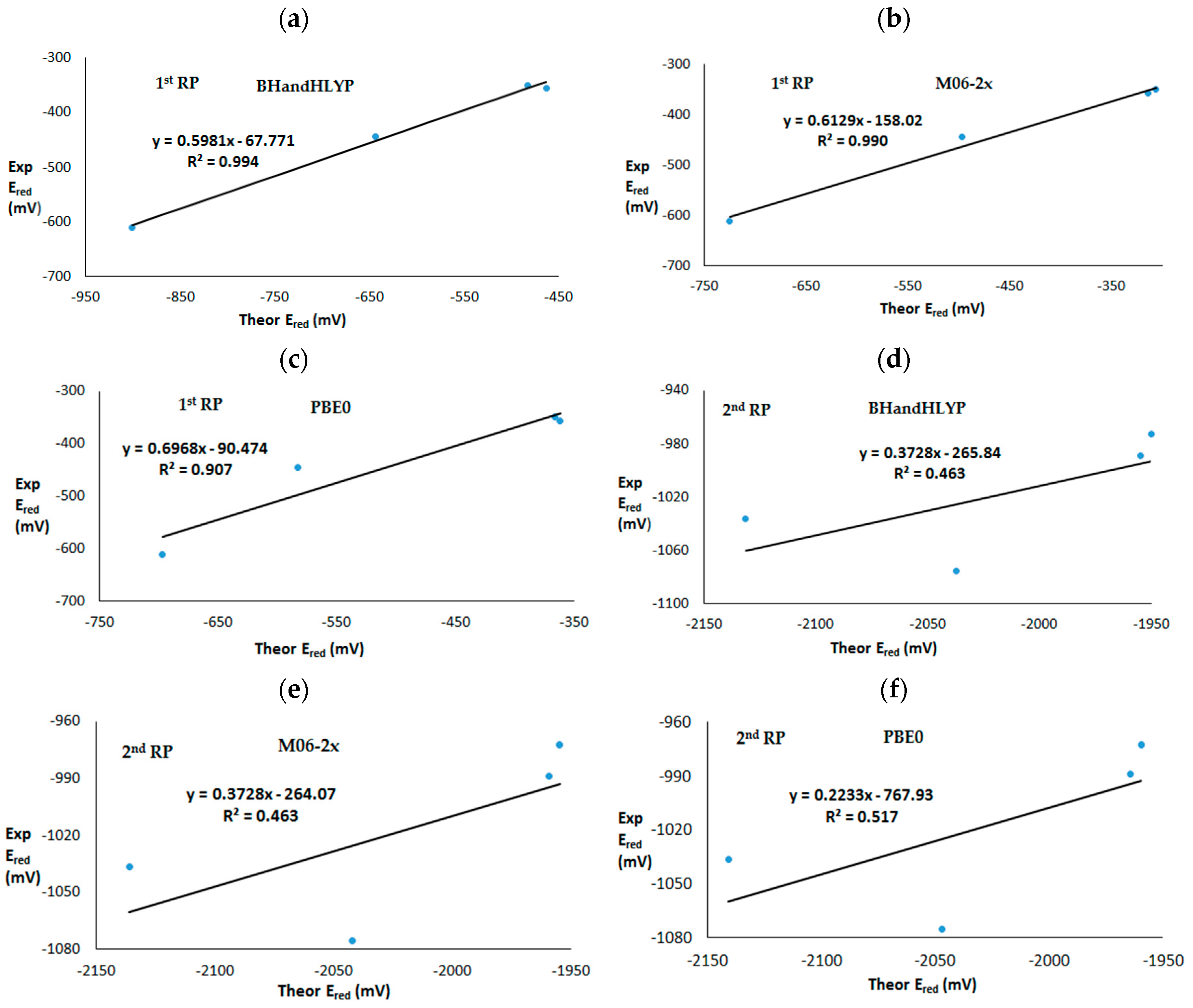
3. Results and Discussion
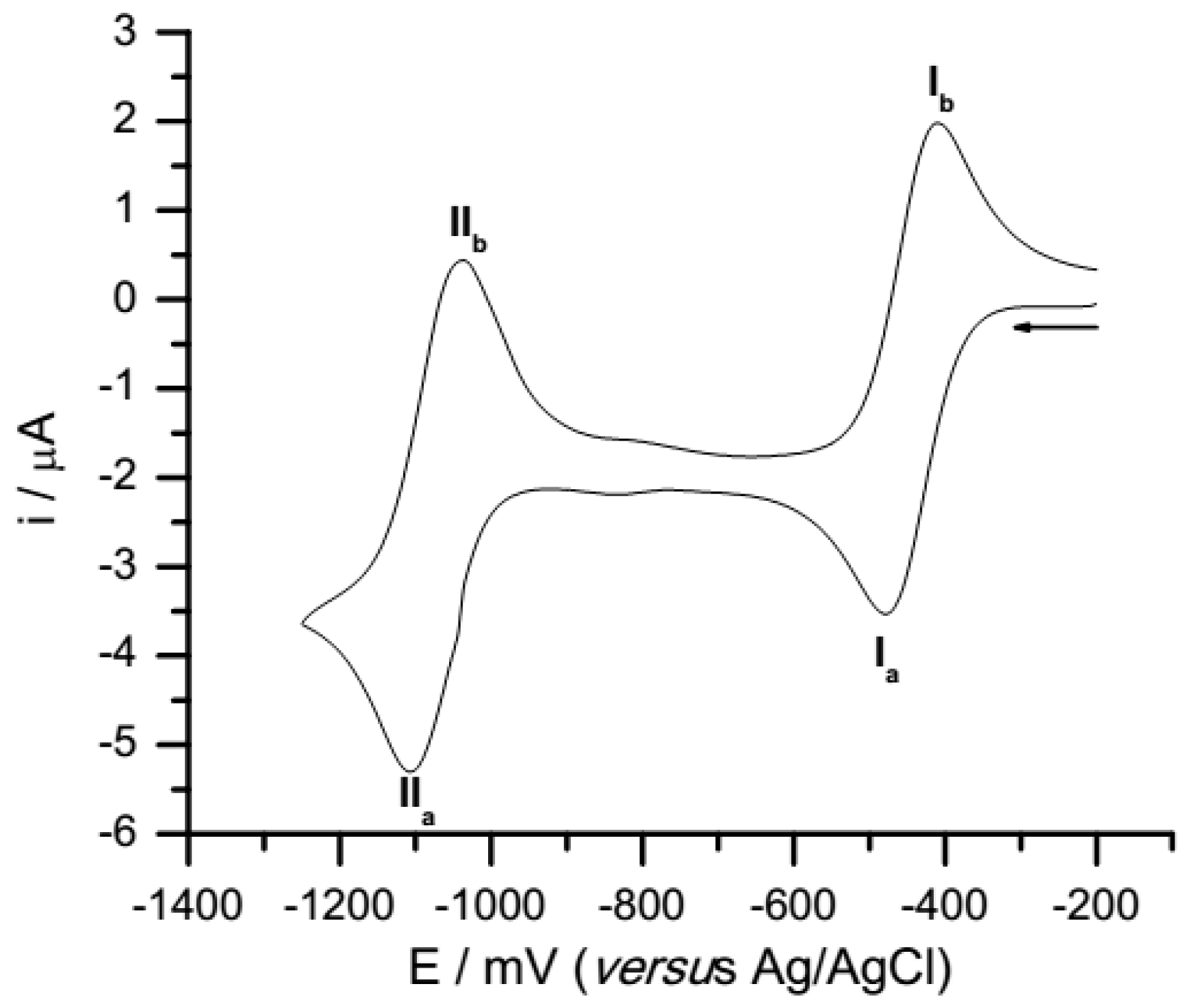
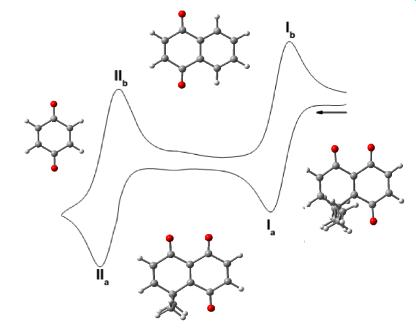
3.1. Cyclic Voltammetry
3.2. Theoretical First and Second One-Electron Reduction Potentials
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Hirst, J. Mitochondrial complex I. Annu. Rev. Biochem. 2013, 82, 551–575. [Google Scholar] [CrossRef] [PubMed]
- Oh, E.T.; Park, H.J. Implications of NQO1 in cancer therapy. BMB Rep. 2015, 48, 609–617. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Buettner, G.R. Thermodynamic and kinetic considerations for the reaction of semiquinone radicals to form superoxide and hydrogen peroxide. Free Radic. Biol. Med. 2010, 49, 919–962. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Singh, A. A review on mitochondrial restorative mechanism of antioxidants in Alzheimer’s disease and other neurological conditions. Front. Pharmacol. 2015, 6, 206. [Google Scholar] [CrossRef] [PubMed]
- Davidson, V.L. Protein-derived cofactors. Expanding the scope of post-translational modifications. Biochemistry 2007, 46, 5283–5292. [Google Scholar] [CrossRef] [PubMed]
- Berenjian, A.; Mahanama, R.; Kavanagh, J.; Dehghani, F. Vitamin K series: Current status and future prospects. Crit. Rev. Biotechnol. 2015, 35, 199–208. [Google Scholar] [CrossRef] [PubMed]
- Urra, F.A.; Martínez-Cifuentes, M.; Pavani, M.; Lapier, M.; Jaña-Prado, F.; Parra, E.; Maya, J.D.; Pessoa-Mahana, H.; Ferreira, J.; Araya-Maturana, R. An ortho-carbonyl substituted hydroquinone derivative is an anticancer agent that acts by inhibiting mitochondrial bioenergetics and by inducing G2/M-phase arrest in mammary adenocarcinoma TA3. Toxicol. Appl. Pharmacol. 2013, 267, 218–227. [Google Scholar] [CrossRef] [PubMed]
- Urra, F.A.; Córdova-Delgado, M.; Lapier, M.; Orellana-Manzano, A.; Acevedo-Arévalo, L.; Pessoa-Mahana, H.; González-Vivanco, J.M.; Martínez-Cifuentes, M.; Ramírez-Rodróguez, O.; Millas-Vargas, J.P.; et al. Small structural changes on a hydroquinone scaffold determine the complex I inhibition or uncoupling of tumoral oxidative phosphorylation. Toxicol. Appl. Pharmacol. 2016, 291, 46–57. [Google Scholar] [CrossRef] [PubMed]
