1. Hydrogen Bonds in Protein Structures
A hydrogen bond (H bond) is established through the sharing of an H atom that is covalently attached to an electronegative donor (D), with a different electronegative acceptor atom (A), and can be written as D-H…A. H bonds are one of the major drivers that participate in the correct folding and organization of secondary and tertiary structural elements in proteins. In addition to their fundamental role in structure, H bonds are also intimately involved in protein:ligand binding interactions and also form the basis for most types of enzyme-catalyzed reactions [
1]. Proteins contain many different kinds of H bond donors and acceptors, from the backbone amide and carbonyl groups, solvent, to the side chains of most amino acids residues. H bonds are directional, which is the driving force behind the specificity of molecular recognition processes in ligand binding [
2].
Studies in the 90s aimed at investigating the H bond frequency between H bond donors/acceptors and have been carried out on a representative set of protein crystal structures available at the time. It was observed that most donors and acceptors engage in H bonds, with only a small percentage remaining as unsatisfied atoms. Many amino acid side chains can and do make multiple H bonds, e.g., Lys, Arg, Asp, while others do not. For protein structure refinement and prediction calculations this is an important consideration to include [
3].
From a theoretical chemistry point of view, the H bond interaction energy can essentially be described as being composed of a combination of covalent and weak interactions, e.g., van der Waals forces [
2,
4]. The energy associated with an H bond is 2–10 kcal/mol. While this may seem insignificant it is important to remember that these bonds are flexible and dynamic in that they are constantly broken and reformed under a range of temperatures and physiological conditions [
2]. There is quite a large variation in H bond properties as observed in protein structures, especially in bond angles when comparing secondary structure elements to side chain characteristics. The D…A distances are typically between 1.7 and 2.4 Å, and the angle at the H atom can be anywhere from 130° to 170°, compared to ~150° at the acceptor [
5]. As H bonds are difficult to directly observe, they are generally inferred when the donor-acceptor distance is less than ~3.5 Å, and the angles at the donor/acceptor are >90° [
1]. Simple H atom sharing between two electronegative atoms represents the most common type of H bond, however, it is now accepted that there are additional types of weaker H bonds; e.g., C-H groups as donors, N-H…S, and C-H…π interactions with aromatic amino acid residues as acceptors [
1,
6]. A study conducted using NMR spectroscopy, DFT calculation, and data mining of high resolution protein crystal structures revealed that C-H…π type interactions are in fact quite common in proteins. These interactions are H bond-like in nature and occur between the methyl groups of Ile, Leu, and Val and the delocalized π electrons in aromatic residues. The results showed an average of four of these for every one hundred residues and are a functionally important part of protein structures [
7]. H bonds in macromolecules are challenging to observe and classify, accordingly the criteria for identifying them in proteins are quite broad, as explained above. Another type of H bond that is commonly found in protein structures is the bifurcated H bond. In this case the slightly positively charged H atom is delocalized between two acceptor atoms. These are most often seen between Ser/Thr side chains with Asp/Glu as acceptors [
8]. This indicates that H bonds cannot be identified only based on geometric criteria and that some additional structural or chemical knowledge is needed to fully understand all the H bonds in a protein. In recent years, bifurcated H bonds have also been directly observed in neutron protein crystal structures between Tyr and solvent, and Lys and Thr [
9,
10].
H atoms are abundant in macromolecules, comprising roughly half of the elements that proteins are composed of (C, H, N, O, S). However, despite their abundance and structural-functional importance, including but not limited to H bonding, electrostatic interactions, solvent coordination and solvent activation, H atoms and their interactions are very difficult to directly observe. This is especially true for X-ray crystallographic studies where information regarding H bonds are usually derived based on their heavy atom neighbor positions and the distances between them. The direct determination of H atom positions in protein crystal structures is restricted mainly due to their low X-ray scattering power, their inherent mobile nature, and limited crystallographic resolution [
11].
2. Neutron Crystallography and Hydrogen Atoms
Neutron protein crystallography (NPX) is well-suited to the determination of the three-dimensional structures of proteins due to their sensitivity to the light atom H and lack of radiation damage at room temperature. NPX is conceptually very similar to standard X-ray crystallography, and crystallographic resolution is understood in the same way. The main difference between the methods lies in how neutrons are scattered from atoms. X-rays are scattered from the clouds of electrons around atomic nuclei, and the magnitude depends on the atomic Z number leading to heavy atoms scattering X-rays better than light atoms (
Table 1). In practice this most often means that H atoms are invisible, except for ultra-high resolution structures that are exceptionally well-ordered, i.e., display low crystallographic B factors. Neutrons are scattered from atomic nuclei and the magnitude is independent of the Z number.
Table 1 shows neutron scattering lengths for the most common atom types found in proteins. Coherent scattering lengths of neutrons are very similar in magnitude for C, N,
1H (and
2H or D), and O, making it as easy to observe C as H (or D) [
11,
12].
In practice this means that even at medium (2.0–2.5 Å crystallographic resolution) it is routine to readily observe the three dimensional positions of H atoms. By extension this means that neutrons are also used to study H bonds [
14]. The isotope of
1H, deuterium (
2H or D), has superior neutron scattering characteristics, making it optimal to replace H with D whenever possible (
Table 1). The presence of
1H presents several practical problems for crystallographic data collection. The large incoherent cross section (
Table 1) contributes to background as the incoherently scattered neutrons do not contribute to measured Bragg reflections. In addition, the coherent scattering length is negative and leads to troughs instead of peaks and can produce cancellation of positive peaks of adjacent atoms, complicating and confusing analysis of nuclear density maps when
1H is present. These effects can be minimized or even eliminated by D exchange (partial deuteration) or expression of the target protein under fully deuterated conditions (perdeuteration). Deuteration in general leads to higher quality maps and enables less ambiguous data analysis and structure interpretation [
11,
13].
Out of all H atoms in a protein, ~25% are titratable and can be exchanged by vapor H/D exchange or by soaking crystals in D
2O-containing solutions. These include H found in the bulk solvent, ordered solvent, polar and charged amino acid side chains, and the protein backbone. Neutron structures derived from H/D exchanged proteins represent the vast majority of deposited structures in the PDB [
11]. The remaining 75% of H atoms are non-exchangeable, typically found attached to carbons and have to be incorporated as D during protein expression. This is routinely done by growing
E. coli cultures for protein expression in deuterated minimal media, such as M9, composed of stock solutions and components dissolved in D
2O [
11]. For the highest level D incorporation (~99%) it is best to use a perdeuterated carbon source, e.g., perdeuterated glycerol or glucose. This is very costly however and ~85% incorporation can be achieved by using deuterated M9 minimal media but supplementing with an unlabeled carbon source [
15].
Cultures can be adapted in progressively increasing amounts of D
2O or grown to high densities and switched to deuterated media just prior to induction of protein expression. All proteins are then extracted and purified as usual, under hydrogenous conditions, with lost D atoms back-exchanged at a later point. Depending on the strategy chosen the resulting protein will have varying degrees and distribution of D incorporation. While perdeuteration is preferable from a theoretical point of view, there are practical drawbacks that make it difficult to achieve in practice and the low numbers of neutron structures determined from perdeuterated proteins reflects this. Although perdeuterated proteins are nearly identical in structure to their hydrogenated counterparts, the proteins are often found to have altered chemical properties, such as reduced solubility and stability. Deuterated cultures also grow slowly, and perdeuterated protein yields are generally lower. Perdeuteration also introduces the need to fine-screen or rescreen crystallization conditions under perdeuterated conditions [
16].
As there is no radiation damage with neutrons used for protein crystallography applications, it is routine to collect data at room temperature. This feature makes sample preparation for data collection simple as crystals can be mounted in quartz capillaries and subjected to H/D exchange in the capillary prior to exposure to neutrons. To achieve this, crystals are mounted in capillaries and a liquid plug of deuterated mother liquor is injected on either side of the crystal prior to sealing the capillary. The pitfalls of finding protective cryogenic solutions and the actual freezing of crystals are also avoided [
11,
16].
Globally there are only a handful of instruments available that are specifically optimized for macromolecular crystallography. In the USA there are currently two instruments, both located at Oak Ridge National Laboratory (ORNL, Oak Ridge, TN, USA). The IMAGINE instrument is located at the HFIR reactor source, while MaNDi is found at the Spallation Neutron Source. In Japan there is the iBIX instrument at the Japan Proton Accelerator Research Complex (J-PARC, Tokai, Japan). In Europe there are currently two instruments operating at reactor neutron sources (
Figure 1). Depending on the source and how the instrument is set up, data can be collected with monochromated (single wavelength) method or with a white Laue (multi-wavelength) method. The LADI-III (ILL, Grenoble, France) instrument uses a quasi-Laue spectrum, while BIODIFF (MLZ, Munich, Germany) uses a monochromated beam. However data is collected, the resulting neutron diffraction data sets from different instruments are remarkably comparable and can be refined using standard crystallographic software packages. After careful crystallographic refinement and analysis one can readily observe the three-dimensional structural details of H/D atoms, H bonds, water molecules and their interactions, the charged state of amino acids residues and their interactions, and the details of ligand binding. All this information is essential for the study of enzyme mechanism and drug binding, making neutron protein crystallography a powerful tool in structural biology [
17,
18].
