
Exploring the Pivotal Role of the CK2 Hinge Region Sub-Pocket in Binding with Tricyclic Quinolone Analogues by Computational Analysis

 ,
,
Abstract
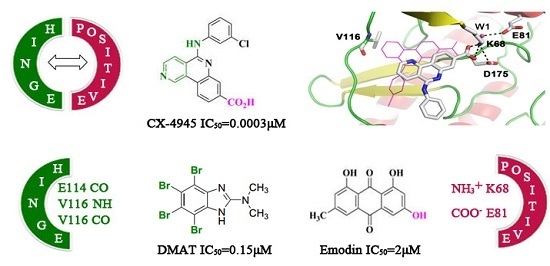
:
1. Introduction
2. Results and Discussion
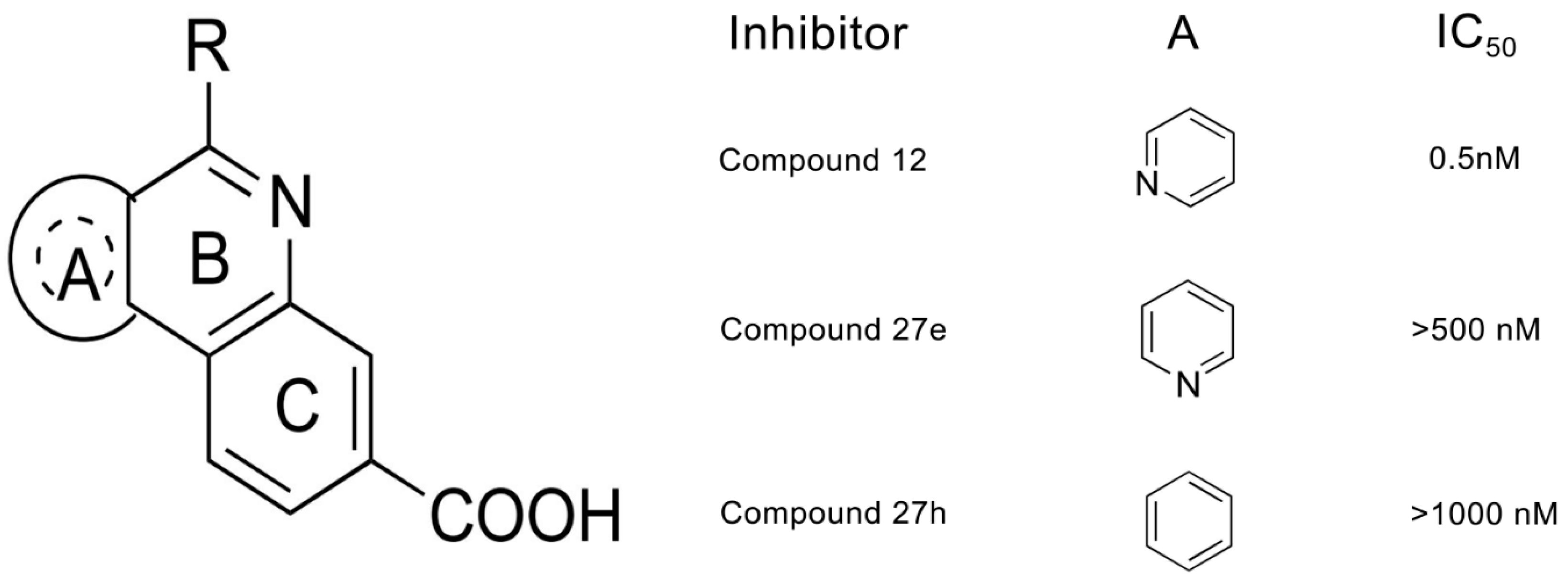
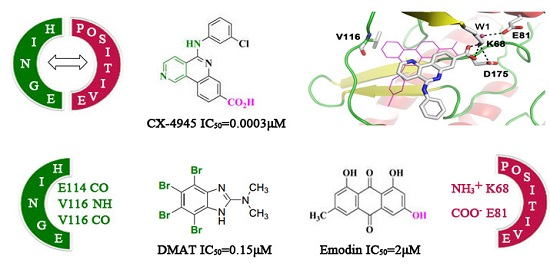
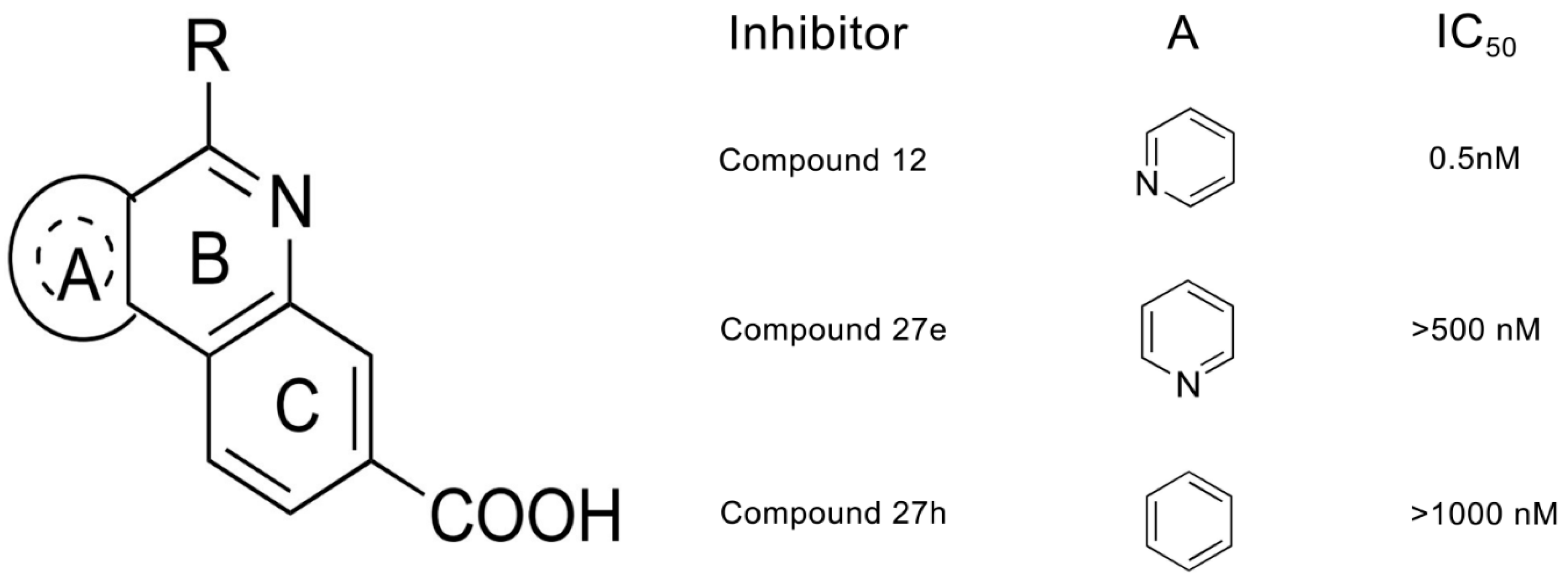
2.1. Molecular Docking Analysis
2.2. Molecular Dynamics Simulationstudies
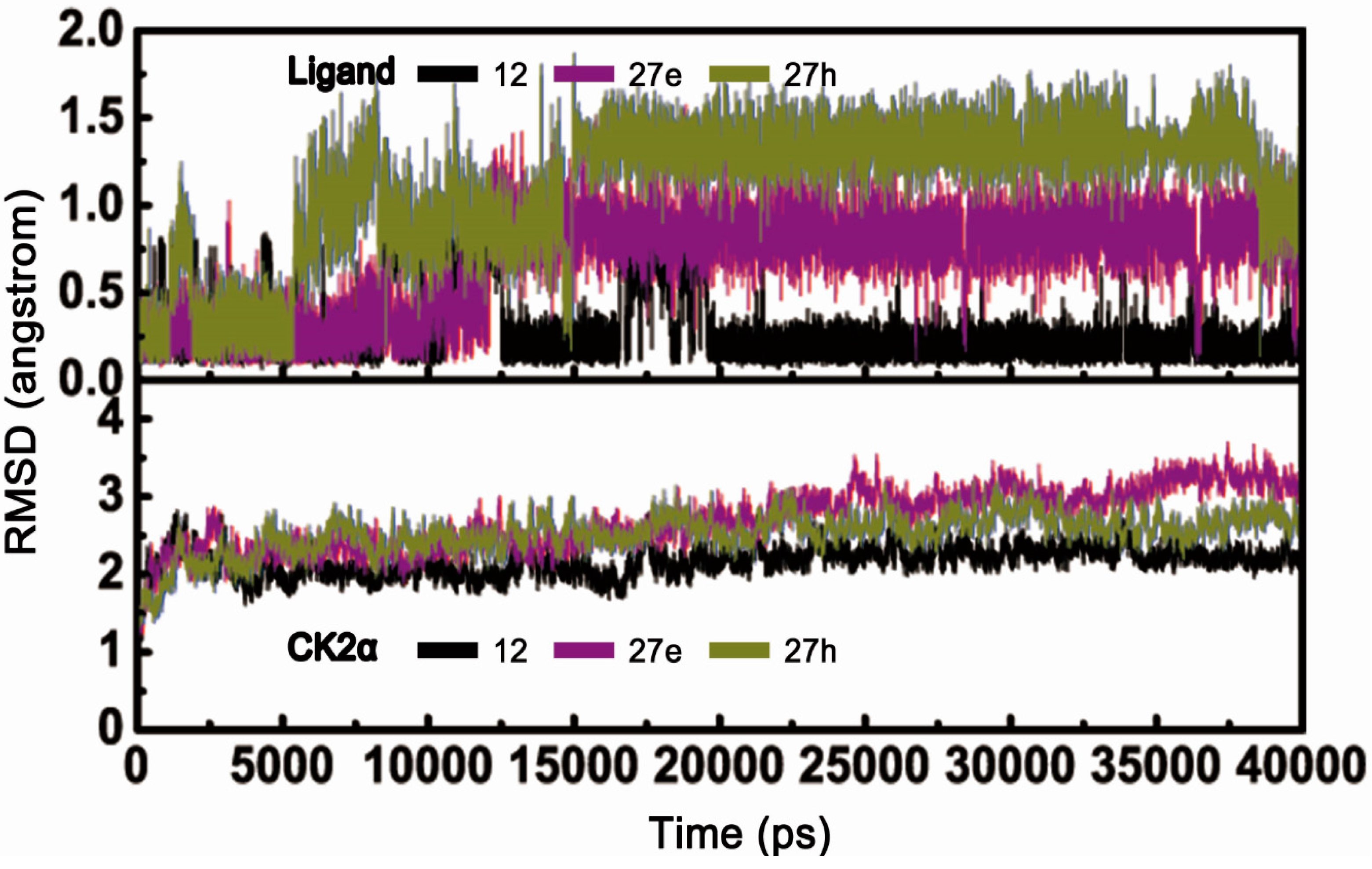
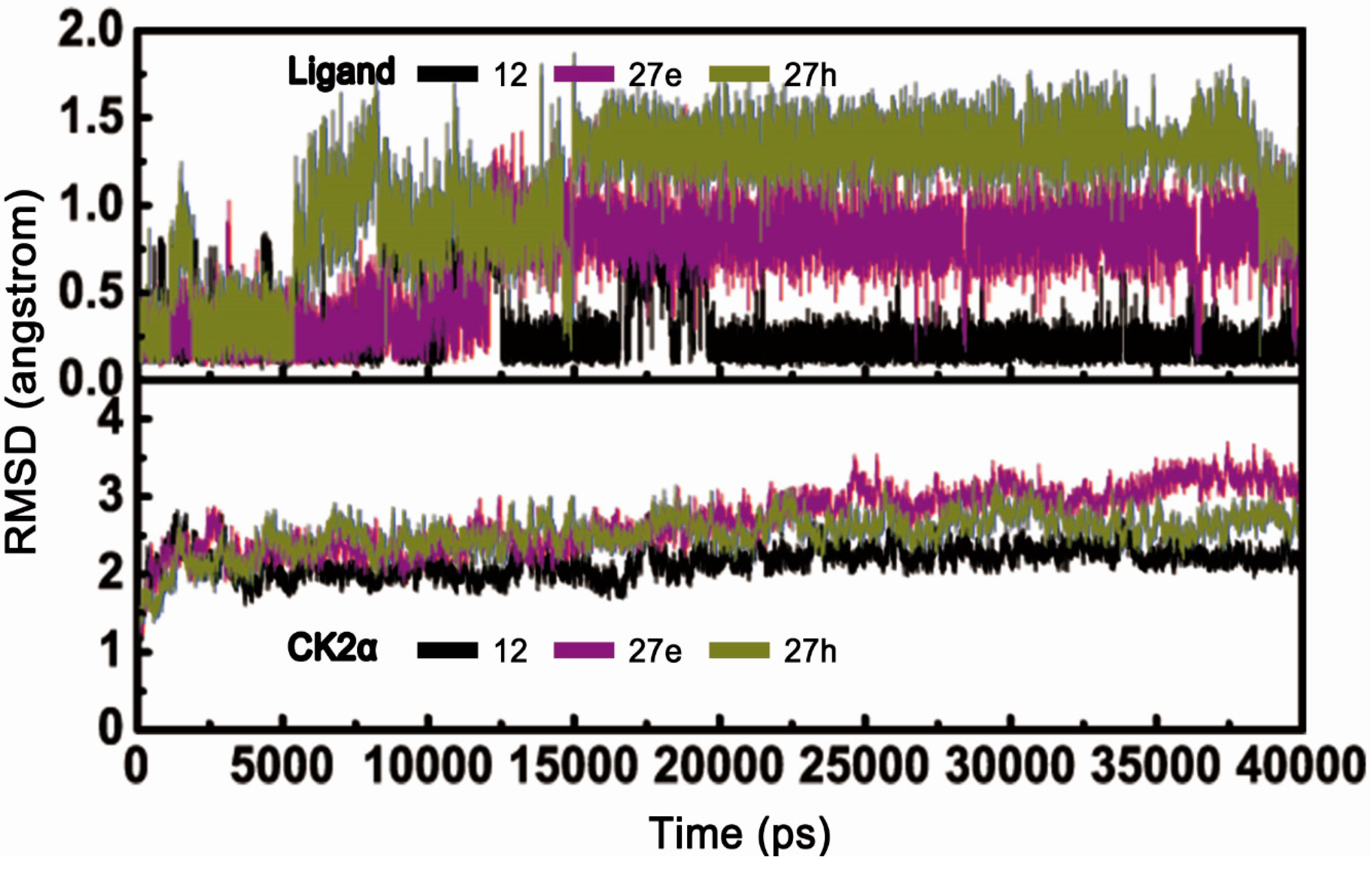
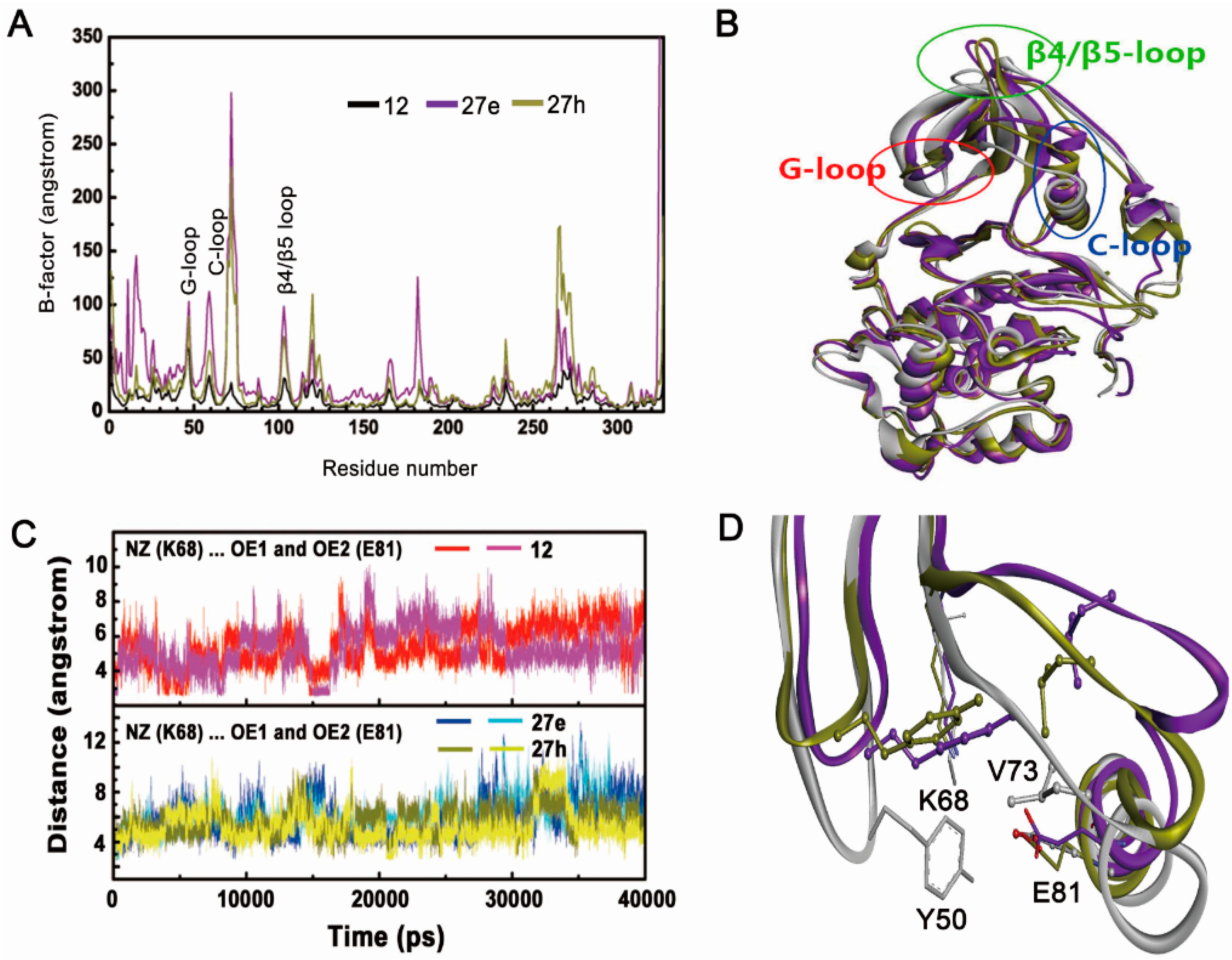
2.2.1. Overall Features of Dynamic Behaviors
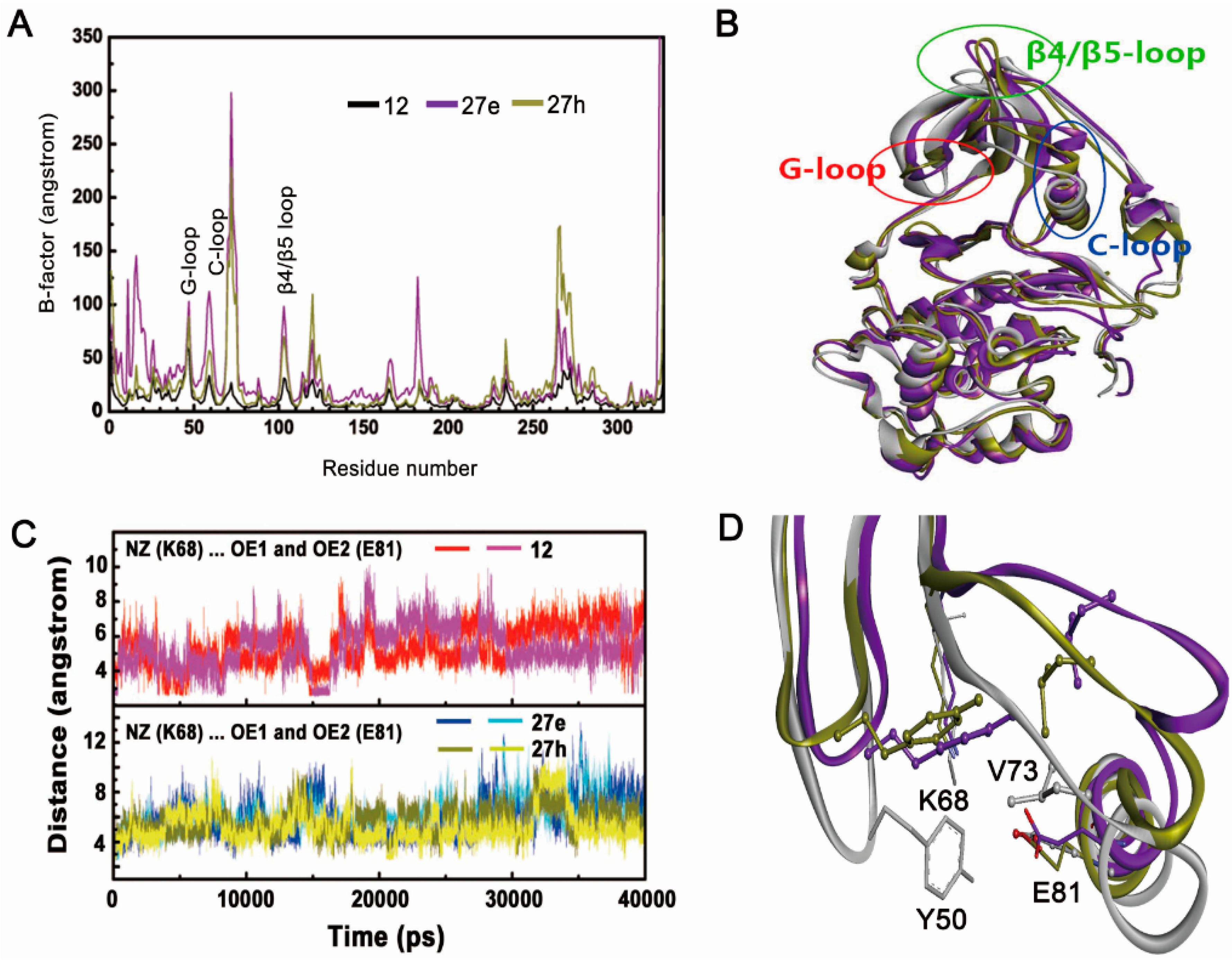
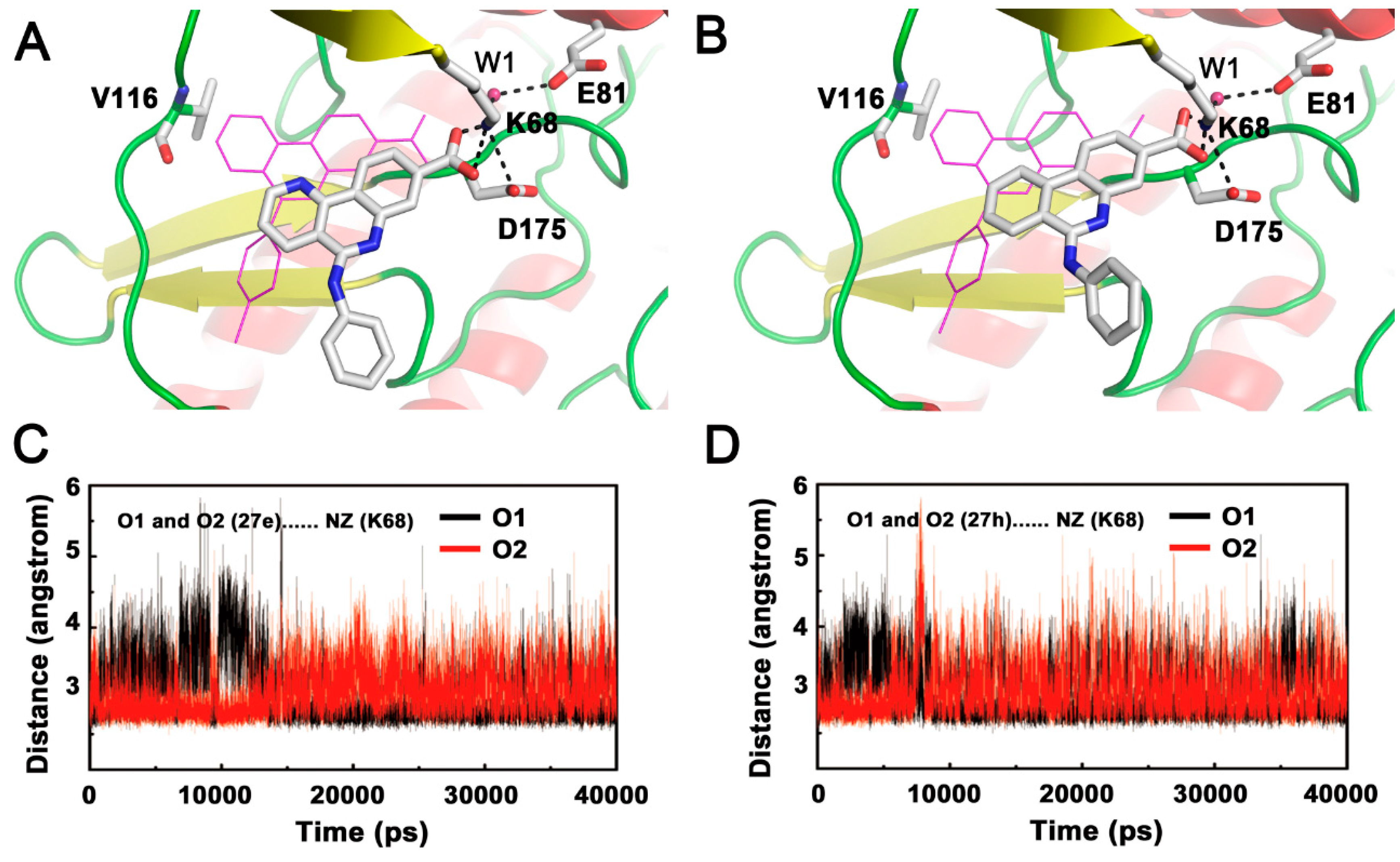
2.2.2. Inappropriate Recognition Mechanisms
2.3. Energy Examination
3. Materials and Methods
3.1. Ligands Preparation and Molecular Docking
3.2. Molecular Dynamics Simulations
3.3. MM/PBSA and MM/GBSA Calculations
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Ruzzene, M.; Pinna, L.A. Addiction to protein kinase CK2: A common denominator of diverse cancer cells? Biochim. Biophys. Acta 2010, 1804, 499–504. [Google Scholar] [CrossRef] [PubMed]
- Trembley, J.H.; Wang, G.; Unger, G.; Slaton, J.; Ahmed, K. Protein kinase CK2 in health and disease: CK2: A key player in cancer biology. Cell. Mol. Life Sci. 2009, 66, 1858–1867. [Google Scholar] [CrossRef] [PubMed]
- Meggio, F.; Pinna, L.A. One-Thousand-and-One substrates of protein kinase CK2? FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2003, 17, 349–368. [Google Scholar] [CrossRef] [PubMed]
- Filhol, O.; Martiel, J.L.; Cochet, C. Protein kinase CK2: A new view of an old molecular complex. EMBO Rep. 2004, 5, 351–355. [Google Scholar] [CrossRef] [PubMed]
- Cozza, G.; Pinna, L.A. Casein kinases as potential therapeutic targets. Expert Opin. Ther. Targets 2016, 20, 319–340. [Google Scholar] [CrossRef] [PubMed]