- Dandawate, P.R.; Vyas, A.C.; Padhye, S.B.; Singh, M.W.; Baruah, J.B. Perspectives on medicinal properties of benzoquinone compounds. Mini Rev. Med. Chem. 2010, 10, 436–454. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.J.; Zhou, F.; Al-Kareef, A.M.Q.; Wang, H. Anticancer agents from marine sponges. J. Asian Nat. Prod. Res. 2015, 17, 64–88. [Google Scholar] [CrossRef] [PubMed]
- Mendoza, L.; Araya-Maturana, R.; Cardona, W.; Delgado-Castro, T.; Garcia, C.; Lagos, C.; Cotoras, M. In vitro sensitivity of Botrytis cinerea to anthraquinone and anthrahydroquinone derivatives. J. Agric. Food Chem. 2005, 53, 10080–10084. [Google Scholar] [CrossRef] [PubMed]
- Mendoza, L.; Ribera, A.; Saavedra, A.; Silva, E.; Araya-Maturana, R.; Cotoras, M. Action mechanism for 3 β-hydroxykaurenoic acid and 4,4-dimethylanthracene-1,9,10(4H)-trione on Botrytis cinerea. Mycologia 2015, 107, 661–666. [Google Scholar] [CrossRef] [PubMed]
- Pinto, A.V.; de Castro, S.L. The trypanocidal activity of naphthoquinones: A review. Molecules 2009, 14, 4570–4590. [Google Scholar] [CrossRef] [PubMed]
- Kumagai, Y.; Shinkai, Y.; Miura, T.; Cho, A.K. The chemical biology of naphthoquinones and its environmental implications. In Annual Review of Pharmacology and Toxicology; Insel, P.A., Amara, S.G., Blaschke, T.F., Eds.; Annual Reviews: Palo Alto, CA, USA, 2012; Volume 52, pp. 221–247. [Google Scholar]
- Kaledin, A.L.; Lian, T.Q.; Hill, C.L.; Musaev, D.G. A hybrid quantum mechanical approach: Intimate details of electron transfer between type-I CdSe/ZnS quantum dots and an anthraquinone molecule. J. Phys. Chem. B 2015, 119, 7651–7658. [Google Scholar] [CrossRef] [PubMed]
- Fuentes, N.; Martin-Lasanta, A.; de Cienfuegos, L.A.; Ribagorda, M.; Parra, A.; Cuerva, J.M. Organic-based molecular switches for molecular electronics. Nanoscale 2011, 3, 4003–4014. [Google Scholar] [CrossRef] [PubMed]