In general the technique is still underutilized, with only ~100 PDB entries resulting from neutron diffraction studies. This is largely due to three major bottlenecks: (1) neutron scattering is a flux limited technique and accordingly requires long data collections times, ~5–20 days depending on the instrument; (2) quite large crystal volumes are required for most instruments (>0.5 mm
3 on average); and (3) there are only a handful of instruments available worldwide to external users. Advances in sources, beamlines, and detector technologies are easing the way forward with much shorter data collection times from smaller crystals [
11,
16,
17,
18].
This review presents a number of topical examples of different kinds of structural and functional information that can be obtained with neutron crystallography, specifically how they relate to the study and importance of H bonds. Examples included are meant to span a broad area in structural biology and include systems where ordered solvent and H bonded solvent network are intimately linked to catalysis. Fluorescent and chromoproteins are also shown as excellent cases where optical properties are solely determined by changes in H bonding around a chromophore. Several enzymology examples are discussed in a later section, highlighting the importance of knowledge of H bonds and being able to determine how they change during catalysis. Due to the strong scattering of D, neutrons have also been used to “see” exotic solvent species such as the elusive hydronium ion (H3O+ or D3O+) and protons (H+ or D+ containing no electrons and therefore invisible with X-rays), an important proposed partner in certain kinds of enzyme-catalyzed reactions. Concluding remarks offer a forward-looking perspective on the place of neutrons in current-day structure biology.
4. Unusual H Bonds and Solvent Species as Observed in Neutron Crystal Structures
4.1. Photoactive Yellow Protein: Short H Bonds vs. Low-Barrier H Bonds
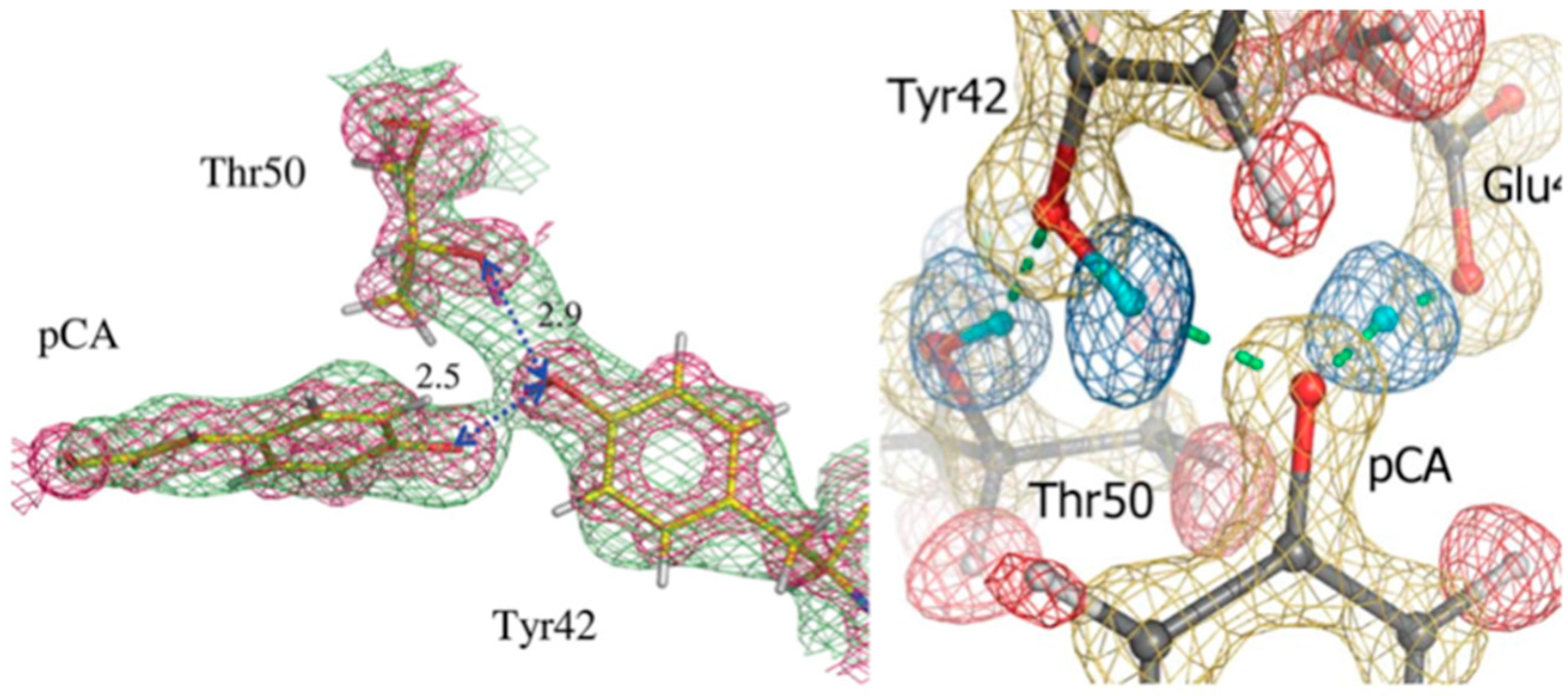
Photoactive yellow protein (PYP) is a soluble 14 kDa protein from
Halorhodospira halophila that absorbs light via its para-coumaric acid (pCA) chromophore. The absorption spectra for PYP is equivalent to the negative photo-dependent response of the organism, implicating PYP in its biological behavior. PYP has a reversible photocycle during which pCA undergoes
trans-to-
cis isomerization, causing remodeling of the H bonded network and proton transfer processes. Isomerization of pCA culminates in large structural changes of N-terminal regions of the protein. This means that H bonding is essential for photocycle kinetics in the protein. In the ground state pCA is stabilized by two H bonds between the pCA phenolate oxygen atom and residues Tyr42 and Glu46. High-resolution X-ray structural studies revealed these bonds to be rather short at 2.49 Å and 2.58 Å, respectively. It was suggested that these bonds represent short H bonds (SHB) [
27].
As the H bonds become shorter there is a lengthening of the donor O-H covalent bond. The barrier decreases as the disorder over two protonation sites collapse to a centered single-well potential, the so-called low-barrier H bond (LBHB). In an LBHB the proton is shared between donor and acceptor atoms, making it behave like a covalent bond [
28].
Neutron crystallography was used to study PYP in ground state in order to investigate these short H bonds and to elucidate H atom positions in the area of the hydrophobic core [
28,
29]. The first neutron crystal structure of PYP was determined to 2.5 Å resolution, from a ~0.8 mm
3 crystal that was H/D exchanged. From this first structure it was possible to elucidate some of the details around the pCA pocket, with pCA being deprotonated and the phenolate oxygen accepting H bonds from Glu46 and Tyr42. Tyr42 in turn is H bonded to Arg52. The observed H bond lengths to pCA were consistent with the high-resolution X-ray work, remarkable given the moderate resolution (2.5 Å) of the neutron structure (
Figure 5) [
29].
In 2009 a much higher resolution neutron structure of PYP was reported, to 1.5 Å resolution. Now it was possible to fully investigate the short H bonds around pCA to see if they could correspond to LBHB [
28]. From detailed investigations of omit F
o − F
c and 2F
o − F
c nuclear density maps it was found that the SHB between Glu46 and pCA could be a LBHB, while the Tyr42-pCA H bond was better classified as a short ionic H bond (SIHB).
It is thought that the deprotonated/charged pCA represents an isolated charge in the otherwise hydrophobic pocket of PYP with Arg52 acts as a counter-ion. However, in the high-resolution neutron structure this Arg was neutral. It was postulated that the SIHB and LBHB compensate for this and provide structural stability to PYP. LBHBs have been implicated in other systems for being part of catalysis and/or stabilization of intermediates. In this case the proposed LBHB may stabilize an isolated charge and participate in proton transfer during the photocycle of PYP [
28]. Comparison of the two studies also shows, unsurprisingly, that higher resolution neutron crystallographic data allows for more accurate and detailed studies.
4.2. Unusual H Bonds as Seen with Neutrons
Recent neutron structures have revealed a wide variety of H bonding details, including LBHBs, SIHBs, and resonance-assisted H bonds. Despite over 40 years of structural, mechanistic, and computational work, there remains much to understand about this fundamental, yet varied interaction, found in protein structures and enzyme mechanisms.
To study whether a LBHB was involved in catalysis, neutron studies were carried out on a serine protease, porcine pancreatic elastase (PPE), in complex with a peptidic inhibitor FR130180 [
30]. The residues His57-Asp102-Ser195 compose the catalytic triad and are conserved in the serine proteases. In the first step of catalysis, the -OD1 of Ser195 makes a nucleophilic attack on a substrate carbonyl group. Next, a tetrahedral intermediate is formed through a covalent bond to Ser195 and the substrate carbonyl group. The tetrahedral intermediate is stabilized through electrostatic interactions that involve H bonds to backbone amides of Ser195 and Gly193. Together this forms a proposed oxyanion hole. Ser195 is activated for nucleophilic attack through a SSHB between side chains of His57 and Asp102 [
31]. The tetrahedral intermediate is resolved through proton transfer from His57 to the leaving group, leaving an acyl-enzyme intermediate. The H bond between Ser195, His57, and Asp102 has been proposed to be a LBHB and has been investigated with high resolution X-ray crystallography and NMR spectroscopy, in different types of proteases such as trypsin and subtilisin. The studies were inconclusive and in some cases instead showed a SIHB where the short H bond (~2.6 Å) is observed. However, the shared H atom remains with the donor, and is not found equidistant between donor/acceptor as expected in a LBHB [
32,
33].