- Guerra, B.; Issinger, O.G. Protein kinase CK2 in human diseases. Curr. Med. Chem. 2008, 15, 1870–1886. [Google Scholar] [CrossRef] [PubMed]
- Ortega, C.E.; Seidner, Y.; Dominguez, I. Mining CK2 in cancer. PLoS ONE 2014, 9, e115609. [Google Scholar]
- Filhol, O.; Giacosa, S.; Wallez, Y.; Cochet, C. Protein kinase CK2 in breast cancer: The CK2β regulatory subunit takes center stage in epithelial plasticity. Cell. Mol. Life Sci. 2015, 72, 3305–3322. [Google Scholar] [CrossRef] [PubMed]
- Swider, R.; Maslyk, M.; Martin-Santamaria, S.; Ramos, A.; de Pascual-Teresa, B. Multisite-Directed inhibitors of protein kinase CK2: New challenges. Mol. Cell. Biochem. 2011, 356, 117–119. [Google Scholar] [CrossRef] [PubMed]
- Swider, R.; Maslyk, M.; Zapico, J.M.; Coderch, C.; Panchuk, R.; Skorokhyd, N.; Schnitzler, A.; Niefind, K.; de Pascual-Teresa, B.; Ramos, A. Synthesis, biological activity and structural study of new benzotriazole-based protein kinase CK2 inhibitors. Rsc Adv. 2015, 5, 72482–72494. [Google Scholar] [CrossRef]
- Cozza, G.; Bortolato, A.; Moro, S. How druggable is protein kinase CK2? Med. Res. Rev. 2010, 30, 419–462. [Google Scholar] [CrossRef] [PubMed]
- Battistutta, R. Protein kinase CK2 in health and disease: Structural bases of protein kinase CK2 inhibition. Cell. Mol. Life Sci. 2009, 66, 1868–1889. [Google Scholar] [CrossRef] [PubMed]
- Perez, D.I.; Gil, C.; Martinez, A. Protein kinases CK1 and CK2 as new targets for neurodegenerative diseases. Med. Res. Rev. 2011, 31, 924–954. [Google Scholar] [CrossRef] [PubMed]
- Pagano, M.A.; Bain, J.; Kazimierczuk, Z.; Sarno, S.; Ruzzene, M.; Di Maira, G.; Elliott, M.; Orzeszko, A.; Cozza, G.; Meggio, F.; et al. The selectivity of inhibitors of protein kinase CK2: An update. Biochem. J. 2008, 415, 353–365. [Google Scholar] [CrossRef] [PubMed]