- Bunz, U.H.F. The larger linear n-heteroacenes. Acc. Chem. Res. 2015, 48, 1676–1686. [Google Scholar] [CrossRef] [PubMed]
- Van Noorden, R. A better battery. Nature 2014, 507, 26–28. [Google Scholar] [CrossRef] [PubMed]
- Er, S.; Suh, C.; Marshak, M.P.; Aspuru-Guzik, A. Computational design of molecules for an all-quinone redox flow battery. Chem. Sci. 2015, 6, 885–893. [Google Scholar] [CrossRef]
- Nagao, M.; Kobayashi, K.; Yamamoto, Y.; Hibino, T. Rechargeable PEM fuel-cell batteries using quinones as hydrogen carriers. J. Electrochem. Soc. 2015, 162, F410–F418. [Google Scholar] [CrossRef]
- Wang, H.; Hu, P.F.; Yang, J.; Gong, G.M.; Guo, L.; Chen, X.D. Renewable-juglone-based high-performance sodium-ion batteries. Adv. Mater. 2015, 27, 2348–2354. [Google Scholar] [CrossRef] [PubMed]
- Liehn, C.; Bouvet, M.; Meunier-Prest, R. Proton transfer versus hydrogen bonding: The reduction of ubiquinone Q2 incorporated in a self-assembled monolayer in unbuffered aqueous solution. ChemElectroChem 2014, 1, 2116–2123. [Google Scholar] [CrossRef]
- Staley, P.A.; Newell, C.M.; Pullman, D.P.; Smith, D.K. The effect of glassy carbon surface oxides in non-aqueous voltammetry: The case of quinones in acetonitrile. Anal. Chem. 2014, 86, 10917–10924. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.G.; Liu, N.; Liu, X.F.; Wang, Y.T.; Sun, L.J.; Xu, X.K.; Zhu, Z.H. The impact of “Effective pH” On the voltammetric behavior of p-benzoquinone and hydroquinone in acetonitrile. J. Electrochem. Soc. 2016, 163, H201–H204. [Google Scholar] [CrossRef]
- Namazian, M.; Zare, H.R.; Yousofian-Varzaneh, H. Electrochemical behavior of tetrafluoro-p-benzoquinone at the presence of carbon dioxide: Experimental and theoretical studies. Electrochim. Acta 2016, 196, 692–698. [Google Scholar] [CrossRef]
- Kim, R.S.; Chung, T.D. The electrochemical reaction mechanism and applications of quinones. Bull. Korean Chem. Soc. 2014, 35, 3143–3155. [Google Scholar] [CrossRef]