FR130180 binds to PPE with the same tetrahedral transition state intermediate as seen with substrate. The nuclear density maps show that His57 is protonated (charged) and involved in a short H bond with Asp102, however the shared D density is clearly located on -ND1 of His57, and is most likely a SIHB. In addition the neutron data showed the carboxyl oxygen (O32) of FR130180 is present as an oxygen anion (oxyanion) and was the first oxyanion in a tetrahedral intermediate observed at an oxyanion hole. At a substrate binding subsite, S4, a π-cation interaction is observed between Arg217 and the benzoyl ring of FR130180. Finally, there is an H bond between the –OH group of Thr175 and the carboxyl group of benzoic acid in the inhibitor. In this interaction, the carboxyl group is protonated and the O-D distance is longer than expected (1.21 vs. 1.02 Å), while being only 1.41 Å away from the -OD1 of Thr175. This configuration is consistent with another type of very strong H bond, the resonance assisted H bond (RAHB) [
30].
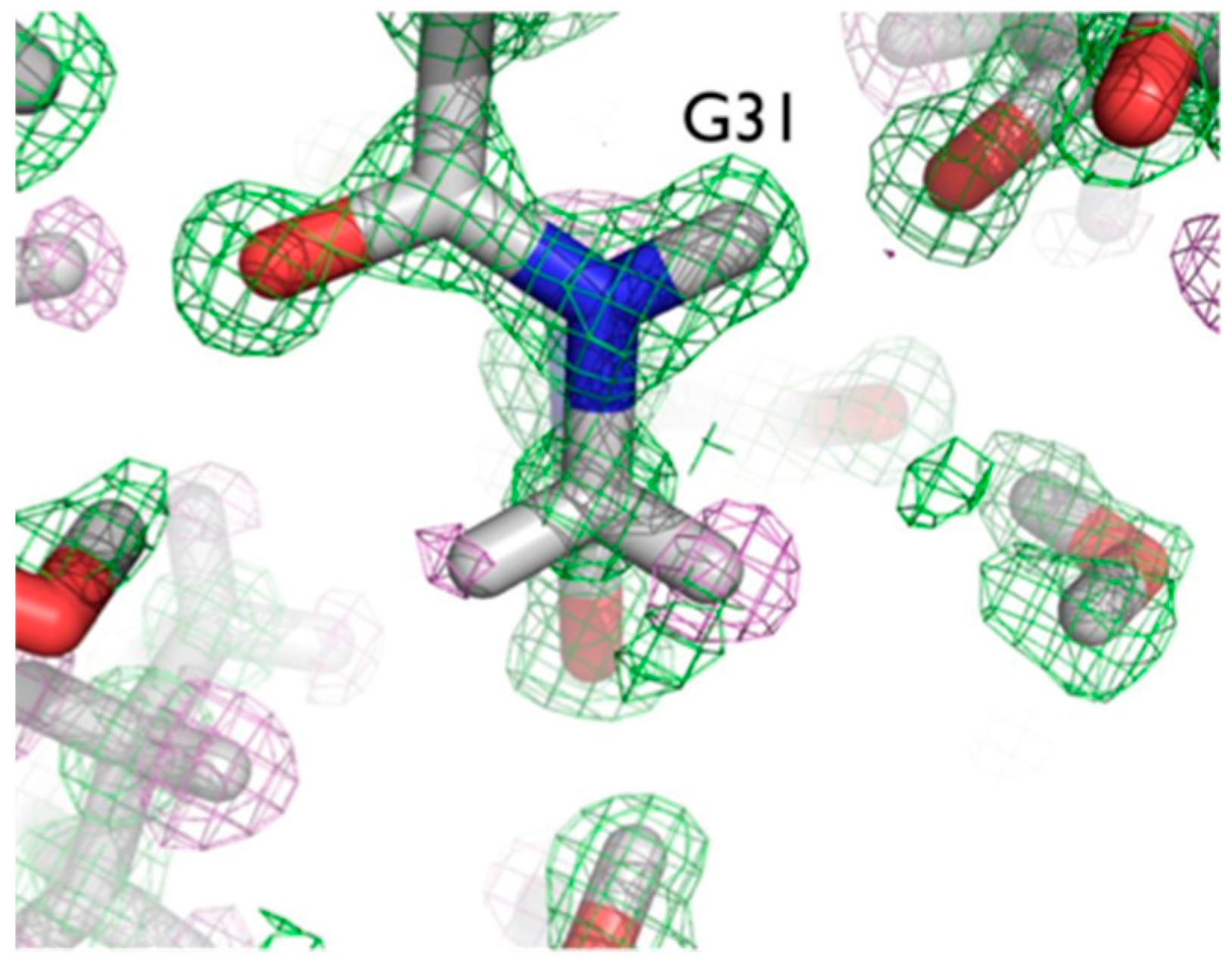
The ultra-high resolution (1.1 Å) of the crambin neutron structure made it possible to clearly resolve H atom positions and the degree of exchange with D [
20]. More accurately than in other neutron structures resolved to date, the relative occupancies of H/D at exchangeable positions could be refined, and solvent molecules and side chains could be identified and oriented. Of note were two particular regions of the protein with unexpected characteristics. In Gly31, the two Hs appear to have different degrees of exchange, based on the observed nuclear density for the two H atoms (
Figure 6).
One interpretation of this is that there was partial exchange of the Cα-H against the deuterated solvent, leading to partial D occupancy, and the possibility of Cα-H…O H bonding. This can be attributed to differing chemical environments in the crystal, corroborated by solution state NMR studies.
The two Cα-H atoms in Gly31 were observed to have distinct NMR proton chemical shifts of 3.96 and 5.56 ppm in solution, reflecting different chemical environments for the two alpha hydrogens [
34]. Since Gly does not have a side chain, they are relatively strong carbon acids (p
Ka ~ 22), comparable to the side chain hydroxyl groups of Ser and Thr (p
Ka ~ 18–20).
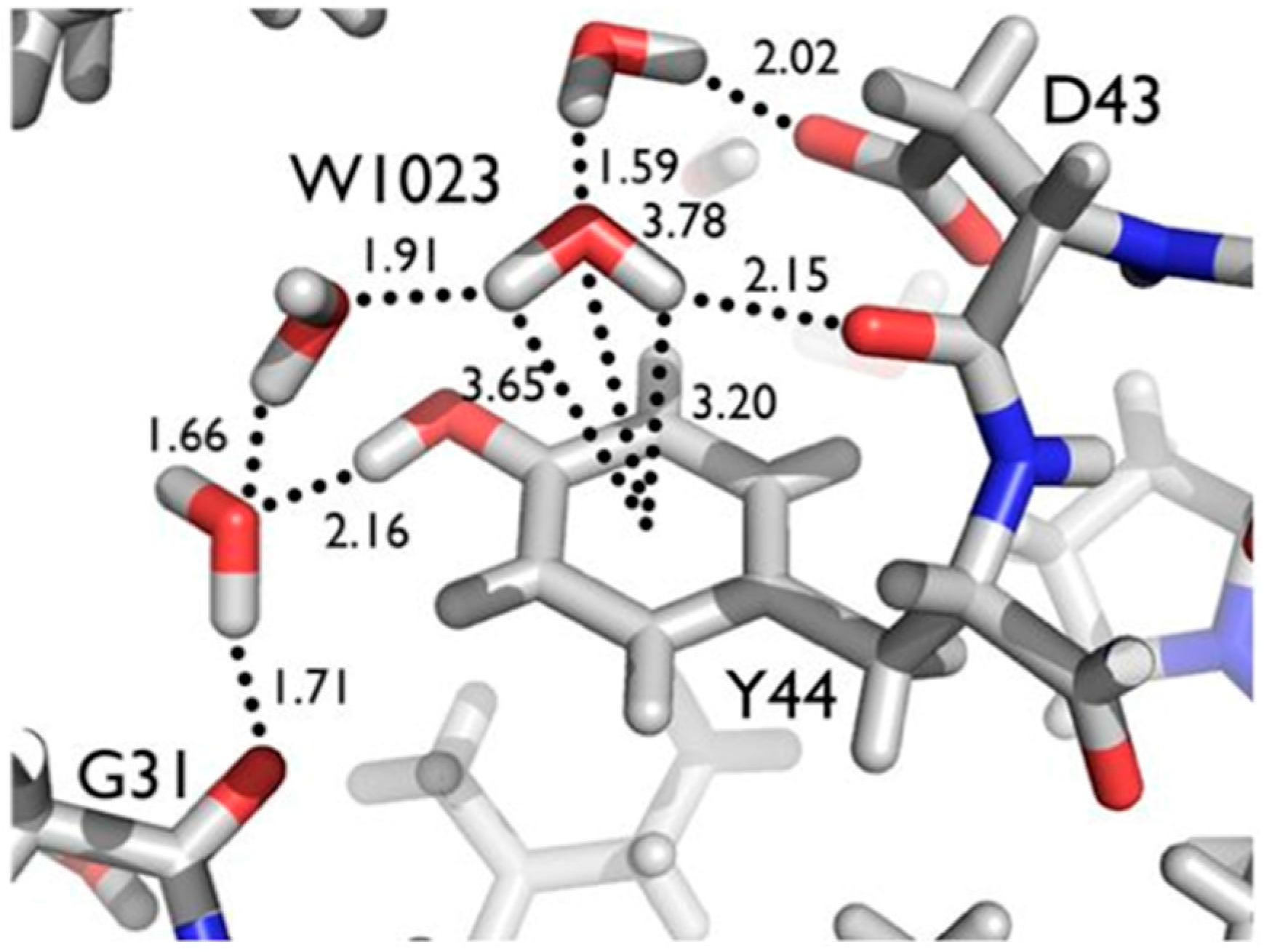
Another series of potential Cα-H…O H bonds were observed in the β-strand region of crambin, e.g., between the carbonyl oxygen of Thr1 and the Cα-H of Ile34. These observations add to the evidence that these weak, but numerous, stabilizing H bond interactions serve to contribute to secondary and tertiary structure stability. Secondly, Tyr44 in crambin is a well-ordered surface residue that is solvated by a network of waters on the one side, and makes a van der Waals interaction with Ile33 on the other side. As shown in
Figure 7, one of these ordered water molecules, W1023, has its D1 atom pointing towards the center of the aromatic ring, indicating a potential O-H…π H bonding interaction [
20]. With improvements in X-ray and neutron instrumentation and crystallization approaches, ultra high-resolution crystallography will allow for H bonding interactions to be studied in greater detail and to better accuracy.