- Sarno, S.; Papinutto, E.; Franchin, C.; Bain, J.; Elliott, M.; Meggio, F.; Kazimierczuk, Z.; Orzeszko, A.; Zanotti, G.; Battistutta, R.; et al. ATP site-directed inhibitors of protein kinase CK2: An update. Curr. Top. Med. Chem. 2011, 11, 1340–1351. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Luan, Y.; Qi, X.; Li, M.; Gong, L.; Xue, X.; Wu, X.; Wu, Y.; Chen, M.; Xing, G.; et al. Emodin triggers DNA double-strand breaks by stabilizing topoisomerase II-DNA cleavage complexes and by inhibiting ATP hydrolysis of topoisomerase II. Toxicol. Sci. 2010, 118, 435–443. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui-Jain, A.; Drygin, D.; Streiner, N.; Chua, P.; Pierre, F.; O’Brien, S.E.; Bliesath, J.; Omori, M.; Huser, N.; Ho, C.; et al. CX-4945, an orally bioavailable selective inhibitor of protein kinase CK2, inhibits prosurvival and angiogenic signaling and exhibits antitumor efficacy. Cancer Res. 2010, 70, 10288–10298. [Google Scholar] [CrossRef] [PubMed]
- Senhwa Biosciences, Inc. Senhwa Biosciences CX-4945 Granted Orphan Drug Designation by the US FDA in Cholangiocarcinoma. Available online: http://www.prnewswire.com/ news-releases/senhwa-biosciences-cx-4945-granted-orphan-drug-designation-by-the-us-fda-in-cholangiocarcinoma-300385278.html (accessed on 19 May 2017).
- Zanin, S.; Borgo, C.; Girardi, C.; O’Brien, S.E.; Miyata, Y.; Pinna, L.A.; Donella-Deana, A.; Ruzzene, M. Effects of the CK2 inhibitors CX-4945 and CX-5011 on drug-resistant cells. PLoS ONE 2012, 7, e49193. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui-Jain, A.; Bliesath, J.; Macalino, D.; Omori, M.; Huser, N.; Streiner, N.; Ho, C.B.; Anderes, K.; Proffitt, C.; O’Brien, S.E.; et al. CK2 inhibitor CX-4945 suppresses DNA repair response triggered by DNA-targeted anticancer drugs and augments efficacy: Mechanistic rationale for drug combination therapy. Mol. Cancer Ther. 2012, 11, 994–1005. [Google Scholar] [CrossRef] [PubMed]