- Armendariz-Vidales, G.; Hernandez-Munoz, L.S.; Gonzalez, F.J.; de Souza, A.A.; de Abreu, F.C.; Jardim, G.A.M.; da Silva, E.N.; Goulart, M.O.F.; Frontana, C. Nature of electrogenerated intermediates in nitro-substituted nor-β-lapachones: The structure of radical species during successive electron transfer in multiredox centers. J. Org. Chem. 2014, 79, 5201–5208. [Google Scholar] [CrossRef] [PubMed]
- Guin, P.S.; Das, S.; Mandal, P.C. Electrochemical reduction of sodium 1,4-dihydroxy-9,10-anthraquinone-2-sulphonate in aqueous and aqueous dimethyl formamide mixed solvent: A cyclic voltammetric study. Int. J. Electrochem. Sci. 2008, 3, 1016–1028. [Google Scholar]
- Gupta, N.; Linschitz, H. Hydrogen-bonding and protonation effects in electrochemistry of quinones in aprotic solvents. J. Am. Chem. Soc. 1997, 119, 6384–6391. [Google Scholar] [CrossRef]
- Aguilar-Martinez, M.; Cuevas, G.; Jimenez-Estrada, M.; Gonzalez, I.; Lotina-Hennsen, B.; Macias-Ruvalcaba, N. An experimental and theoretical study of the substituent effects on the redox properties of 2-(R-phenyl)amine-1,4-naphthalenediones in acetonitrile. J. Org. Chem. 1999, 64, 3684–3694. [Google Scholar] [CrossRef] [PubMed]
- Iordache, A.; Maurel, V.; Mouesca, J.M.; Pecaut, J.; Dubois, L.; Gutel, T. Monothioanthraquinone as an organic active material for greener lithium batteries. J. Power Sources 2014, 267, 553–559. [Google Scholar] [CrossRef]
- Sanchez-Cruz, P.; Santos, A.; Diaz, S.; Alegria, A.E. Metal-independent reduction of hydrogen peroxide by semiquinones. Chem. Res. Toxicol. 2014, 27, 1380–1386. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.C.; Liu, T.Y.; Lee, S.W.; Jang, S.S. First-principles density functional theory modeling of Li binding: Thermodynamics and redox properties of quinone derivatives for lithium-ion batteries. J. Am. Chem. Soc. 2016, 138, 2374–2382. [Google Scholar] [CrossRef] [PubMed]
- Isegawa, M.; Neese, F.; Pantazis, D.A. Ionization energies and aqueous redox potentials of organic molecules: Comparison of DFT, correlated Ab Initio theory and pair natural orbital approaches. J. Chem. Theory Comput. 2016, 12, 2272–2284. [Google Scholar] [CrossRef] [PubMed]