4.3. Protons (H+), Hydroxides (OH−), and Hydronium Ions (H3O+) in Proteins
Water (H2O), protons (H+), hydroxide (OH−) and hydronium ions (H3O+) are important players in many chemical and biochemical processes. They may be involved in a variety of different enzyme catalytic processes but are difficult, or in the case of H+, impossible to directly observe using X-rays. In electron density maps derived from X-ray crystallography, most of these hydrated proton forms are indistinguishable from each other as only the oxygen peak of these species are visible in electron density maps. Neutrons provide the appropriate tool to “see” the light atoms in solvent and make it possible to discern exotic solvent species from ordinary water.
Structure-function studies of xylose isomerase (XI) using neutrons made it possible to observe the first hydronium, hydroxyl, and proton ions in a biological system [
35,
36]. XI from
Streptomyces rubiginosus is a 172 kDa homotetramer with 2 metal sites that bind divalent cations, usually magnesium, in the active site. XI catalyzes the interconversion of
d-glucose to
d-fructose, or
d-xylose to
d-xylulose, and is of considerable industrial interest for production of sweeteners and in the biofuel field [
37]. Metal site M1 is coordinated by a number of waters, and Glu and Asp amino acid residues. Metal site M2 is coordinated by His, Asp, Glu side chains and a catalytic water molecule. During catalysis there are multiple steps: substrate binding and coordination adjacent to the two metal sites, sugar ring opening, movement of a H from sugar C2 to sugar C1 through a proposed hydride shift or donation of a hydride anion, and ring closure. The process culminates with the product leaving the active site. These steps necessarily involve complex changes in water and amino side chain positions, as well as movement of H atoms through protonation/deprotonation of water and active site residues [
35,
38].
Using a combination of different metal substitutions and by soaking in substrate or product molecules, it was possible to trap various intermediate stages that occur during catalysis by XI. In one such structure of the natural magnesium-containing enzyme in complex with the product xylulose, a hydroxyl anion was observed in place of the catalytic water at the M2 site. This observation supports the idea that the catalytic water molecule donated a proton during the isomerization step of catalysis and gave important clues as to the molecular events that occur upon isomerization [
35].
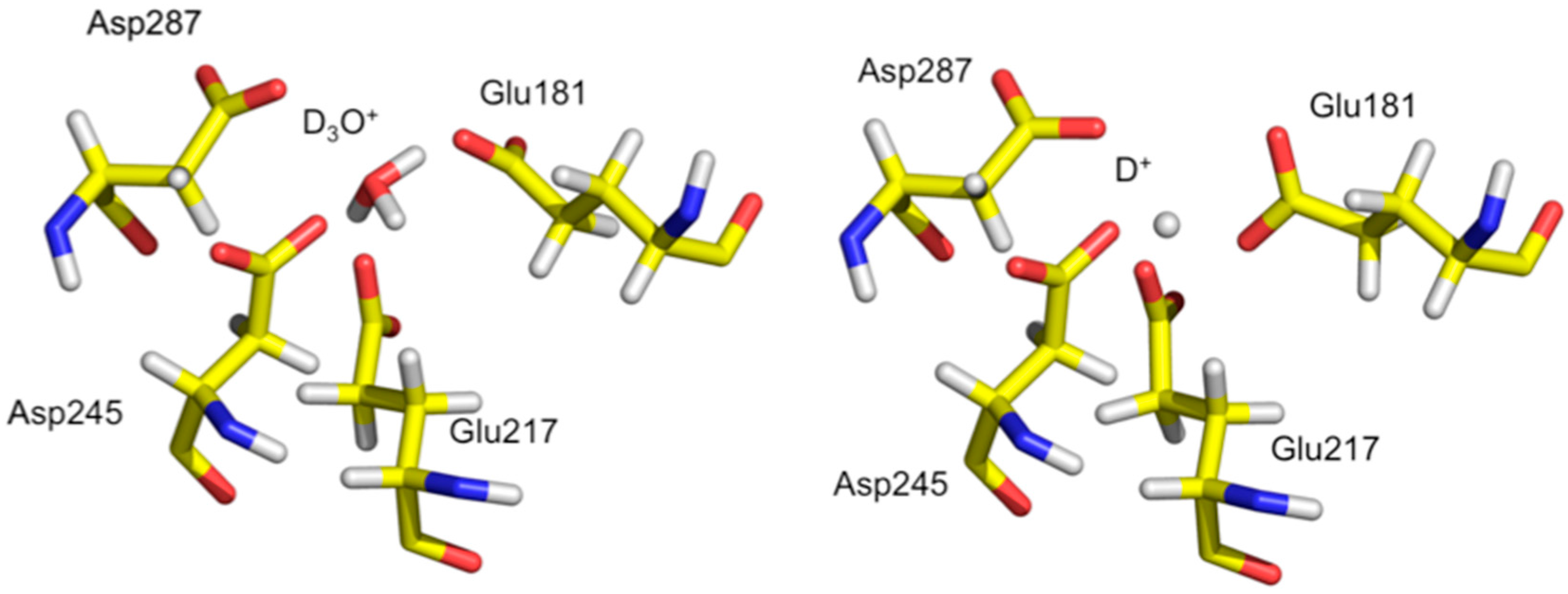
Subsequent neutron studies looked at metal free XI at both pH 5.9 and 7.7 to help understand why the enzyme is inactive at low pH. XI is most active at ~pH 8, however for industrial applications it is desirable to engineer the enzyme to also be active at lower pH. It is known that at low pH the metals from M1 and M2 are expelled due to electrostatic changes in the area and this was also observed in the neutron crystal structures. Neutron studies were conducted to study how the active site looks in the absence of metal, and how the charged state of residues change as a function of pH.
Analysis of the neutron structure at pH 7.7 showed that the metal at position M1 is replaced by a hydronium molecule (
Figure 8) [
36]. The hydronium ion templates the appropriate size and shape for metal binding at M1 and is coordinated by Glu181, Glu217, Asp245, and Asp287. In the pH 5.9 structure the hydronium ion has been dehydrated to leave a proton only in the M1 site. The proton is involved in a very short trifurcated H bond to coordinating residues and located closest to Glu217. At low pH the amino acid residues have collapsed around the site and does not have the proper geometry to accept an incoming metal ligand. Soaking in solutions of metal salts were also unsuccessful, providing further evidence that the low pH form of M1 is not able to bind metal, thus explaining the lack of activity of XI at low pH [
37].
4.4. The Role of D3O+ Ions in Redox Processes
Rubredoxins are highly thermostable, small, mononuclear iron-sulfur cluster proteins found in both prokaryotes and eukaryotes. As redox proteins they serve as an important model system for the study of proton transfer in catalysis. Iron-sulfur proteins vary greatly in their redox potentials, reflecting differences in solvation, electrostatics, and H bonding. Neutron studies were initiated to obtain high resolution structures of both oxidized and reduced forms on rubredoxin, in order to study water molecules, H bonding, and to investigate the charged state of chemical groups in the protein as a function of oxidation state [
39].
Inspection of the ~1.3 and 1.4 Å resolution nuclear density maps revealed several interesting structural features involved with protein stability in both oxidized and reduced forms of the protein. Four hydronium ions were observed in both oxidized and reduced rubredoxin structures. The first molecule, HYD1, was located near Leu51 amide group and involved with amide-imide tautomerization. The second one, HYD2, is found by Pro44-Pro19-Ser46. HYD2 is ~10 Å away from the iron-sulfur cluster and forms water-mediated H bonds to iron-sulfur coordinating Cys and adjacent Ser residues. In this way HYD2 is at the center of a tight H bonded group that connects water to the metal cluster coordinating residues. HYD3 also found near the iron-sulfur cluster (~9 Å), but near Pro25-Ser24. The last one, HYD4, is H bonded to Ala16 carbonyl group. Occupancy refinement of one of the D atoms indicates water/hydronium equilibrium.
In summary then, three of the four hydronium ions are observed close to the protein main chain and another one forms the core of an intricate H bonded water network. One of these hydronium ions is located near main chain amide of Leu51 and is probably involved with redox driven tautomerization of amide to imide forms of the residue. In addition to the several hydronium ions seen in rubredoxin, there were two additional important protonation shifts observed, in residues Glu47, Asp13, and Asp15. Glu47 was seen to be unprotonated in the reduced form while being protonated in the oxidized form. This shifting D atom is also associated with HYD2. Asp13 on the other hand is unprotonated in the oxidized form while Asp15 is protonated. The observation of four hydronium ions and three shifting residues in close proximity to the iron-sulfur cluster suggest that these entities and surrounding solvent play important roles in charge transfer processes that occur in the change between oxidization and reduction in rubredoxin and associated redox processes [
39].