- Chilin, A.; Battistutta, R.; Bortolato, A.; Cozza, G.; Zanatta, S.; Poletto, G.; Mazzorana, M.; Zagotto, G.; Uriarte, E.; Guiotto, A.; et al. Coumarin as attractive casein kinase 2 (CK2) inhibitor scaffold: An integrate approach to elucidate the putative binding motif and explain structure-activity relationships. J. Med. Chem. 2008, 51, 752–759. [Google Scholar] [CrossRef] [PubMed]
- Battistutta, R.; Mazzorana, M.; Sarno, S.; Kazimierczuk, Z.; Zanotti, G.; Pinna, L.A. Inspecting the structure-activity relationship of protein kinase CK2 inhibitors derived from tetrabromo-benzimidazole. Chem. Boil. 2005, 12, 1211–1219. [Google Scholar] [CrossRef] [PubMed]
- Haddach, M.; Pierre, F.; Regan, C.F.; Borsan, C.; Michaux, J.; Stefan, E.; Kerdoncuff, P.; Schwaebe, M.K.; Chua, P.C.; Siddiqui-Jain, A.; et al. Synthesis and SAR of inhibitors of protein kinase CK2: Novel tricyclic quinoline analogs. Bioorg. Med. Chem. Lett. 2012, 22, 45–48. [Google Scholar] [CrossRef] [PubMed]
- Battistutta, R.; Cozza, G.; Pierre, F.; Papinutto, E.; Lolli, G.; Sarno, S.; O’Brien, S.E.; Siddiqui-Jain, A.; Haddach, M.; Anderes, K.; et al. Unprecedented selectivity and structural determinants of a new class of protein kinase CK2 inhibitors in clinical trials for the treatment of cancer. Biochemistry 2011, 50, 8478–8488. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zhang, N.; Zhong, R.G. Exploring the crucial structural elements required for tricyclic quinoline analogs as protein kinase CK2 inhibitors by a combined computational analysis. Med. Chem. Res. 2013, 22, 4410–4422. [Google Scholar] [CrossRef]
- Zhou, Y.; Li, X.; Zhang, N.; Zhong, R. Structural basis for low-affinity binding of non-R2 carboxylate-substituted tricyclic quinoline analogs to CK2alpha: Comparative molecular dynamics simulation studies. Chem. Biol. Drug Des. 2015, 85, 189–200. [Google Scholar] [CrossRef] [PubMed]