- Namazian, M.; Almodarresieh, H.A.; Noorbala, M.R.; Zare, H.R. DFT calculation of electrode potentials for substituted quinones in aqueous solution. Chem. Phys. Lett. 2004, 396, 424–428. [Google Scholar] [CrossRef]
- Shamsipur, M.; Alizadeh, K.; Arshadi, S. Computational electrochemistry of aqueous two-electron reduction potentials of some amino-9,10-anthraquinone derivatives. J. Mol. Struct.-Theochem. 2006, 758, 71–74. [Google Scholar] [CrossRef]
- Bachman, J.E.; Curtiss, L.A.; Assary, R.S. Investigation of the redox chemistry of anthraquinone derivatives using density functional theory. J. Phys. Chem. A 2014, 118, 8852–8860. [Google Scholar] [CrossRef] [PubMed]
- Baik, M.H.; Friesner, R.A. Computing redox potentials in solution: Density functional theory as a tool for rational design of redox agents. J. Phys. Chem. A 2002, 106, 7407–7412. [Google Scholar] [CrossRef]
- Schmidt am Busch, M.; Knapp, E.W. One-electron reduction potential for oxygen- and sulfur-centered organic radicals in protic and aprotic solvents. J. Am. Chem. Soc. 2005, 127, 15730–15737. [Google Scholar] [CrossRef] [PubMed]
- Jaque, P.; Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Computational electrochemistry: The aqueous Ru3+ |Ru2+ reduction potential. J. Phys. Chem. C 2007, 111, 5783–5799. [Google Scholar] [CrossRef]
- Hodgson, J.L.; Namazian, M.; Bottle, S.E.; Coote, M.L. One-electron oxidation and reduction potentials of nitroxide antioxidants: A theoretical study. J. Phys. Chem. A 2007, 111, 13595–13605. [Google Scholar] [CrossRef] [PubMed]
- Marenich, A.V.; Majumdar, A.; Lenz, M.; Cramer, C.J.; Truhlar, D.G. Construction of Pourbaix diagrams for ruthenium-based water-oxidation catalysts by density functional theory. Angew. Chem. Int. Ed. 2012, 51, 12810–12814. [Google Scholar] [CrossRef] [PubMed]