With the growing use of neutron protein crystallography, it can be expected that more of these unusual solvent types will be directly observed and shed light on intricate and complex protein functions.
5. Structural Enzymology and H Bonds
Hydrogen atoms are central to enzyme mechanisms and catalysis often relies on the movement of H atoms, either through proton shuttling or hopping or through the breaking/making of H bonds. There are many examples of neutron crystallography helping to elucidate enzyme mechanistic questions and all cannot be covered here. In this section a few examples are discussed where using neutrons made a significant contribution with regards to hydrogen atoms and their interactions.
5.1. Cytochrome C Peroxidase
Heme peroxidases are widespread in nature and support a wide range of biological functions. They employ peroxide to oxidize a number of different substrates. Catalysis by heme peroxidases require the formation of two transient, highly oxidized ferryl (Fe(IV)) intermediates, the so-called Compounds
I and
II. These intermediates are applied widely in other oxygen-dependent catalytic heme-containing enzymes, including the cytochrome P450s, nitric oxide synthases, terminal oxidases and the heme dioxygenases [
40]. It is important to understand the protonation state around the heme as this determines the reactivity and involvement of the enzyme.
Compound
I and Compound
II form sequentially during catalysis and represent different Fe-IV (ferryl) species. They are different in their oxidation state, specifically the porphyrin ring. It is thought that the species are Fe(IV)=O or Fe(IV)-OH and it has been challenging to characterize the identities definitively [
40,
41]. Photo-reduction of the iron has been observed in both Raman spectroscopy and X-ray crystallography, rendering the interpretation of the oxidation states derived from these methods unreliable. Neutron crystallography can provide the high resolution, unambiguous structural information required to characterize the iron oxidation state, without inducing photo-reduction in the samples [
40,
41]. To unravel the mechanism of the peroxidases, two related heme peroxidases have been studied with neutron crystallography. Cytochrome c peroxidase (Ccp) oxidizes the small protein cytochrome c by hydrogen peroxide, and the mechanistically and structurally related ascorbate peroxidase (APX), the oxidation of ascorbate [
40,
41].
Cytochrome c peroxidase is a well-studied model system for heme-containing oxidases, which play a key role in many metabolic processes. Various species along the reaction path can be characterized by spectroscopic methods and trapped by cryo-cooling for crystallographic structure determination. However, the protonation states—which are crucial for understanding the electronic structure—are not easy to infer even with the combination of spectroscopy and crystallography.
The structure from neutron diffraction study of the ferric state of the heme, determined at 2.4 Å resolution, showed a heavy water molecule (i.e., D
2O) bound at the start of the reaction (
Figure 9, left). Crystals were then treated with peroxide, known to induce formation of Compound
I. The treated crystals were cryo-cooled to trap the intermediate and studied with neutrons. The neutron structure, determined to 2.5 Å resolution, clearly shows the oxygen to be unprotonated, consistent with the H bonding structure of the active site (
Figure 9, right). The ferryl iron is then double bonded to an oxygen (Fe(IV)=O). The Trp 191 radical is protonated, so the radical species is a (protonated) π-cation radical [
41].
Another recent neutron crystallographic study focused on determining the oxidation and coordination state of the Compound
II intermediate in APX [
40]. APX catalyzes the peroxide-dependent oxidation of ascorbate by using both Compound
I and Compound
II intermediates. APX and Ccp are highly conserved and Ccp has served as a model system for mechanistic studies in heme enzyme catalysis. The experimental challenges of studying Compound
II are more easily overcome in APX compared to Ccp. In APX Compound
I appears as a ferryl heme and a porphyrin π-cation radical and is readily distinguishable from Compound
II, which contains only a ferryl species. This is different from Ccp where Compound
I is a ferryl heme and a Trp radical. Careful X-ray analysis and microspectrophotometry, along with neutron crystallography, revealed the heme ligand to be hydroxyl. At the resolution limits of the crystallographic studies, it is not justified to assert that the refined distance (1.88 Å) corresponds to a hydroxyl bound to the heme. However, analysis of omit maps and lack of residual density led to the determination that Compound
II in APX is indeed Fe(IV)-OH [
40]. Together these results offer new insights on the mechanism of heme peroxidases, with implications for the entire family of enzymes. Importantly, for the first time, they offer unambiguous insights into the coordination and protonation state around the heme groups where other techniques have been unsuitable.
5.2. Urate Oxidase
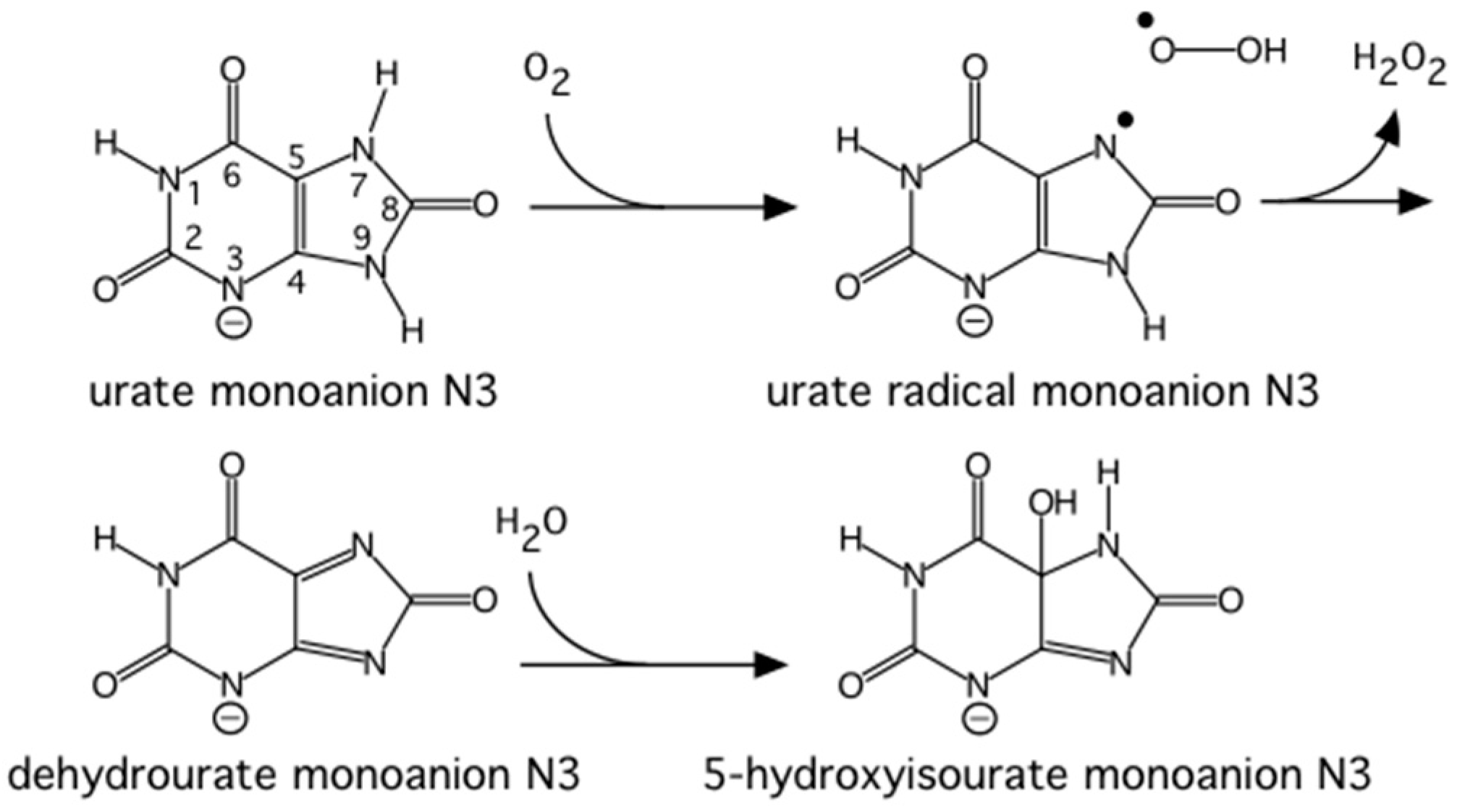
Urate oxidase (EC 1.7.3.3) is an enzyme involved in purine metabolism. It is a co-factorless oxidase that catalyzes the reaction between uric acid and molecular oxygen to form 5-hydroxy-isourate (5-HIU), which in turn is hydrolyzed to allantoin.
The reaction between oxygen and a urate dianion is spontaneous in solution, but the predominant protonation state of uric acid in solution is a monoanion with N3 deprotonated (
Figure 10) [
42]. Therefore the principal role of the enzyme is to deprotonate the urate substrate in proximity of the oxygen. Numerous X-ray structures were determined both with inhibitors that mimic urate as well as oxygen at atomic resolution. The results were difficult to reconcile with the biochemical data, since the site of the next deprotonation in solution, N7, is H bonded to a main chain amide and no feasible general bases can be found in the vicinity. The neutron structure at 2.35 Å resolution (
Figure 11) showed that in fact the species in the active site was the enol tautomer of urate, 8-hydroxyxanthine.