- Pierre, F.; Chua, P.C.; O’Brien, S.E.; Siddiqui-Jain, A.; Bourbon, P.; Haddach, M.; Michaux, J.; Nagasawa, J.; Schwaebe, M.K.; Stefan, E.; et al. Discovery and SAR of 5-(3-chlorophenylamino)benzo[c][2,6]naphthyridine-8-carboxylic acid (CX-4945), the first clinical stage inhibitor of protein kinase CK2 for the treatment of cancer. J. Med. Chem. 2011, 54, 635–654. [Google Scholar] [CrossRef] [PubMed]
- Borhani, D.W.; Shaw, D.E. The future of molecular dynamics simulations in drug discovery. J. Comput.-Aided Mol. Des. 2012, 26, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Durrant, J.D.; McCammon, J.A. Molecular dynamics simulations and drug discovery. BMC Biol. 2011, 9, 71. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Du, Y.; Liu, X.; Shen, Q.C.; Huang, A.L.; Zheng, M.Y.; Luo, X.M.; Jiang, H.L. Binding sensitivity of adefovir to the polymerase from different genotypes of HBV: Molecular modeling, docking and dynamics simulation studies. Acta Pharmacol. Sin. 2013, 34, 319–328. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Zhong, R. Structural basis for decreased affinity of Emodin binding to Val66-mutated human CK2α as determined by molecular dynamics. J. Mol. Model. 2010, 16, 771–780. [Google Scholar] [CrossRef] [PubMed]
- Karplus, M.; McCammon, J.A. Molecular dynamics simulations of biomolecules. Nat. Struct. Biol. 2002, 9, 646–652. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.X.; Wu, T.X.; Jiang, Y.J.; Zou, J.W.; Zhuang, S.L.; Mao, X.; Yu, Q.S. Role of phosphorylated Thr-197 in the catalytic subunit of cAMP-dependent protein kinase. J. Mol. Struct. THEOCHEM 2007, 805, 9–15. [Google Scholar] [CrossRef]
- Welburn, J.P.; Tucker, J.A.; Johnson, T.; Lindert, L.; Morgan, M.; Willis, A.; Noble, M.E.; Endicott, J.A. How tyrosine 15 phosphorylation inhibits the activity of cyclin-dependent kinase 2-cyclin A. J. Biol. Chem. 2007, 282, 3173–3181. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Jiang, Y.; Zou, J.; Yu, Q.; Zhao, W. Structural basis for the complete loss of GSK3β catalytic activity due to R96 mutation investigated by molecular dynamics study. Proteins 2009, 75, 671–681. [Google Scholar] [CrossRef] [PubMed]