- Cramer, C.J.; Truhlar, D.G. Implicit solvation models: Equilibria, structure, spectra, and dynamics. Chem. Rev. 1999, 99, 2161–2200. [Google Scholar] [CrossRef] [PubMed]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum mechanical continuum solvation models. Chem. Rev. 2005, 105, 2999–3093. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Revision A.01, Gaussian 09; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Sasaki, K.; Kashimura, T.; Ohura, M.; Ohsaki, Y.; Ohta, N. Solvent effect in the electrochemical reduction of p-quinones in several aprotic solvents. J. Electrochem. Soc. 1990, 137, 2437–2443. [Google Scholar] [CrossRef]
- Rongfeng, Z.; Evans, D.H. The current for a two-electron reaction is not necessarily twice that of a one-electron reaction. J. Electroanal. Chem. 1995, 385, 201–207. [Google Scholar] [CrossRef]
- Leventis, N.; Gao, X.R. In the presence of very fast comproportionation, sampled current voltammetry and rotating disk electrode voltammetry yield equal two versus one-electron limiting current ratios. Reconciliation through analysis of concentration profiles. J. Electroanal. Chem. 2001, 500, 78–94. [Google Scholar] [CrossRef]
- Aguilar-Martinez, M.; Macias-Ruvalcaba, N.A.; Bautista-Martinez, J.A.; Gomez, M.; Gonzalez, F.J.; Gonzalez, I. Review: Hydrogen bond and protonation as modifying factors of the quinone reactivity. Curr. Org. Chem. 2004, 8, 1721–1738. [Google Scholar] [CrossRef]
- Kirilyuk, I.A.; Bobko, A.A.; Semenov, S.V.; Komarov, D.A.; Irtegova, I.G.; Grigor'ev, I.A.; Bagryanskaya, E. Effect of sterical shielding on the redox properties of imidazoline and imidazolidine nitroxides. J. Org. Chem. 2015, 80, 9118–9125. [Google Scholar] [CrossRef] [PubMed]
- Vega, A.; Ramirez-Rodriguez, O.; Martinez-Cifuentes, M.; Ibanez, A.; Araya-Maturana, R. 8,8-diethyl-1,4,5,8-tetrahydronaphthalene-1,4,5-trione. Acta Cryst. E 2009, 65, o345. [Google Scholar] [CrossRef] [PubMed]
- Marenich, A.V.; Ho, J.M.; Coote, M.L.; Cramer, C.J.; Truhlar, D.G. Computational electrochemistry: Prediction of liquid-phase reduction potentials. Phys. Chem. Chem. Phys. 2014, 16, 15068–15106. [Google Scholar] [CrossRef] [PubMed]
- Barone, V.; Cossi, M. Quantum calculation of molecular energies and energy gradients in solution by a conductor solvent model. J. Phys. Chem. A 1998, 102, 1995–2001. [Google Scholar] [CrossRef]