It was shown by quantum chemical calculations that despite the enol tautomer being unstable in solution, the active site can stabilize it. Interestingly, the O-H was clearly pointing away from the purine plane, even though there was no H bonding partner observed to explain this behavior. There was however enough empty space for a water molecule, even though none were observed in the X-ray maps. Further quantum mechanics/molecular mechanics calculations were performed to understand this. It was not possible to obtain good geometry consistent with the high-resolution X-ray structure unless a water molecule was placed in proper H bonding geometry with respect to the O-H. This illustrates the synergies between crystallography and computational chemistry where computational methods can predict or fill in details when crystal structures give incomplete information. A water molecule was indeed observed in this position in an X-ray structure from anaerobically grown crystals [
43].
This result allowed a more plausible mechanism to be postulated where the deprotonation occurs on the oxygen instead of the nitrogen. One of the unsolved questions in the mechanism was always how an H atom from the N7 position can reach the oxygen site above the ring plane. A surprising observation in the neutron structure was that an H bonded network extended from N7 all the way to the oxygen site (that contains Cl
− in this structure), and the H atoms were disordered (
Figure 12).
Nevertheless the X-ray structure showed that the oxygen atoms were well ordered. This is indicative of a proton relay chain that can easily pass the proton through three water molecules, a Lys and a Ser side chain. The existence of such a relay chain also explains how the oxygen site can be provided with a second proton to eventually form H2O2.
5.3. β-Lactamase
Bacteria have developed antibiotic resistance to β–lactam antibiotics by expressing an enzyme, β-lactamase, which breaks the amide bond within the lactam ring and renders the compound ineffective. This class of antibiotics inhibits cell wall synthesis by binding to bacterial penicillin-binding proteins (PBPs). The β-lactamases are divided into classes A through D. All but class B use a serine-reactive mechanism. The class A CTX-M extended-spectrum-β-lactamase (ESBL) has high hydrolytic activity against first, second, and third generation cephalosporins and monobactams. The enzymes work by using a general base mechanism to break the amide bond within the β-lactam ring [
44]. One class A enzyme, CTX-M-44, was chosen for neutron studies to further investigate the role of active site Glu166, Ser70, and Lys73 in catalysis. CTX-M-44 is composed of 262 residues and has two conserved domains, with the active site located at the interface between the domains. It has been proposed that Glu166 acts as a general catalytic base in the acylation part of the reaction through deprotonation of Ser70. Ser70 then attacks the carbonyl carbon of the β-lactam ring. Glu166 is also thought to be involved in the second part de-acylation part of the reaction by activating a hydrolytic water molecule. Interestingly, the CTX-M-44 Glu166Ala (E166A) mutant is still able to degrade β-lactams, but with the rates several orders of magnitude slower [
45]. This suggests there is more than one way to degrade β-lactams, including some that have been proposed to use Lys73 [
46]. Neutron studies were conducted on the E166A mutant as it degrades the β-lactam slow enough to allow crystallographic investigation of the acyl-intermediate. Studies were done on both the ligand-free enzyme and in complex with cefotaxime. The data on the complex revealed that upon inhibitor binding, an oxyanion hole is formed involving the backbone amides of Ser237 and Ser70 with stabilizing H bonds to the carbonyl oxygen of cefotaxime. This interaction serves to stabilize the negative charge that is formed in the tetrahedral intermediates during the acylation and deacylation stages of hydrolysis.
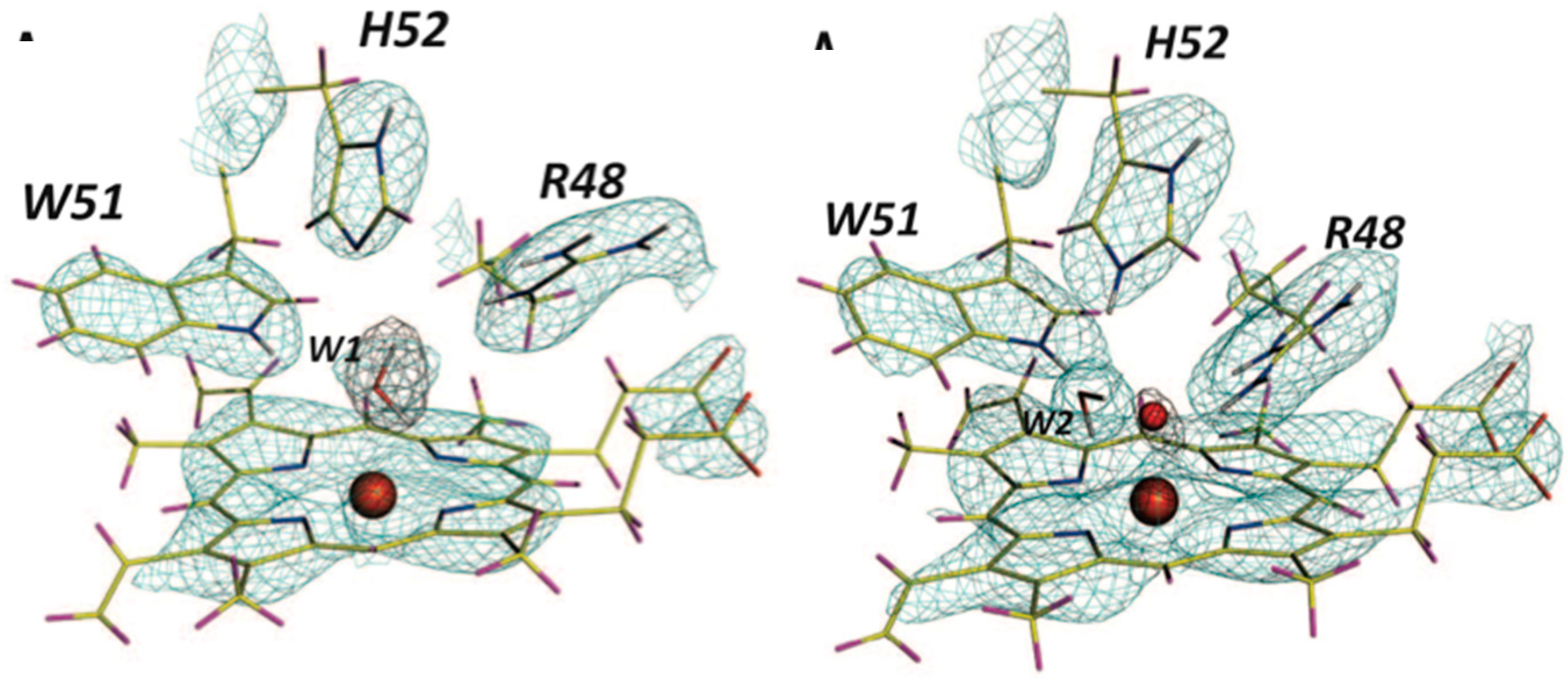
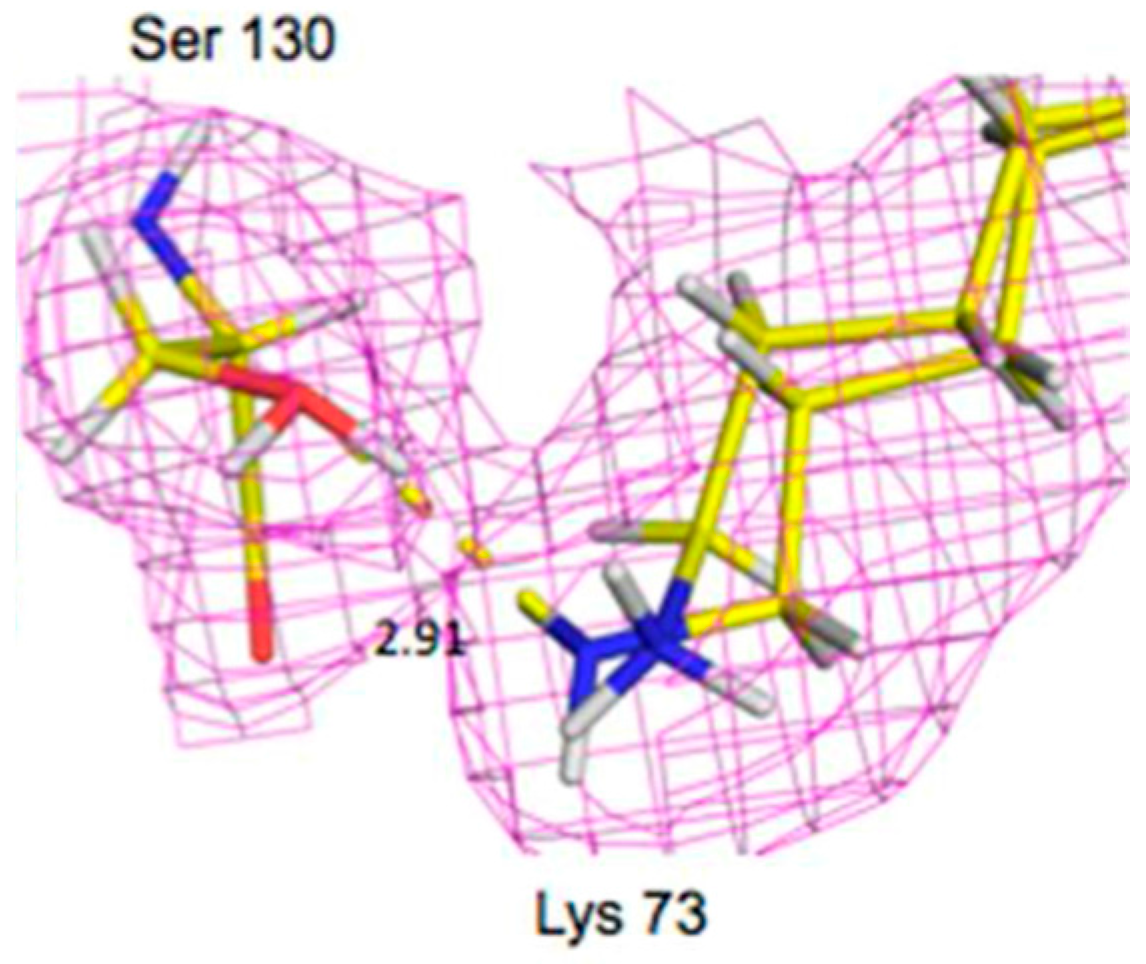
Lys73 is highly conserved in class A β-lactamases and has been thought to act as general base, in competition with Glu166, by abstracting a proton from Ser70. The ability of E166A mutant to still produce the acyl-enzyme complexes, which supports the idea that Lys73 is involved, albeit less effectively than Glu166. In the ligand-free neutron structure, it is seen that Lys73 is charged (-D
3+) and occupies a single conformation that is identical to that observed in the form where Glu166 is present. In the cefotaxime-complex structures however, Lys73 is observed in two alternative conformations that refine to near equal occupancy, presumably due to different charged states (
Figure 13). These observations together support a role for Ly73 in proton transfer and represents a viable alternative pathway when Glu166 is mutated [
45].