- Papinutto, E.; Ranchio, A.; Lolli, G.; Pinna, L.A.; Battistutta, R. Structural and functional analysis of the flexible regions of the catalytic α-subunit of protein kinase CK2. J. Struct. Biol. 2012, 177, 382–391. [Google Scholar] [CrossRef] [PubMed]
- Nie, Z.; Perretta, C.; Erickson, P.; Margosiak, S.; Almassy, R.; Lu, J.; Averill, A.; Yager, K.M.; Chu, S. Structure-Based design, synthesis, and study of pyrazolo[1,5-a][1,3,5]triazine derivatives as potent inhibitors of protein kinase CK2. Bioorg. Med. Chem. Lett. 2007, 17, 4191–4195. [Google Scholar] [CrossRef] [PubMed]
- Dowling, J.E.; Chuaqui, C.; Pontz, T.W.; Lyne, P.D.; Larsen, N.A.; Block, M.H.; Chen, H.; Su, N.; Wu, A.; Russell, D.; et al. Potent and selective inhibitors of CK2 kinase identified through structure-guided hybridization. ACS Med. Chem. Lett. 2012, 3, 278–283. [Google Scholar] [CrossRef] [PubMed]
- Pagano, M.A.; Meggio, F.; Ruzzene, M.; Andrzejewska, M.; Kazimierczuk, Z.; Pinna, L.A. 2-Dimethylamino-4,5,6,7-tetrabromo-1H-benzimidazole: A novel powerful and selective inhibitor of protein kinase CK2. Biochem. Biophys. Res. Commun. 2004, 321, 1040–1044. [Google Scholar] [CrossRef] [PubMed]
- Yim, H.; Lee, Y.H.; Lee, C.H.; Lee, S.K. Emodin, an anthraquinone derivative isolated from the rhizomes of Rheum palmatum, selectively inhibits the activity of casein kinase II as a competitive inhibitor. Planta Med. 1999, 65, 9–13. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita, T.; Sekiguchi, Y.; Fukada, H.; Nakaniwa, T.; Tada, T.; Nakamura, S.; Kitaura, K.; Ohno, H.; Suzuki, Y.; Hirasawa, A.; et al. A detailed thermodynamic profile of cyclopentyl and isopropyl derivatives binding to CK2 kinase. Mol. Cell. Biochem. 