- Cossi, M.; Rega, N.; Scalmani, G.; Barone, V. Energies, structures, and electronic properties of molecules in solution with the C-PCM solvation model. J. Comput. Chem. 2003, 24, 669–681. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Goodson, T.; Zimmerman, P.M. Achieving accurate reduction potential predictions for anthraquinones in water and aprotic solvents: Effects of inter- and intramolecular H-bonding and ion pairing. J. Phys. Chem. C 2016, 120, 22235–22247. [Google Scholar] [CrossRef]
- Fry, A. Computational studies of ion pairing. 9. The “Steric” Effect of tetraalkylammonium ions with electrochemically generated anions is not steric. Electrochem. Commun. 2013, 35, 88–90. [Google Scholar] [CrossRef]
- Marcus, Y.; Hefter, G. Ion pairing. Chem. Rev. 2006, 106, 4585–4621. [Google Scholar] [CrossRef] [PubMed]
- Rene, A.; Evans, D.H. Electrochemical reduction of some o-quinone anion radicals: Why is the current intensity so small? J. Phys. Chem. C 2012, 116, 14454–14460. [Google Scholar] [CrossRef]
- Atifi, A.; Ryan, M.D. Electrochemistry and spectroelectrochemistry of 1,4-dinitrobenzene in acetonitrile and room-temperature ionic liquids: Ion-pairing effects in mixed solvents. Anal. Chem. 2014, 86, 6617–6625. [Google Scholar] [CrossRef] [PubMed]
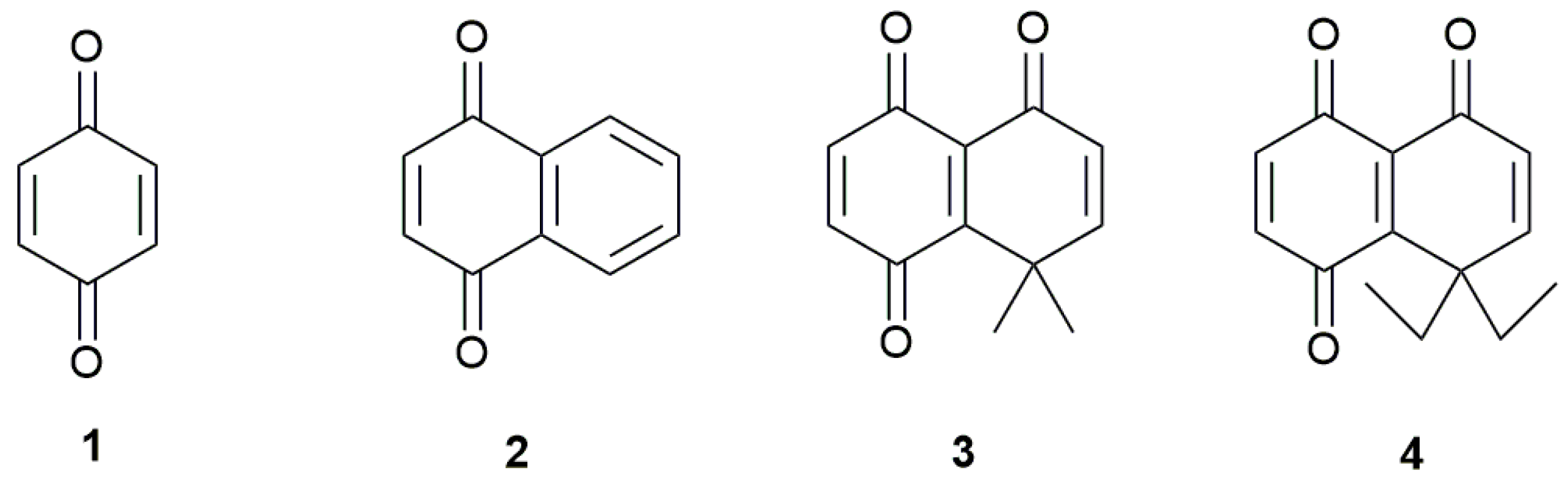
Sample Availability: Samples of the compounds 1, 2, 3 and 4 are available from the authors. |
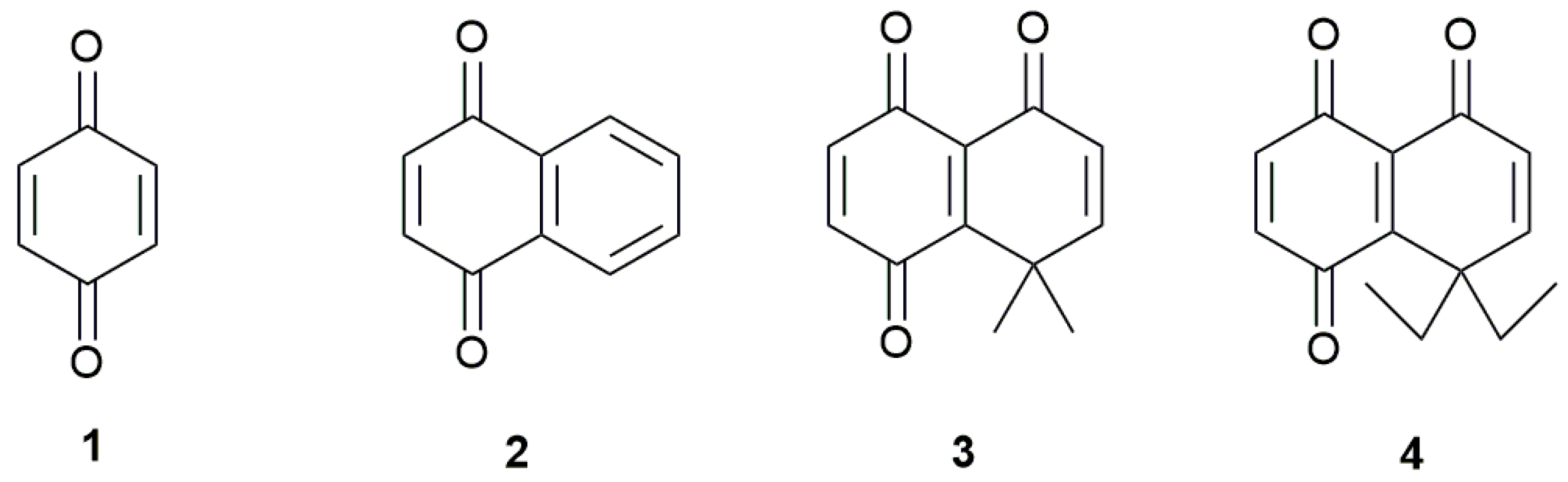

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | Semiquinone | Hidroquinone Dianion | E°I | E°II | ΔEpI | ΔEpII | ||
|---|---|---|---|---|---|---|---|---|
| EpcIa | EpaIb | EpcIIa | EpaIIb | |||||
| 1 | −479 ± 2 | −410 ± 6 | −1107 ± 4 | −1044 ± 4 | −445 | −1076 | −69 | −63 |
| 2 | −650 ± 3 | −571 ± 5 | −1066 ± 4 | −1007 ± 2 | −611 | −1037 | −79 | −59 |
| 3 | −390 ± 1 | −323 ± 9 | −1022 ± 4 | −923 ± 5 | −357 | −973 | −67 | −99 |
| 4 | −382 ± 2 | −317 ± 5 | −1033 ± 3 | −945 ± 3 | −350 | −989 | −65 | −88 |