5.4. Diisopropyl Fluorophosphatase (DFPase)
The enzyme diisopropyl fluorophosphatase (DFPase; 314 residues, 35 kDa), found in the Mediterranean squid
Loligo vulgaris, is a calcium-dependent enzyme that can hydrolyze a variety of toxic organophosphorus compounds through hydrolysis of a P-F bond. The compounds include the nerve agents Sarin, Soman, and cyclosarin, and the pesticide diisopropyl fluorophosphate (DFP). These agents act as irreversible inhibitors of acetylcholinesterase and serine proteases. The neutron structure of DFPase, together with a variety of high-resolution X-ray structures of wild-type and site-directed mutants, and enzymology have greatly clarified the mechanism [
47,
48].
Single- and multiple-turnover studies utilizing an
18O substrate, together with mass spectroscopy, showed a
16O-containing product under single-turnover conditions, and incorporation of
18O under multiple turnover conditions. After multiple turnovers, the enzyme was digested to determine where
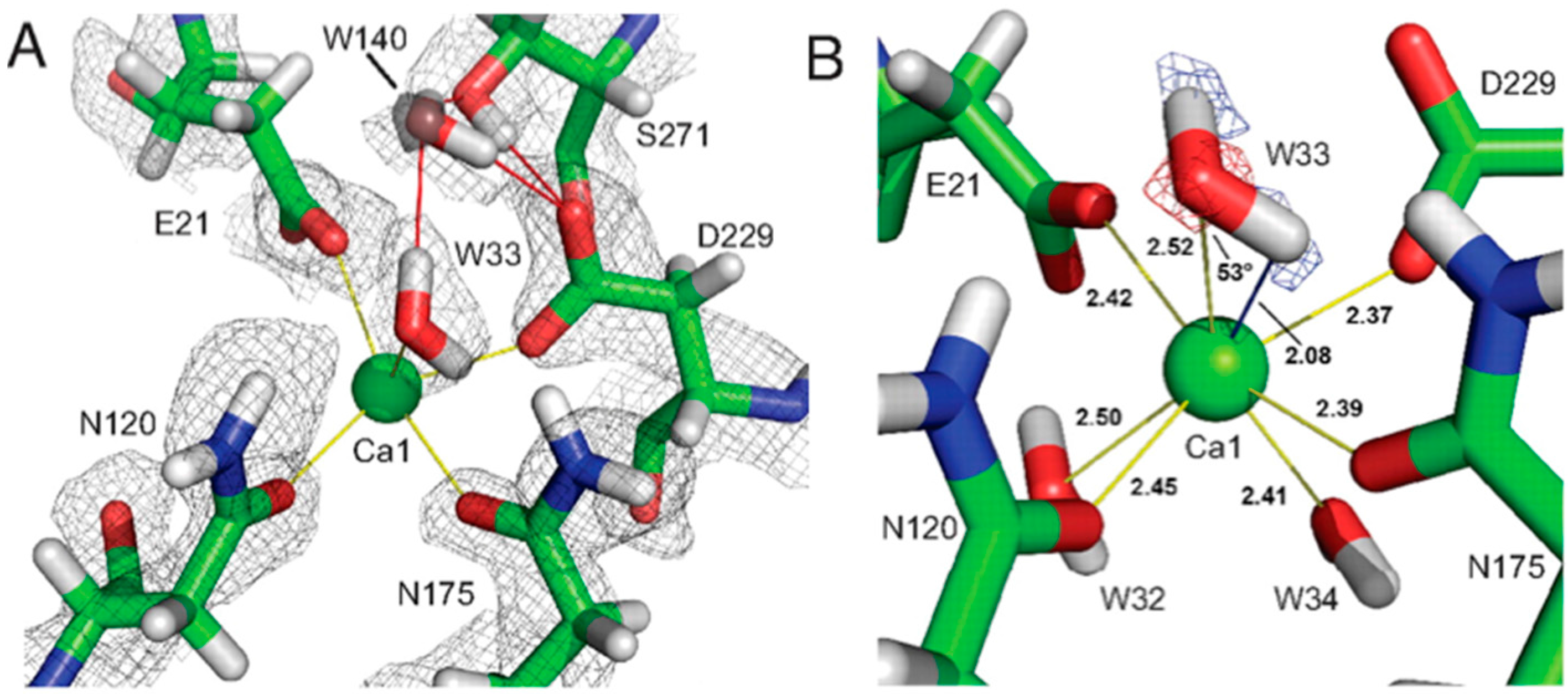
18O had been incorporated and results showed that the proteolytic peptide in question contained the calcium-coordinating residue Asp229. These labeling experiments demonstrated that the reaction proceeded through a direct attack of an amino acid on the substrate, and not through a metal-activated water as previously thought. In the neutron structure, Asp229 was found to be deprotonated, consistent with the proposed mechanism (
Figure 14A) [
49].
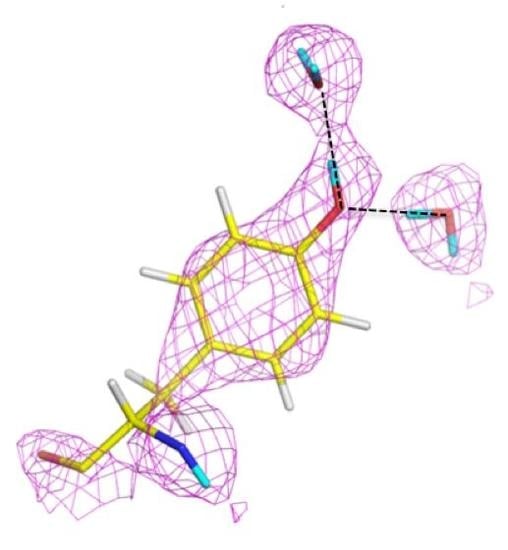
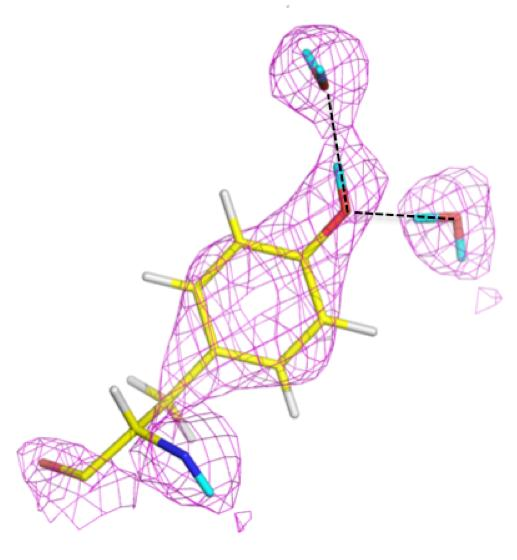
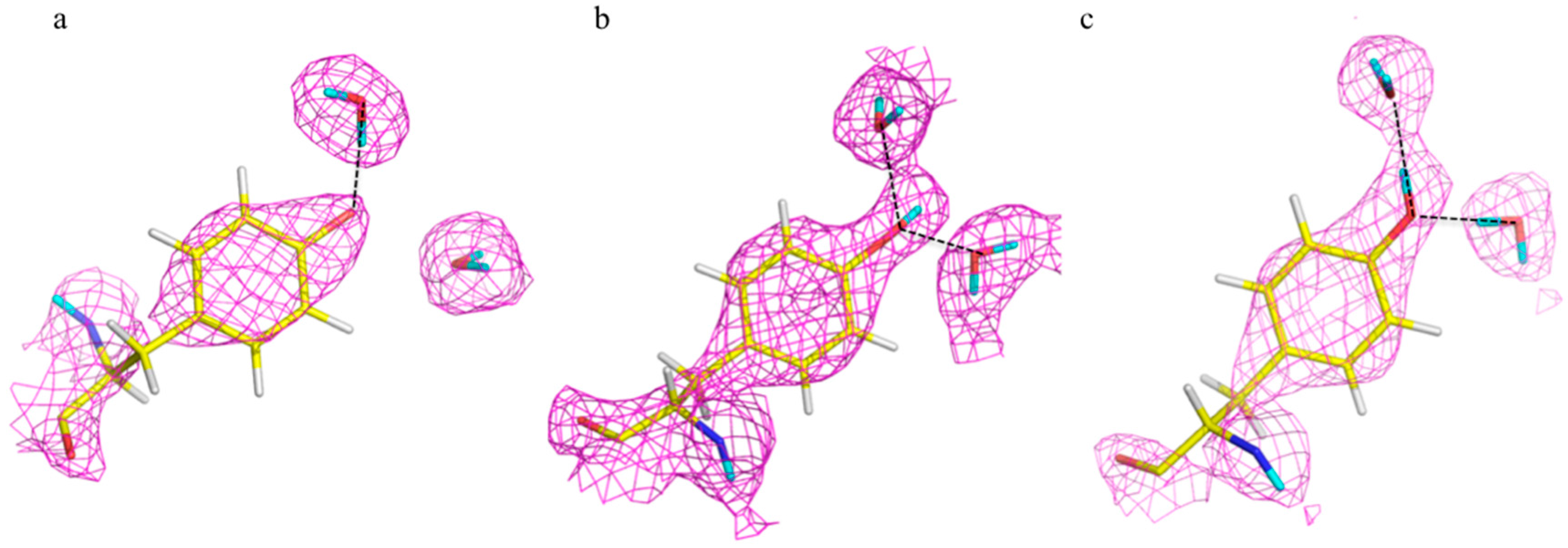
The catalytic calcium ion is involved in 7-fold coordination, with a solvent molecule bound in the active site pocket. While X-ray structures were not able to discern the species of the solvent molecule, the neutron structure revealed it to be a water molecule, and not a hydroxyl, ascertained by the elongated nuclear density and F
o − F
c omit maps (
Figure 14B) [
47]. This unambiguously showed that the mechanism did not involve direct water activation by the calcium ion, as had been previously suggested by other investigators, but was consistent with the Asp229 in the role as nucleophile. Of note was the orientation of the bound water (D
2O) molecule in relation to the position of the catalytic calcium ion, with a very close distance (2.08 Å) between one of the D atoms of the water and the calcium ion, and a Ca-O-D angle of 53 degrees.