2011, 356, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, I.; Murata, K.; Nagata, N.; Kurono, M.; Kinoshita, T.; Yasue, M.; Miyazaki, T.; Takei, Y.; Nakamura, S.; Sakurai, A.; et al. Identification of protein kinase CK2 inhibitors using solvent dipole ordering virtual screening. Eur. J. Med. Chem. 2015, 96, 396–404. [Google Scholar] [CrossRef] [PubMed]
- Ohno, H.; Minamiguchi, D.; Nakamura, S.; Shu, K.; Okazaki, S.; Honda, M.; Misu, R.; Moriwaki, H.; Nakanishi, S.; Oishi, S.; et al. Structure-Activity relationship study of 4-(thiazol-5-yl)benzoic acid derivatives as potent protein kinase CK2 inhibitors. Bioorg. Med. Chem. 2016, 24, 1136–1141. [Google Scholar] [CrossRef] [PubMed]
- Dowling, J.E.; Alimzhanov, M.; Bao, L.; Chuaqui, C.; Denz, C.R.; Jenkins, E.; Larsen, N.A.; Lyne, P.D.; Pontz, T.; Ye, Q.; et al. Potent and selective CK2 kinase inhibitors with effects on Wnt pathway signaling in vivo. ACS Med. Chem. Lett. 2016, 7, 300–305. [Google Scholar] [CrossRef] [PubMed]
- Ostrynska, O.V.; Balanda, A.O.; Bdzhola, V.G.; Golub, A.G.; Kotey, I.M.; Kukharenko, O.P.; Gryshchenko, A.A.; Briukhovetska, N.V.; Yarmoluk, S.M. Design and synthesis of novel protein kinase CK2 inhibitors on the base of 4-aminothieno[2,3-d]pyrimidines. Eur. J. Med. Chem. 2016, 115, 148–160. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Chen, W.J.; Zhou, Y.; Zhao, H.; Zhong, R.G. Rational design of coumarin derivatives as CK2 inhibitors by improving the interaction with the hinge region. Mol. Inform. 2016, 35, 15–18. [Google Scholar] [CrossRef] [PubMed]
- Clark, M.; Cramer, R.D.; Vanopdenbosch, N. Validation of the general-purpose Tripos 5.2 force-field. J. Comput. Chem. 1989, 10, 982–1012. [Google Scholar] [CrossRef]
- SYBYL, V. SYBYL, Version 8.1; Tripos Inc.: 1699 South Hanley Rd., St. Louis, MO, USA, 2008. Available online: https://www.certara.com/ (accessed on 19 May 2017).