| Compounds | ΔE°I–II (mV) | ln K |
|---|---|---|
| 1 | 631 | 25.2 |
| 2 | 426 | 17.0 |
| 3 | 616 | 24.6 |
| 4 | 639 | 25.6 |
| Compound | Ered | ||||
|---|---|---|---|---|---|
| BHandHLYP | M06 | PBE0 | E°(exp) | ||
| 1st RP | 1 | −644 | −497 | −583 | −445 |
| 2 | −901 | −725 | −697 | −611 | |
| 3 | −463 | −314 | −362 | −357 | |
| 4 | −483 | −306 | −366 | −350 | |
| 2nd RP | 1 | −2037 | −1769 | −1843 | −1076 |
| 2 | −2131 | −1842 | −1934 | −1037 | |
| 3 | −1950 | −1651 | −1624 | −973 | |
| 4 | −1954 | −1669 | −1660 | −989 | |
| Molecule | Q | SQ•− | HQ−2 | |||
|---|---|---|---|---|---|---|
| EHOMO | ELUMO | ESOMO | ELUMO | EHOMO | ELUMO | |
| 1 | −9.58 | −2.59 | −1.37 | 2.98 | 3.99 | 7.36 |
| 2 | −8.79 | −2.28 | −1.31 | 2.72 | 3.58 | 5.94 |
| 3 | −8.55 | −2.69 | −1.87 | 2.23 | 2.93 | 5.71 |
| 4 | −8.51 | −2.66 | −1.91 | 2.19 | 2.82 | 5.48 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martínez-Cifuentes, M.; Salazar, R.; Ramírez-Rodríguez, O.; Weiss-López, B.; Araya-Maturana, R. Experimental and Theoretical Reduction Potentials of Some Biologically Active ortho-Carbonyl para-Quinones. Molecules 2017, 22, 577. https://doi.org/10.3390/molecules22040577
Martínez-Cifuentes M, Salazar R, Ramírez-Rodríguez O, Weiss-López B, Araya-Maturana R. Experimental and Theoretical Reduction Potentials of Some Biologically Active ortho-Carbonyl para-Quinones. Molecules. 2017; 22(4):577. https://doi.org/10.3390/molecules22040577
Chicago/Turabian StyleMartínez-Cifuentes, Maximiliano, Ricardo Salazar, Oney Ramírez-Rodríguez, Boris Weiss-López, and Ramiro Araya-Maturana. 2017. "Experimental and Theoretical Reduction Potentials of Some Biologically Active ortho-Carbonyl para-Quinones" Molecules 22, no. 4: 577. https://doi.org/10.3390/molecules22040577
APA StyleMartínez-Cifuentes, M., Salazar, R., Ramírez-Rodríguez, O., Weiss-López, B., & Araya-Maturana, R. (2017). Experimental and Theoretical Reduction Potentials of Some Biologically Active ortho-Carbonyl para-Quinones. Molecules, 22(4), 577. https://doi.org/10.3390/molecules22040577