5.5. Fungal Lytic Polysaccharide Monooxygenase
Fungal lytic polysaccharide monooxygenases (LPMO) are Cu-containing metalloenzymes involved with the degradation of polysaccharides through the insertion of one O into the glycosidic bond carbon, leading to chain cleavage. There is significant interest in the mechanism of these enzymes as there is potential application in cellulose degradation for biofuel production. The general mechanism starts with a single electron reducing the resting state Cu(II) to Cu(I). Cu(I) has a high affinity for molecular oxygen (O
2). Oxygen binding to Cu(I) quickly regenerates Cu(II) through oxidation with the formation of Cu-superoxide. From this intermediate there are multiple possible pathways that may involve superoxide, hydroperoxyl, or oxyl that are responsible for abstracting an H atom from the glycosidic carbon. In the absence of substrate the superoxide species can leave Cu(II) and generate peroxide. There are many uncertainties in how these enzymes activate oxygen and the mechanism of peroxide production when no substrate is present [
50,
51].
Neurospora crassa LPMO is a 223 residue (23.3 kDa) enzyme with one Cu per monomer. High resolution X-ray crystallography and DFT calculations were supplemented with neutron crystallography in order to better understand mono-Cu activation of molecular oxygen and H2O2 production in the absence of substrate.
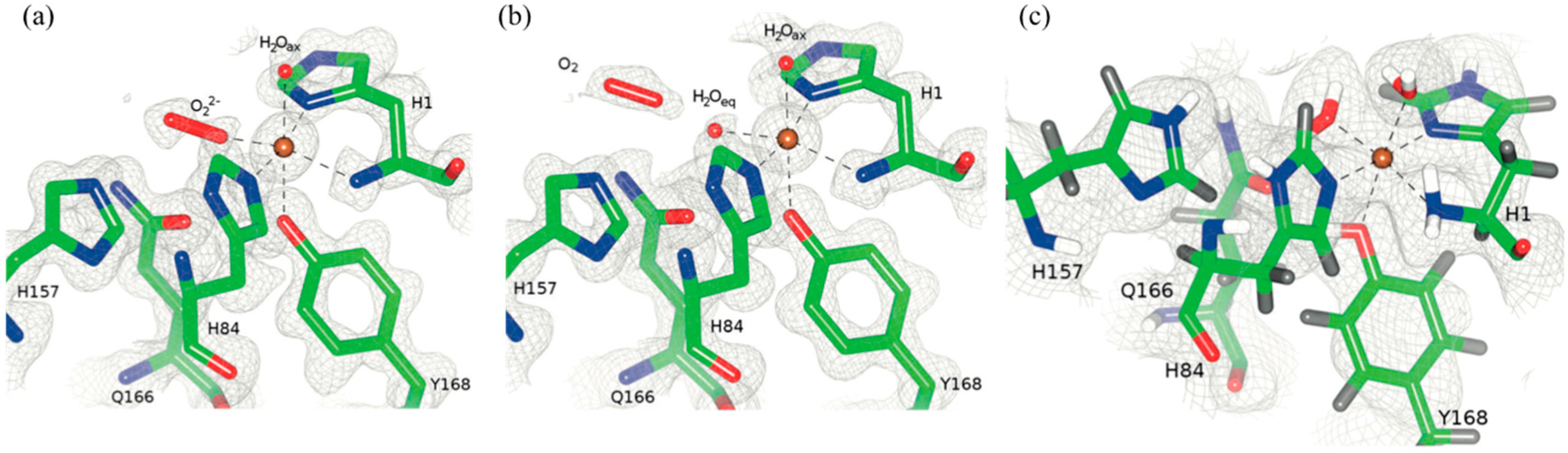
X-ray crystal structures of the resting Cu(II) state showed that Cu is coordinated by a conserved “His brace” made of the backbone amide and imidazole of His1 and the imidazole of His84. The hydroxyl group of Tyr168 is pointing towards with Cu site with the remaining axial and equatorial coordination positions occupied by two waters, H
2O
ax and H
2O
eq (
Figure 15) [
51].
The coordination sphere for Cu indicates the presence of Cu(II), despite the possibility of photoreduction upon X-ray exposure. Treating the crystals with ascorbate in the presence of atmospheric oxygen prior to freezing leads to reduction to Cu(I) and formation of the Cu(II)-dioxo complex. The crystal structure shows that H
2O
eq is displaced by a peroxo (O
22−) species in the equatorial position (bond length ~1.44 Å). This configuration is consistent with displacement by water to release peroxide in the absence of substrate. In the non-crystallographic symmetry related monomer molecular oxygen was instead observed, adjacent to H
2O
ax. The molecular oxygen in this position was proposed to be in a “pre-bound” position and not yet activated as was seen in the adjacent molecule [
51].
The neutron crystal structure of the resting state showed a nearby His157 to be neutral. At lower pH this His157 would be protonation and positioned to coordinate molecular oxygen in this “pre-bound” location. This implies that His157 could possibly promote oxygen binding and activation. DFT calculations were performed on different His157 protonation states and conformations and showed a strong thermodynamically favored addition of oxygen when His157 is charged. Overall these studies revealed additional details about oxygen binding and activation not considered before and the observation of “pre-bound” molecular oxygen indicate a mechanism for H
2O
2 formation in the absence of substrate [
51].
7. Conclusions
This review highlights specific areas of structural enzymology and general protein biochemistry where neutrons have made a large impact. Due to their unique nature and interactions with biological materials, neutrons have the ability to give high resolution, detailed, unambiguous information on H atoms and their all-important interaction, the H bond. Neutron crystallography has provided the opportunity to “see” into the heart of the enzyme active sites or ligand binding regions, and often brings new insights. Neutrons have contributed direct evidence of amino acid involvement in catalysis or drug binding, how waters are organized and oriented, and even how some waters are activated to perform catalysis. Arguably the most important thing neutrons can show us is the H bond and how it ties the protein, solvent, and/or ligand together. As illustrated in this review, neutron crystallography complements results from NMR spectroscopy, X-ray crystallography, modeling, and computational chemistry. In recent years, significant advances have been made in the fields of DFT calculations and
1H-NMR spectroscopy for the study of H bonds [
62,
63]. It can be expected that theoretical and experimental information gained from all these methods will impact how protein structures are refined and analyzed.
Neutron crystallography is under-utilized by the general structural biology community due to perceived issues with sample preparation and acquiring beam time. With current molecular biology tools and equipment for protein production, purification and crystallization, there are now far fewer technical limitations to preparing sufficient material to grow large enough crystals for neutron diffraction experiments. Current instruments routinely collect high quality data from crystals that are 0.1–1 mm3 and smaller in volume, a range that is readily attainable for many systems. The low number of instruments and highly competitive nature of being awarded beam time, especially compared to macromolecular X-ray beamlines, represents the true bottleneck for current and new users. With the advent of the new European Spallation Source (Lund, Sweden), and the planned Macromolecular Diffractometer NMX there, data collection and sample requirements will dramatically improve for the user community.




















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}