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 1997, 267, 727–748. [Google Scholar] [CrossRef] [PubMed]
- Case, D.A.; Cheatham, T.E., 3rd; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M., Jr.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The Amber biomolecular simulation programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef] [PubMed]
- Cornell, W.D.; Cieplak, P.; Bayly, C.I.; Gould, I.R.; Merz, K.M.; Ferguson, D.M.; Spellmeyer, D.C.; Fox, T.; Caldwell, J.W.; Kollman, P.A. A second generation force-field for the simulation of proteins, nucleic-acids, and organic-molecules. J. Am. Chem. Soc. 1995, 117, 5179–5197. [Google Scholar] [CrossRef]
- Gaussian 03, R.C. Gaussian 03, Revision C.02; Gaussian Inc.: Wallingford, CT, USA, 2003. Available online: http://gaussian.com/ (accessed on 19 May 2017).
- Besler, B.H.; Merz, K.M.; Kollman, P.A. Atomic charges derived from semiempirical methods. J. Comput. Chem. 1990, 11, 431–439. [Google Scholar] [CrossRef]
- Fox, T.; Kollman, P.A. Application of the RESP methodology in the parametrization of organic solvents. J. Phys. Chem. B 1998, 102, 8070–8079. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Postma, J.P.M.; Vangunsteren, W.F.; Dinola, A.; Haak, J.R. Molecular-Dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef]
- Ryckaert, J.-P.; Ciccotti, G.; Berendsen, H.J.C. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N⋅log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Massova, I.; Kollman, P.A. Combined molecular mechanical and continuum solvent approach (MM-PBSA/GBSA) to predict ligand binding. Perspect. Drug Discov. Des. 2000, 18, 113–135. [Google Scholar] [CrossRef]
- Kollman, P.A.; Massova, I.; Reyes, C.; Kuhn, B.; Huo, S.; Chong, L.; Lee, M.; Lee, T.; Duan, Y.; Wang, W.; et al. Calculating structures and free energies of complex molecules: Combining molecular mechanics and continuum models. Acc. Chem. Res. 2000, 33, 889–897. [Google Scholar] [CrossRef] [PubMed]
- Sitkoff, D.; Sharp, K.A.; Honig, B. Accurate calculation of hydration free energies using macroscopic solvent models. J. Phys. Chem. 1994, 98, 1978–1988. [Google Scholar] [CrossRef]
- Wang, W.; Kollman, P.A. Computational study of protein specificity: The molecular basis of HIV-1 protease drug resistance. Proc. Natl. Acad. Sci. USA 2001, 98, 14937–14942. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Morin, P.; Wang, W.; Kollman, P.A. Use of MM-PBSA in reproducing the binding free energies to HIV-1 RT of TIBO derivatives and predicting the binding mode to HIV-1 RT of efavirenz by docking and MM-PBSA. J. Am. Chem. Soc. 2001, 123, 5221–5230. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Not Available. |




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Energy Term (kcal/mol) | 12 | 27e | 27h |
|---|---|---|---|
| ΔEele | −100.93 ± 4.0 | −112.33 ± 3.3 | −93.01 ± 4.0 |
| ΔEvdw | −30.55 ± 3.0 | −37.76 ± 3.1 | −36.28 ± 2.5 |
| ΔEgas a | −131.48 ± 6.0 | −150.10 ± 5.7 | −129.29 ± 3.4 |
| ΔGnonpolar | −4.84 ± 0.12 | −5.59 ± 0.20 | −5.20 ± 0.10 |
| ΔGpolar | 89.96 ± 3.9 | 110.84 ± 3.6 | 92.73 ± 5.0 |
| ΔGsol b | 85.12 ± 3.9 | 105.25 ± 2.2 | 87.52 ± 3.5 |
| ΔGele c | −10.97 ± 2.7 | −1.50 ± 3.1 | −0.28 ± 2.2 |
| ΔGbinding d | −46.36 ± 3.2 | −44.85 ± 3.1 | −41.76 ± 3.0 |
| ΔΔGbinding | 0 | 1.51 | 4.60 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, Y.; Zhang, N.; Tang, S.; Qi, X.; Zhao, L.; Zhong, R.; Peng, Y. Exploring the Pivotal Role of the CK2 Hinge Region Sub-Pocket in Binding with Tricyclic Quinolone Analogues by Computational Analysis. Molecules 2017, 22, 840. https://doi.org/10.3390/molecules22050840
Zhou Y, Zhang N, Tang S, Qi X, Zhao L, Zhong R, Peng Y. Exploring the Pivotal Role of the CK2 Hinge Region Sub-Pocket in Binding with Tricyclic Quinolone Analogues by Computational Analysis. Molecules. 2017; 22(5):840. https://doi.org/10.3390/molecules22050840
Chicago/Turabian StyleZhou, Yue, Na Zhang, Shan Tang, Xiaoqian Qi, Lijiao Zhao, Rugang Zhong, and Yongzhen Peng. 2017. "Exploring the Pivotal Role of the CK2 Hinge Region Sub-Pocket in Binding with Tricyclic Quinolone Analogues by Computational Analysis" Molecules 22, no. 5: 840. https://doi.org/10.3390/molecules22050840